

ABSTRACT
Neutrophils are short-lived, abundant peripheral blood leukocytes that provide a first line of defense against bacterial and fungal infections while also being a key part of the inflammatory response. Chemokines induce neutrophil recruitment to inflammatory sites, where neutrophils perform several diverse functions that are aimed at fighting infections. Neutrophil effector functions are tightly regulated processes that are governed by an array of intracellular signaling pathways and initiated by receptor-ligand binding events. Dysregulated neutrophil activation can result in excessive inflammation and host damage, as is evident in several autoimmune diseases. Rho family small GTPases and agonist-activated phosphoinositide 3-kinases (PI3Ks) represent 2 classes of key regulators of the highly specialized neutrophil. Here we review cross-talk between these important signaling intermediates in the context of neutrophil functions. We include PI3K-dependent activation of Rho family small GTPases and of their guanine nucleotide exchange factors and GTPase activating proteins, as well as Rho GTPase-dependent regulation of PI3K.
KEYWORDS: Cdc42; chemotaxis; FcγR; GPCR; NADPH oxidase; neutrophil; PI3K; PtdIns(3,4,5)P3; polarization; phagocytosis; RhoA; Rac
Introduction
Neutrophils are the most abundant peripheral blood leukocytes in humans. These terminally differentiated, highly specialized phagocytes represent a first line of defense against bacterial and fungal infections.1,2 As part of the innate immune system, neutrophils do not distinguish between host and intruder but use their destructive force indiscriminately. Neutrophils need to be tightly controlled. Insufficient neutrophil activity renders the host susceptible to repeated infections, which can be life-threatening. Yet, dysregulated neutrophil activation can result in excessive collateral damage to the host, as exemplified by a range of autoimmune diseases, such as rheumatoid arthritis.
Upon activating conditions, adhesion molecules (selectins and integrins) undergo changes that permit the circulating neutrophil to interact with the vessel wall. Neutrophils roll along the vessel wall, slow down and crawl along the inside of the vessel until they adhere firmly and extravasate. Following gradients of chemokines and chemoattractants neutrophils travel through tissue to reach sites of infection or insult. Once arrived at their destination, neutrophils phagocytose pathogens. They degranulate, releasing an arsenal of cytotoxic enzymes and generate reactive oxygen species (ROS) to kill pathogens (Fig. 1). Moreover, neutrophils can release their chromatin to act as extracellular traps (NETs), for example aiding the destruction of pathogens that are too large to be engulfed. Neutrophils also produce cytokines, to recruit and cross-talk with other immune cells thus playing a part in orchestrating the immune response. Circulating neutrophils are only short-lived, with estimated half-lives ranging from hours to days depending on the method used. Neutrophil lifespan is, however, significantly extended under inflammatory conditions. To end their short lives, aged circulating neutrophils home to liver, spleen and bone marrow for clearance by resident macrophages. The majority of the longer-lived post-migrated, inflammatory neutrophils eventually undergo apoptosis, and display ‘eat-me’ signals. By doing so they, too, induce their own clearance by phagocytic macrophages in a process termed efferocytosis. In this way, neutrophil apoptosis helps to keep the fine balance between the generation and resolution of inflammation, enabling immunity while avoiding excessive inflammation.
Figure 1.
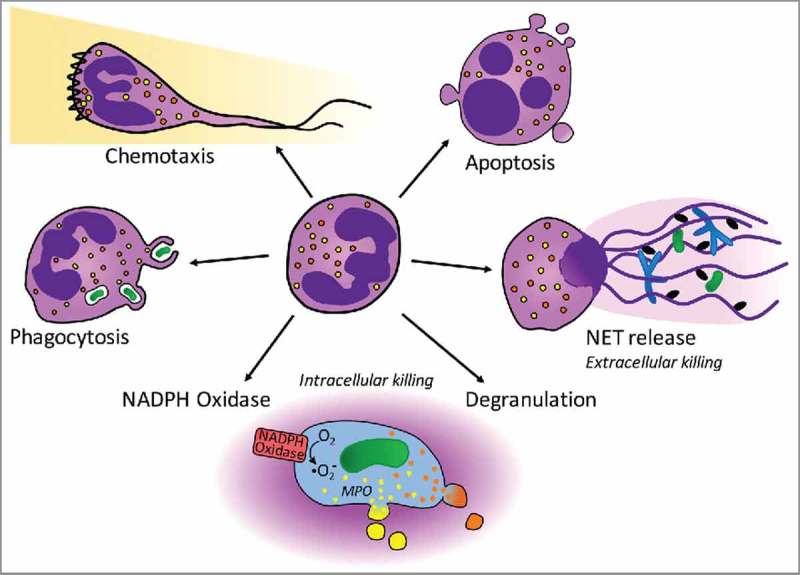
Neutrophil function involves numerous processes that are regulated by Rho GTPases. Circulating neutrophils (center) leave the circulation and chemotax along gradients of chemokines/chemoattractants to reach inflammatory sites (top left). In contrast to the round, circulating cells, chemotaxing neutrophils are polarized and characterized by a leading edge with actin-rich lamellipodium (indicated here by a zigzag line) and a trailing end. As professional phagocytes, neutrophils recognize and engulf small pathogens (e.g., bacteria and yeast; left). Phagocytosis involves the formation of a phagocytic cup which closes around the particle, forming the phagosome. Once engulfed, pathogens are killed intracellularly, in a process that uses ROS, antimicrobial peptides and proteases. Killing depends on 2 distinct processes, the assembly and activation of the NADPH oxidase (bottom left), as well as degranulation (bottom right). The NADPH oxidase catalyzes the generation of oxygen radicals, while degranulation ensures delivery of enzymes required for their conversion to biocidal ROS [in particular myeloperoxidase (MPO), a component of primary/azurophil granules; shown here in yellow]. Secondary/specific granules (shown in orange) deliver antimicrobial peptides and proteases into the phagosomes, which also contribute to intracellular killing. Under certain conditions killing occurs extracellularly, for example when pathogens are too large to be engulfed (e.g., parasites or fungal hyphae) or in conditions of sepsis, neutrophils release NETs (right). NETs consist of decondensed chromatin and antimicrobial proteins, and trap and kill pathogens. At the end of their short lives, neutrophils undergo apoptosis (top right). To limit the inflammation generated, they display ‘eat-me’ signals, thus triggering their own uptake by pro-resolution macrophages in a process termed efferocytosis.
