

Abstract
Neutrophils, the front line defenders against infection, express four serine proteases (NSPs) that play roles in the control of cell signaling pathways, defense against pathogens, and whose imbalance leads to pathological conditions. Dissecting the roles of individual NSPs in humans is problematic because neutrophils are end stage cells with a short half life and minimal ongoing protein synthesis. To gain insight into the regulation of NSPs activity we have generated a small molecule chemical toolbox consisting of activity-based probes with different fluorophore detecting groups with minimal wavelength overlap, and highly selective natural and unnatural amino acid recognition sequences. The key feature of these activity-based probes is the ability to use them for simultaneous observation and detection of all four individual NSPs by fluorescence microscopy, a feature never achieved in previous studies. Using these probes we demonstrate uneven distribution of NSPs in neutrophil azurophil granules, such that they seem to be mutually excluded from each other, suggesting the existence of unknown granule targeting mechanisms.
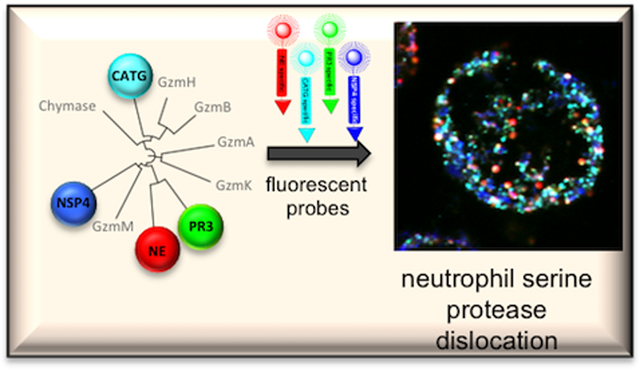
Graphical Abstract
1. INTRODUCTION
Neutrophils are terminal effector cells that constitute the first line innate immune defense against infection. They contain specialized sets of vesicles, or granules, that incorporate a number of proteins important for pathogen killing and other immune cell functions1. The most abundant of these are the unique azurophil (primary) granules that accommodate four serine proteases found mainly in neutrophils (NSPs), namely neutrophil elastase (NE), cathepsin G (CATG), proteinase 3 (PR3), and neutrophil proteinase 4 (NSP4) – also known as PRSS571–2. These proteases have been implicated in the control of cellular signaling by chemokine activation, activation of cells receptors and non-oxidative pathway of intracellular pathogen destruction, and the generation of Neutrophil Extracellular Traps (NETs)3–4. Mice knockouts of NE and CATG present with a phenotype consistent with a role for these enzymes in clearance of Gram-negative bacteria and fungal pathogens5–7. A mutation of one of these proteases, NE, is associated with episodes of cyclic neutropenia8–9 and severe congenital neutropenia10.
NSPs constitute a subset of the hematopoietic serine proteases (HeSPs)11 related by around 40% amino acid sequence identity, that also encompasses the granzymes and chymase12. Catalytic (proteolytic) and non-catalytic roles have been proposed, and several endogenous substrates have been identified, although it is not entirely clear how important proteolysis of individual substrates is for cell physiological functions. However, it is evident that the activity of these proteases presents a major proteolytic burden that must be closely regulated in vivo, primarily by endogenous potent protease inhibitors.
Dissecting the roles of individual NSPs in humans is problematic because neutrophils are end stage cells with a short half life and minimal ongoing protein synthesis – precluding genetic manipulations. Antibody related techniques yield information on total protein (protease) amount, however they lack the ability to differentiate between active and non-active enzyme forms and cannot be used in live cell imaging. To overcome these shortcomings we developed a set of highly selective NSP small molecule chemical tools (substrates, inhibitors and fluorescent activity-based probes) that allow us to monitor each NSP acting individually or in parallel in neutrophils. We sought to generate probes that would enable simultaneous imaging of all four NSPs, with the criteria that these probes should be highly selective and penetrate to the cellular location where the active proteases are stored.