Ligation of neutrophil cell surface receptors triggers a large number of signaling pathways, that are involved in the tight control of neutrophil behavior. Key regulators include Rho family small GTPases as well as agonist-activated PI3Ks.
Small GTPases cycle between a GDP- and a GTP-bound form; only when GTP-bound can they interact with and activate effector molecules. Small GTPases are themselves subject to regulation. The intrinsic GTPase activity of many GTPases is low; it is activated by GTPase activating proteins (GAPs), while exchange of GDP for GTP is catalyzed by guanine nucleotide exchange factors (GEFs). A third regulator, Rho-GDP dissociation inhibitor (RhoGDI) prevents GTP exchange and sequesters the GTPase in the cytosol, thereby aiding the spatial regulation of Rho GTPase signaling (reviewed in ref 3). Major Rho family small GTPases expressed by human neutrophils are Rac proteins (Rac1, Rac2 and RhoG), RhoA and Cdc42, with several other Rho GTPases expressed at minor levels.4 Rho GTPases are best known for their regulatory function in dynamic actin rearrangements, but they also control a host of other cellular functions by regulating several effector proteins each.
Few reliable, cell permeable drugs for small GTPases, and their regulators have been developed yet, and primary neutrophils are not amenable to culture, transfection or transduction. Most of our current understanding of small GTPases and of their regulators in the neutrophil therefore stems from genetically manipulated mice, or from (human) leukemia cell lines that can be induced to become neutrophil-like. These models have shown that most if not all neutrophil functions are subject to regulation by Rho family small GTPases and their GEFs and GAPs (see Table 1 for some examples). Rho GTPase are particularly involved during neutrophil recruitment, which includes several distinct steps that are themselves Rho GTPase-dependent, e.g. polarization, transendothelial migration (TEM) and chemotaxis. These processes have been the subject of particularly thorough investigation by many groups. A second important regulatory contribution to neutrophil function is found during microbial killing, which again comprise distinct processes that are subject to regulation by Rho GTPases, e.g., phagocytosis, the NADPH oxidase and degranulation.
Table 1.
Regulation of neutrophil effector functions by Rho GTPases and their regulators. Many neutrophil functions are subject to regulation by Rho family GTPases. This table summarizes recent developments, mostly drawing from mouse models and placing a focus on GEFs and GAPs. Please note that due to space constraints not all papers could be cited.
| Neutrophil Function | Small GTPases involved | GEFs / GAPs involved |
|---|---|---|
| Adhesion | Rac2 (flow conditions)11 | Vav1/3 (static and flow conditions)17 |
| RhoA (tail retraction37) | ArhGAP25 (flow conditions)28 | |
| P-Rex1/Vav1 (static and flow conditions)19 | ||
| ARAP3 (static and flow conditions)39,40 | ||
| Spreading | Rac211 | Vav1/3, Vav1–317,19 |
| ARAP339,40 | ||
| Polarization | Cdc4230,31 | Cdc42GAP29 |
| Rac213 | DOCK2, DOCK2/523,24 | |
| RhoG24 | ARAP339 | |
| Chemotaxis | Rac1 (directionality)13 | P-Rex1/Vav119 |
| Rac 2 (migration and speed)11,13 | DOCK2, DOCK5, DOCK2/5 22,23 | |
| Cdc4230,31 | ArhGAP15 (directionality)26 | |
| Rho37 | Cdc42GAP29 | |
| ARAP3 (migration and directionality)39,40 | ||
| Recruitment | Rac1 (sterile peritonitis)12 | P-Rex1/Vav1 or -3 (to inflamed peritoneum or lung)20 |
| Rac2 (sterile peritonitis)11 | ArhGAP25 (sterile peritonitis)28 | |
| Rho (acute lung injury)37 | ArhGAP15 (air pouch, bacterial peritonitis and abdominal sepsis)26 | |
| Cdc42GAP (sterile peritonitis)29 | ||
| ARAP3 (sterile peritonitis and arthritic joint)39 | ||
| Phagocytosis | Rac2 | Vav family (IgG and complement opsonized)17 |
| (Cdc42?) | ARHGAP2527 | |
| ArhGAP15 (serum opsonized only)26 | ||
| NADPH oxidase | Rac2 but not Rac111,12 | Vav1–3 and ARHGAP25 (to opsonized particles)19,27 |
| RhoG (to fMLF and C5a)15,24 | P-Rex1 (LPS-primed to fMLF)19 | |
| P-Rex1/Vav1 (unprimed or primed to fMLF) 19 | ||
| DOCK2, DOCK2/5 (to PMA)23 | ||
| ArhGAP15 (to fMLF or C5a but not opsonized zymosan)26 | ||
| NET release | Rac2 | DOCK2, DOCK5, DOCK2/5 23 |
| Apoptosis | RhoG (in sterile peritonitis)15 | |
| Cdc42 (immune complex induced apoptosis; primary human neutrophils)8 |
Class I (also known as agonist-activated) phosphoinositide 3-kinases (PI3Ks) represent a second class of key regulators in the neutrophil.5 These PI3Ks phosphorylate the membrane lipid phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] in the D3 position to generate the lipid second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3). PI3Ks are heterodimeric enzymes that consist of a catalytic subunit (p110α,β,δ; class IA or p110γ; class IB) and a regulatory subunit (a p85/p55-style adaptor for class IA, and a p101 or p84 adaptor for class IB PI3Ks). PI3Kα and δ act downstream of receptor tyrosine kinases (RTK; with phosphopeptide binding of the p85 adaptor). PI3Kγ is activated downstream of G protein coupled receptors (GPCRs) by G protein βγ subunits, and PI3Kβ is synergistically activated by both phosphopeptide and Gβγ. In addition, PI3Ks are subject to regulation by Ras proteins, with GTP-bound Ras binding to the Ras binding domain (RBD) of p110α,δ and γ while GTP-bound Rac or Cdc42 can activate p110β by binding to its RBD.6 While PI3Kα and PI3Kβ are ubiquitously expressed, most of PI3Kδ and PI3Kγ is expressed in leukocytes. Neutrophils express all 4 PI3K isotypes. The analysis of isotype usage in any biological scenario is aided by the availability of mouse knockouts and catalytic dead knock-ins, as well as the development of isoform-selective PI3K inhibitors. The analysis of PI3K isoforms in N-Formyl-methionyl-leucyl-phenylalanine (fMLF)-stimulated mouse and human neutrophils has revealed significant cross-talk between isoforms following neutrophil stimulation as well as distinct differences between signaling pathways in mouse and human neutrophils.7,8
Agonist-activated PI3Ks signal through multiple effector proteins, with an average cell estimated to express between 25 and 50 PI3K effectors. Such effectors include enzymes and adapters, that are activated catalytically and/or recruited to the plasma membrane by PIP3. A notable fraction of PI3K effectors in the neutrophil comprises regulators of small GTPases,9 indicative of the large amount of cross-talk between these 2 classes of regulators. Here, we discuss cross-talk between PI3K and Rho family small GTPases in the neutrophil. There are numerous examples of Rho GTPase-dependent regulation of neutrophil function, involving PI3K-regulated GEFs and GAPs. As indicated, many studies analyzed this in the context of neutrophil polarization and chemotaxis. PI3K (in conjunction with Rac) was thought to be a key to the neutrophil's ‘chemotactic compass’ for some time. However, this view has since been revised and PI3K's role in chemotaxis is now thought to be context-dependent.5,10
Rac
Rac is best known as the regulator of actin polymerization (Arp2/3-dependent, branched meshworks) as found in actin-rich lamellipodia, which are very important for neutrophil function. Neutrophils express the leukocyte-enriched Rac2 as well as the ubiquitous Rac1 and the Rac-related RhoG. Rac proteins have been extensively studied in neutrophils, in particular genetically in the mouse. To summarize, both Rac1 and Rac2 have important functions in the neutrophil (e.g., refs. 11–13). Much of these data are not very recent; since it has been extensively reviewed previously, it is not covered in-depth here. In-line with the well-documented function of Rac in the NADPH oxidase in other cell types, the neutrophil's NADPH oxidase function was found to be dramatically reduced in Rac2-deficient neutrophils. Interestingly, it was not affected by Rac1-deficiency. Similarly, phagocytosis was reduced by Rac2- but not by Rac1-deficiency. In contrast, both Rac isoforms were reported to be involved in chemoattractant-induced dynamic actin rearrangements and were required for efficient transwell chemotaxis in vitro. In keeping with this, lack of either Rac isoform interfered with efficient neutrophil recruitment to sites of inflammation in vivo. Analysis of chemotactic tracks in vitro suggested that Rac1 regulated chemotactic directionality while Rac2 was required for neutrophil migration and speed. RhoG, which functions upstream of Rac1/2,14 regulates the NADPH oxidase following GPCR stimulation, but it was shown to be dispensible for transwell chemotaxis or neutrophil recruitment in vivo.15
PI3K-dependent regulation of Rac
A strong link between PI3K and Rac activation has been observed in many contexts. Since it was established that Rac activation can be PI3K-dependent, several PIP3-activated Rac GEFs have been described (reviewed in ref. 16). Several of these, e.g., Vav, P-Rex, DOCK and Tiam GEFs, are expressed in the neutrophil, and neutrophils from some relevant mouse knockout lines have been analyzed. Vav GEFs were shown genetically to regulate integrin dependent processes,17 while P-Rex1 regulated GPCR-dependent processes such as ROS production.18 Individually, neither of these GEFs were found to be major regulators of neutrophil migration. In combination, however, Vav1/3 and P-Rex1 deletion significantly impaired neutrophil chemotaxis in vitro and neutrophil recruitment to sites of sterile inflammation in vivo.19,20 Contrasting with the mild chemotaxis defects of Vav1/3 and P-Rex1-deficient neutrophils, loss of Tiam2 (expression of which is abrogated in ATF3 transcription factor knockout neutrophils) has been shown to interfere with neutrophil chemotaxis in vitro and recruitment to the inflamed lung in vivo.21 Given the important function of Tiam1 in cancer cell adhesion, migration, invasion and polarity, it will be interesting to analyze any potential Tiam1 function in neutrophils. Neutrophils also express members of the atypical (non Dbl-domain containing) DOCK GEFs, which are thought to be regulated by PIP3 and also phosphatidic acid, and which can function as bipartite GEFs together with ELMO proteins. The analysis of DOCK2, DOCK5 and DOCK2/5-deficient neutrophils identified these DOCK family GEFs as important regulators of neutrophil polarization, chemotactic speed and persistent directionality.22,23 In an interesting twist, P-Rex1 has recently been shown to activate RhoG, which in turn regulates the DOCK2-ELMO complex to activate Rac signaling following GPCR activation of neutrophils.24
While PI3K-regulated Rac activation is well established, more recently, PI3K was also shown to drive Rac-inactivation.25 Recently, the function of 2 PIP3 activated Rac GAPs, ArhGAP15 and ArhGAP25, were analyzed in myeloid cells. ArhGAP15-deficient neutrophils chemotaxed with improved directionality in vitro, and were recruited more efficiently in a model of sepsis in vivo; they phagocytosed and killed pathogens more efficiently.26 Phagocytosis in ArhGAP25 knock-down PLB-985 neutrophil-like cells or in monocyte-derived macrophages was mildly upregulated; ArhGAP25-deficient neutrophils exhibited reduced rolling but enhanced crawling under flow conditions. These neutrophils were also characterized by increased TEM and improved recruitment to sites of inflammation in vivo.27,28 These phenotypes are suggestive of a potential role of both of these GAPs in reducing host defense, perhaps to protect the host from neutrophil-inflicted damage.
Cdc42
Apart from its action on the actin cytoskeleton, Cdc42 is a well-established key regulator of polarization in many biological systems. Interestingly, in the neutrophil, Rac rather than Cdc42 was thought for some time to control polarity and directionality (reviewed in ref. 10). The function of Cdc42 function in these processes has since been analyzed in neutrophils from (conditional) knockout mice. This established that neutrophil directionality and polarity were impaired both when Cdc42 was deleted, or when (due to the deletion of Cdc42GAP) too much Cdc42 activity was present.29,30 An important function for Cdc42 in neutrophil polarization and directionality is also supported by the recent analysis of the localized activities of Rac, Ras, Rho and Cdc42 in fMLF-stimulated neutrophil-like PLB-985 cells using fluorescence resonance energy transfer (FRET)-based biosensors.31 Interestingly, Cdc42-GTP became more distinctly localized to the leading edge than Rac-GTP upon chemoattractant stimulation of neutrophil-like PLB-985 cells. Cdc42-GTP redistribution preceded cellular turning, suggestive of a role in polarization. At the same time, RhoA-GTP was excluded from the leading edge, confirming an older report that used FRET imaging to demonstrate RhoA activity at the trailing end of chemotaxing neutrophil-like HL-60 cells.32 Interestingly, this distribution was observed irrespective of PI3K activity, and of the existence of a chemoattractant gradient.
PI3K-dependent regulation of Cdc42
We recently identified an unusual immune complex-induced pro-apoptotic pathway, PI3Kβ/δ-Cdc42-Pak-Mek-Erk, that operates in human neutrophils.8 In this context, immune complex-induced Cdc42 activation was dependent on PI3K. In contrast, GPCR stimulation with the bacterial peptide fMLF caused Cdc42 activation irrespective of PI3K inhibition. This suggested the existence of a (directly or indirectly) PIP3-activated Cdc42 GEF in the neutrophil, that acts downstream of FcγR but not GPCR stimulation. Given that integrins and FcγRs are known to share downstream signaling pathways, the as-yet-to-be-identified Cdc42 GEF is likely to function in adhesion-dependent situations as well. This observation is interesting, as there are only few instances in which Cdc42 has been shown to be activated in a PI3K-dependent fashion in any tissues. For example, EGF-induced activation of Cdc42 in MTLn3 carcinoma cells was shown to be abrogated on inhibition of PI3K.33 No Cdc42 GEF has yet been shown to be directly activated (or recruited to the plasma membrane) by PIP3, but α-PIX was suggested to be involved indirectly in Gβγ, Pak, Cdc42 and PIP3 co-localization to the leading edge of chemotaxing neutrophils.34
Conversely, PI3K has been demonstrated to regulate Cdc42 inactivation. Hence, Cdc42 inactivation was shown to be regulated by PI3K in phagocytosing mouse macrophage-like RAW264.7 cells.35 Three PIP3-activated Rac/Cdc42 GAPs were recently identified in a knock-down based screen analyzing the regulation of phagocytosis in RAW264.7.36 In addition, Cdc42GAP was isolated as a PIP3-binding protein from neutrophils.9 As discussed above, Cdc42GAP was since shown genetically to regulate neutrophil chemotaxis and recruitment, with Cdc42GAP-deficient neutrophils characterized by reduced directionality but increased speed and enhanced TEM. This was associated with altered adhesive (podosome-like) structures and MAPK signaling.29
RhoA
RhoA is another regulator of cell migration, which is best known for controling contractile actin cables (stress fibers), and stable focal adhesions, which anchor the cell to the substratum. Actin cables, along with focal adhesions (and indeed the shorter lived focal contacts), do not actually exist in neutrophils. Nonetheless, RhoA has long been regarded as another important regulator of neutrophil migration. Using FRET microscopy, RhoA-GTP was shown to localize to the leading edge and trailing end of many cell types during cell migration. Such experiments have not yet been reported with primary neutrophils. In neutrophil-like cell lines (HL-60 and PLB-985), however, RhoA-GTP was shown to be restricted to the trailing end.31,32 This distribution is thought to be regulated by a series of regulatory feed-back loops.10,31,32 The major function of RhoA in neutrophil migration is therefore thought to lie in the regulation tail retraction, mediated by its effector ROCK. However, a recent report on RhoA knockout neutrophils has cast doubts on this assumption. Rho-deficient neutrophils displayed enhanced neutrophil integrin activation and were characterized by increased random and directional migration in vitro and by enhanced recruitment to inflammatory sites in vivo.37 This suggests that RhoA may in fact act as a negative regulator of neutrophil migration and activation.
PI3K-dependent regulation of RhoA
No PI3K-activated RhoA GEF has been reported yet in the neutrophil, but observations in other cell types support the existence of a PI3K-regulated Rho GEF. For example, in p110α-deficient or p110α kinase-dead endothelial cells, or upon PI3K inhibition in wild-type endothelial cells, RhoA activation was dramatically reduced, correlating with a migration and tail retraction defect.38
Our understanding of PI3K-driven RhoA inactivation in the neutrophil is greater than that of Rho activation. ARAP3, a dual GAP for RhoA and Arf6, was identified in a screen for PIP3 binding proteins from neutrophils.9 In ARAP3-deficient neutrophils, or in knock-ins carrying a point mutation that uncoupled ARAP3 from the activation by PI3K, adhesion dependent neutrophil functions were upregulated due to increased β2 integrin affinities.39,40 ARAP3-deficient neutrophils were not only characterized by an integrin-dependent migration defect but also by a chemotactic directionality defect. In-line with the reported enhanced chemotaxis of RhoA-deficient neutrophils toward KC (a mouse homolog of human IL8), but not toward fMLF,37 the chemotaxis defect of neutrophils in which ARAP3 had been uncoupled from activation by PI3K that was more pronounced with migration toward MIP2 (another mouse homolog of IL8) than fMLF.39
Regulation of PI3K by Rho family GTPases
Rho family small GTPases have been reported to promote PI3K in several contexts. GTP-bound Rac and Cdc42 were shown to interact with the p110β RBD, directly activating it in a manner akin to Ras-dependent activation of p110α, p110γ and p110δ.6 By analyzing knock-in mice that harbored mutations which interfered with the regulatory input of either Gβγ or Rac/Cdc42, PI3Kβ was shown to be activated synergistically by these 2 classes of activators on concurrent GPCR and RTK stimulation in macrophages (while GPCR stimulation alone relied heavily on PI3Kγ). ROS production and adhesion / spreading assays were used as indirect read-outs of PI3K activity in neutrophils to demonstrate that both Rac/Cdc42 and Gβγ-dependent stimulation of p110β were required in addition to FcγR (or indeed integrin) activation to derive full adhesion-dependent activation of PI3Kβ (ref. 41 and Fig. 2). In a series of elegant experiments, the authors of this study demonstrated that Gβγ and Rac/Cdc42 activated p110β in a cooperative fashion. Interestingly, a paracrine positive feed-back loop operates in FcγR- (or integrin-) stimulated neutrophils to enable this.42 Hence, stimulation of the FcγR / integrin not only causes activation of Rac/Cdc42 via as yet undefined GEF(s) but also induces the production of the neutrophilic lipid mediator leukotriene B4 (LTB4), which binds to its (G protein coupled) receptor, BLT1, thereby generating G protein βγ subunits, that bind to the p110β RBD, driving the synergistic activation of this PI3K together with phosphopeptide. Interestingly, GPCR stimulation (using fMLF) alone relied almost entirely on PI3Kγ rather than PI3Kβ.41 While this was not addressed in the study, it seems likely that BLT1 ligation also leads to the activation of downstream Rac/Cdc42 GEF(s), perhaps in conjunction with the PI3K lipid product PIP3, adding an additional level of cross-talk between PI3K and Rac/Cdc42.
Figure 2.
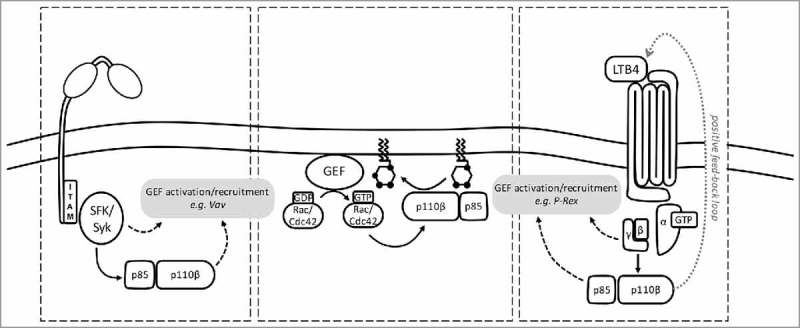
PI3Kβ is activated by phosphopeptide, Gβγ and Rac/Cdc42 in the integrin/immune complex-stimulated neutrophil. (Left) Neutrophil integrin or FcγR ligation causes Src family kinase (SFK)-dependent phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs), triggering the activation of Syk kinase, which in turn recruits the p85 adaptor in an SH2 domain- and phosphotyrosine motif-dependent fashion to activate PI3Kβ. Integrin/FcγR ligation and the PIP3 also activate Rac/Cdc42 GEFs. (Right) In a paracrine feed-forward loop, PI3Kβ drives LTB4 production. LTB4 triggers activation of its GPCR (BLT1), resulting in release of Gβγ subunits activate PI3Kβ by binding to p110β. GPCR ligation and PIP3 also activate Rac/Cdc42 GEFs. (Center panel) Rac/Cdc42 GEFs enable GTP-loading of Rac/Cdc42, which can then bind the p110β RBD, to further activate PI3Kβ. Full activation of PI3Kβ requires phosphotyrosine motifs, Gβγ and Rac/Cdc42.
In chemotaxing mouse neutrophils (and human neutrophil-like cells), Rho family GTPases and PI3K have been reported to regulate one another by using feed-back loops. Using a combination of inhibitors and probes as well as expression of dominant negative / constitutively active small GTPase constructs in HL60 cells showed that PIP3 polarization to the leading edge required actin polymerization and Rac activation (reviewed in ref. 10). Building on these earlier studies, generation and localization of the PI3K lipid product PIP3 were examined by activity assays and monitored using a fluorescent probe in the analysis of DOCK2-deficient neutrophils. Interestingly, in the absence of DOCK2, which is recruited to the neutrophil's leading edge by PIP3, PIP3 was generated, but not subsequently polarized.22 The authors showed that PIP3 was generated by DOCK2-deficient neutrophils, but did not drive persistent signaling. DOCK2 was proposed to be involved in a feed-back loop involving PI3K, Rac and actin which stabilizes the PIP3 signal at the leading edge. In a separate study, RhoA was shown to downregulate PI3K signaling at the trailing end of chemotaxing neutrophils by activating the PIP3 phosphatase PTEN.43
Conclusions and future directions
The examples given above outline significant cross-talk between PI3K and Rho family small GTPases in the neutrophil. This ensures the smooth running of processes that are controlled by these signaling intermediates; key areas studied with mouse knockout neutrophils were polarization and chemotaxis, as well as in vivo neutrophil recruitment. Such studies have shown that the loss of GEFs and GAPs that regulate cross-talk between PI3K and small GTPases (such as CDC42GAP,29 ARAP340 and DOCK222) causes significant chemotaxis and polarization defects. Yet loss of function studies in the PI3K field have arrived at the conclusion that PI3K is dispensible for chemotaxis (reviewed in ref. 5). Could the role of PI3K have been underestimated? Recent findings with patients harboring rare PI3K gain of function mutations provided evidence that too much PI3K signaling can have devastating effects on lymphocyte function (e.g., ref. 44). Such a view is supported by the fact that increased PI3K signaling, due to the absence of the PIP3 phosphatase SHIP1 severely affected neutrophil chemotaxis and polarization.45 SHIP1-deficient neutrophils were characterized by poor migration and enhanced spreading; they failed to polarize PIP3 at the leading edge of neutrophils. This raises the question whether cross-talk between PI3K and small GTPases, perhaps not only downstream of GPCR signaling, could be involved in correct PIP3 (and neutrophil) polarization. This field promises to deliver further interesting insights in the future.
Abbreviations
- FRET
fluorescence resonance energy transfer
- fMLF
N-Formyl-methionyl-leucyl-phenylalanine
- GAP
GTPase activating protein
- GEF
guanine nucleotide exchange factor
- GPCR
G protein coupled receptor
- LTB4
leukotriene B4
- MPO
myeloperoxidase
- NET
neutrophil extracellular trap.
- PI3K
phosphoinositide 3-kinase
- PIP3
phosphatidylinositol-(3,4,5)-trisphosphate
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- RBD
Ras binding domain
- TEM
transendothelial migration
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a Medical Research Council UK project grant (MR/K501293/1) and a Medical Research Council UK studentship.
References
- [1].Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol 2014; 15:602-11; PMID:24940954; http://dx.doi.org/ 10.1038/ni.2921 [DOI] [PubMed] [Google Scholar]
- [2].Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013; 13:159-75; PMID:23435331; http://dx.doi.org/ 10.1038/nri3399 [DOI] [PubMed] [Google Scholar]
- [3].Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 2013; 93:269-309; PMID:23303910; http://dx.doi.org/ 10.1152/physrev.00003.2012 [DOI] [PubMed] [Google Scholar]
- [4].van Helden SF, Anthony EC, Dee R, Hordijk PL. Rho GTPase expression in human myeloid cells. PLoS One 2012; 7:e42563; PMID:22916134; http://dx.doi.org/ 10.1371/journal.pone.0042563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Curr Top Microbiol Immunol 2010; 346:183-202; PMID:20473789 [DOI] [PubMed] [Google Scholar]
- [6].Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, Diefenbacher M, Stamp G, Downward J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013; 153:1050-63; PMID:23706742; http://dx.doi.org/ 10.1016/j.cell.2013.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, et al.. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood 2005; 106:1432-40; PMID:15878979; http://dx.doi.org/ 10.1182/blood-2005-03-0944 [DOI] [PubMed] [Google Scholar]
- [8].Chu JY, Dransfield I, Rossi AG, Vermeren S. Non-canonical PI3K-Cdc42-Pak-Mek-Erk signaling promotes immune-complex-induced apoptosis in human neutrophils. Cell Rep 2016; 17:374-86; PMID:27705787; http://dx.doi.org/ 10.1016/j.celrep.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Krugmann S, Anderson KE, Ridley SH, Risso N, McGregor A, Coadwell J, Davidson K, Eguinoa A, Ellson CD, Lipp P, et al.. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol Cell 2002; 9:95-108; PMID:11804589; http://dx.doi.org/ 10.1016/S1097-2765(02)00434-3 [DOI] [PubMed] [Google Scholar]
- [10].Wang F. The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harb Perspect Biol 2009; 1:a002980; PMID:20066099; http://dx.doi.org/ 10.1101/cshperspect.a002980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, Spaetti A, Pollock JD, Borneo JB, Bradford GB, et al.. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity 1999; 10:183-96; PMID:10072071; http://dx.doi.org/ 10.1016/S1074-7613(00)80019-9 [DOI] [PubMed] [Google Scholar]
- [12].Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol 2003; 170:5652-7; PMID:12759446; http://dx.doi.org/ 10.4049/jimmunol.170.11.5652 [DOI] [PubMed] [Google Scholar]
- [13].Sun CX, Downey GP, Zhu F, Koh AL, Thang H, Glogauer M. Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood 2004; 104:3758-65; PMID:15308574; http://dx.doi.org/ 10.1182/blood-2004-03-0781 [DOI] [PubMed] [Google Scholar]
- [14].Katoh H, Negishi M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature 2003; 424:461-4; PMID:12879077; http://dx.doi.org/ 10.1038/nature01817 [DOI] [PubMed] [Google Scholar]
- [15].Condliffe AM, Webb LM, Ferguson GJ, Davidson K, Turner M, Vigorito E, Manifava M, Chilvers ER, Stephens LR, Hawkins PT. RhoG regulates the neutrophil NADPH oxidase. J Immunol 2006; 176:5314-20; PMID:16621998; http://dx.doi.org/ 10.4049/jimmunol.176.9.5314 [DOI] [PubMed] [Google Scholar]
- [16].Campa CC, Ciraolo E, Ghigo A, Germena G, Hirsch E. Crossroads of PI3K and Rac pathways. Small GTPases 2015; 6:71-80; PMID:25942647; http://dx.doi.org/ 10.4161/21541248.2014.989789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gakidis MA, Cullere X, Olson T, Wilsbacher JL, Zhang B, Moores SL, Ley K, Swat W, Mayadas T, Brugge JS. Vav GEFs are required for beta2 integrin-dependent functions of neutrophils. J Cell Biol 2004; 166:273-82; PMID:15249579; http://dx.doi.org/ 10.1083/jcb.200404166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, et al.. P-Rex1 regulates neutrophil function. Curr Biol 2005; 15:1867-73; PMID:16243035; http://dx.doi.org/ 10.1016/j.cub.2005.09.050 [DOI] [PubMed] [Google Scholar]
- [19].Lawson CD, Donald S, Anderson KE, Patton DT, Welch HC. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. J Immunol 2011; 186:1467-76; PMID:21178006; http://dx.doi.org/ 10.4049/jimmunol.1002738 [DOI] [PubMed] [Google Scholar]
- [20].Pan D, Amison RT, Riffo-Vasquez Y, Spina D, Cleary SJ, Wakelam MJ, Page CP, Pitchford SC, Welch HC. P-Rex and Vav Rac-GEFs in platelets control leukocyte recruitment to sites of inflammation. Blood 2015; 125:1146-58; PMID:25538043; http://dx.doi.org/ 10.1182/blood-2014-07-591040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Boespflug ND, Kumar S, McAlees JW, Phelan JD, Grimes HL, Hoebe K, Hai T, Filippi MD, Karp CL. ATF3 is a novel regulator of mouse neutrophil migration. Blood 2014; 123:2084-93; PMID:24470589; http://dx.doi.org/ 10.1182/blood-2013-06-510909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, Watanabe K, Sanematsu F, Sasazuki T, Sasaki T, et al.. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol 2006; 174:647-52; PMID:16943182; http://dx.doi.org/ 10.1083/jcb.200602142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Watanabe M, Terasawa M, Miyano K, Yanagihara T, Uruno T, Sanematsu F, Nishikimi A, Côté JF, Sumimoto H, Fukui Y. DOCK2 and DOCK5 act additively in neutrophils to regulate chemotaxis, superoxide production, and extracellular trap formation. J Immunol 2014; 193:5660-7; PMID:25339677; http://dx.doi.org/ 10.4049/jimmunol.1400885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Damoulakis G, Gambardella L, Rossman KL, Lawson CD, Anderson KE, Fukui Y, Welch HC, Der CJ, Stephens LR, Hawkins PT. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci 2014; 127:2589-600; PMID:24659802; http://dx.doi.org/ 10.1242/jcs.153049 [DOI] [PubMed] [Google Scholar]
- [25].Costa C, Barberis L, Ambrogio C, Manazza AD, Patrucco E, Azzolino O, Neilsen PO, Ciraolo E, Altruda F, Prestwich GD, et al.. Negative feedback regulation of Rac in leukocytes from mice expressing a constitutively active phosphatidylinositol 3-kinase gamma. Proc Natl Acad Sci U S A 2007; 104:14354-9; PMID:17720808; http://dx.doi.org/ 10.1073/pnas.0703175104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Costa C, Germena G, Martin-Conte EL, Molineris I, Bosco E, Marengo S, Azzolino O, Altruda F, Ranieri VM, Hirsch E. The RacGAP ArhGAP15 is a master negative regulator of neutrophil functions. Blood 2011; 118:1099-108; PMID:21551229; http://dx.doi.org/ 10.1182/blood-2010-12-324756 [DOI] [PubMed] [Google Scholar]
- [27].Csepanyi-Komi R, Sirokmany G, Geiszt M, Ligeti E. ARHGAP25, a novel Rac GTPase-activating protein, regulates phagocytosis in human neutrophilic granulocytes. Blood 2012; 119:573-82; PMID:22096251; http://dx.doi.org/ 10.1182/blood-2010-12-324053 [DOI] [PubMed] [Google Scholar]
- [28].Csepanyi-Komi R, Wisniewski E, Bartos B, Levai P, Nemeth T, Balazs B, , Kurz AR, Bierschenk S, Sperandio M, Ligeti E. Rac GTPase activating protein ARHGAP25 regulates leukocyte transendothelial migration in mice. J Immunol 2016; 197:2807-15; PMID:27566826; http://dx.doi.org/ 10.4049/jimmunol.1502342 [DOI] [PubMed] [Google Scholar]
- [29].Szczur K, Xu H, Atkinson S, Zheng Y, Filippi MD. Rho GTPase CDC42 regulates directionality and random movement via distinct MAPK pathways in neutrophils. Blood 2006; 108:4205-13; PMID:16931627; http://dx.doi.org/ 10.1182/blood-2006-03-013789 [DOI] [PubMed] [Google Scholar]
- [30].Szczur K, Zheng Y, Filippi MD. The small Rho GTPase Cdc42 regulates neutrophil polarity via CD11b integrin signaling. Blood 2009; 114:4527-37; PMID:19752396; http://dx.doi.org/ 10.1182/blood-2008-12-195164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang HW, Collins SR, Meyer T. Locally excitable Cdc42 signals steer cells during chemotaxis. Nat Cell Biol 2016; 18:191-201; PMID:26689677; http://dx.doi.org/ 10.1038/ncb3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wong K, Pertz O, Hahn K, Bourne H. Neutrophil polarization: spatiotemporal dynamics of RhoA activity support a self-organizing mechanism. Proc Natl Acad Sci U S A 2006; 103:3639-44; PMID:16537448; http://dx.doi.org/ 10.1073/pnas.0600092103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].El-Sibai M, Nalbant P, Pang H, Flinn RJ, Sarmiento C, Macaluso F, Cammer M, Condeelis JS, Hahn KM, Backer JM. Cdc42 is required for EGF-stimulated protrusion and motility in MTLn3 carcinoma cells. J Cell Sci 2007; 120:3465-74; PMID:17855387; http://dx.doi.org/ 10.1242/jcs.005942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li Z, Hannigan M, Mo Z, Liu B, Lu W, Wu Y, Smrcka AV, Wu G, Li L, Liu M, et al.. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell 2003; 114:215-27; PMID:12887923; http://dx.doi.org/ 10.1016/S0092-8674(03)00559-2 [DOI] [PubMed] [Google Scholar]
- [35].Beemiller P, Zhang Y, Mohan S, Levinsohn E, Gaeta I, Hoppe AD, Swanson JA. A Cdc42 activation cycle coordinated by PI 3-kinase during Fc receptor-mediated phagocytosis. Mol Biol Cell 2010; 21:470-80; PMID:19955216; http://dx.doi.org/ 10.1091/mbc.E08-05-0494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schlam D, Bagshaw RD, Freeman SA, Collins RF, Pawson T, Fairn GD, Grinstein S. Phosphoinositide 3-kinase enables phagocytosis of large particles by terminating actin assembly through Rac/Cdc42 GTPase-activating proteins. Nat Commun 2015; 6:8623; PMID:26465210; http://dx.doi.org/ 10.1038/ncomms9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jennings RT, Strengert M, Hayes P, El-Benna J, Brakebusch C, Kubica M, Knaus UG. RhoA determines disease progression by controlling neutrophil motility and restricting hyperresponsiveness. Blood 2014; 123:3635-45; PMID:24782506; http://dx.doi.org/ 10.1182/blood-2014-02-557843 [DOI] [PubMed] [Google Scholar]
- [38].Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, et al.. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008; 453:662-6; PMID:18449193; http://dx.doi.org/ 10.1038/nature06892 [DOI] [PubMed] [Google Scholar]
- [39].Gambardella L, Anderson KE, Jakus Z, Kovacs M, Voigt S, Hawkins PT, Stephens L, Mócsai A, Vermeren S. Phosphoinositide 3-OH kinase regulates integrin-dependent processes in neutrophils by signaling through its effector ARAP3. J Immunol 2013; 190:381-91; PMID:23180820; http://dx.doi.org/ 10.4049/jimmunol.1201330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gambardella L, Anderson KE, Nussbaum C, Segonds-Pichon A, Margarido T, Norton L, Ludwig T, Sperandio M, Hawkins PT, Stephens L, et al.. The GTPase-activating protein ARAP3 regulates chemotaxis and adhesion-dependent processes in neutrophils. Blood 2011; 118:1087-98; PMID:21490342; http://dx.doi.org/ 10.1182/blood-2010-10-312959 [DOI] [PubMed] [Google Scholar]
- [41].Houslay DM, Anderson KE, Chessa T, Kulkarni S, Fritsch R, Downward J, Backer JM, Stephens LR, Hawkins PT. Coincident signals from GPCRs and receptor tyrosine kinases are uniquely transduced by PI3Kbeta in myeloid cells. Sci Signal 2016; 9:ra82; PMID:27531651; http://dx.doi.org/ 10.1126/scisignal.aae0453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, Hirose M, Juss J, Oxley D, Chessa TA, et al.. PI3Kbeta plays a critical role in neutrophil activation by immune complexes. Sci Signal 2011; 4:ra23; PMID:21487106; http://dx.doi.org/ 10.1126/scisignal.2001617 [DOI] [PubMed] [Google Scholar]
- [43].Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, et al.. Regulation of PTEN by Rho small GTPases. Nat Cell Biol 2005; 7:399-404; PMID:15793569; http://dx.doi.org/ 10.1038/ncb1236 [DOI] [PubMed] [Google Scholar]
- [44].Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, Baxendale H, Coulter T, Curtis J, Wu C, et al.. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 2013; 342:866-71; PMID:24136356; http://dx.doi.org/ 10.1126/science.1243292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, Balla T, Yamazaki M, Watanabe H, Itoh R, et al.. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol 2007; 9:36-44; PMID:17173042; http://dx.doi.org/ 10.1038/ncb1515 [DOI] [PubMed] [Google Scholar]