NSPs are members of the trypsin/chymotrypsin protease family and thus their primary substrate specificity is dominated by their S1 pockets (Schechter and Berger nomenclature13), an enzyme feature that dictates which amino acids are preferred adjacent to the peptide bond that will be cut. NE and PR3 have a common preference for Val or Ala14–18, CATG prefers Tyr, Phe, or Lys17, 19, and NSP4 prefers Arg20. Accordingly, it should be readily possible to distinguish CATG and NSP4 from each other and the other NSPs, but the overlap in specificity between NE and PR3 presents a problem. To accomplish the goals of selectivity visualizing NSPs we deployed a Hybrid Combinatorial Substrate Library (HyCoSuL) positional scanning approach that has recently proven useful for interrogating specificity of several endopeptidase families21–24. Here we describe a unique toolbox that allows us to investigate the location of individual NSPs in neutrophils.
2. EXPERIMENTAL SECTION
2.1. HyCoSuL P1 Phe library synthesis
The P1-Phe HyCoSuL library was synthesized as previously described21–22, with the exception of the P1 position, where Phe was incorporated. The library contains three sublibraries (P4–P2), with the general structures Ac-P4-mix-mix-Phe-ACC, Ac-mix-P3-mix- Phe-ACC and Ac-mix-mix-P2-Phe-ACC, where mix is an equimolar mixture of natural amino acids (without cysteine, methionine was replaced with norleucine), P4–P2 are natural or unnatural amino acid, ACC is the fluorogenic reporter 7-amino-4-carbamoylmethylcoumarin. Libraries were dissolved in dry, biochemical grade DMSO to a final concentration of 10mM and stored at −20°C until use.
2.2. CATG specificity determination with HyCoSuL screening
Specificity screening was carried out in the Cathepsin G buffer (0.1 mM Tris/HCl, 0.5 M NaCl, pH 7.5) in the same manner as previously described for NE, PR3 and NSP421–22. The final library concentration was 100μM, the final CATG concentration was 100nM, and the reaction volume was 100μL. Library screening was carried out using fMax spectrofluorimeter (Molecular Devices) and 96-well Opaque Corning plates (Corning®). The excitation wavelength was 355 nm and the emission wavelength was 460 nm (cutoff at 455 nm). 1μL of each library component was loaded into each of 96 wells, CATG was preincubated in assay buffer for 15 minutes at 37°C prior to addition to each well. Substrate hydrolysis was recorded in kinetic mode at 37°C for at least 20 minutes and the linear portion of each progress curve was used to calculate Relative Fluorescence Units per second (Rfu/s). All experiments were repeated at least three times and the results are presented as mean values. Specificity was calculated by normalizing Rfu/s to the highest value (set at 100%). Calculations were made in Microsoft Excel.
2.3. General method of individual fluorogenic substrates synthesis (1–4)
Individual substrates were synthesized as previously described21–22, 25 using ACC as a reporter group25. After every coupling and deprotection reaction, efficiency was analyzed by a ninhydrin test.
Synthesis: 100mg of the Fmoc-RA resin was activated with dry DCM, followed by N-terminal amine group deprotection with 25% PIP/DMF. Next, 2.5 eq of Fmoc-ACC-OH (made in house) was dissolved in DMF and activated with 2.5 eq of HOBt and 2.5eq of DICI for 3 minutes in a microfuge tube and added to the resin. After 24 hours of reaction uncoupled components were removed, the resin was washed 3 times with DMF and the coupling reaction was repeated using 1.5 eq of reagents. After Fmoc deprotection 2.5 eq of Fmoc-P1-OH was dissolved in DMF and activated with 2.5 eq of HATU and 2.5 eq of 2,4,6-trimethylpiridine and added to the resin. After 24 hours of agitation, the resin was washed and Fmoc-P1-OH coupling was repeated using 1.5 eq of the above chemicals. Following Fmoc-deprotection the peptide chain was elongated with 2.5 eq of Fmoc-P2-OH, 2.5 eq of HOBT and 2.5 eq of DICI. Elongation was continued until H2N-P4-P3-P2-P1-ACC. Afterwards, a free amine at the N-terminal end of the peptide was acetylated with 5 eq of AcOH mixed with 5 eq of HBTU and 5eq of DIPEA, and, added to the resin after 3 minutes of activation, 45 minutes at room temperature. The resin was washed six times with DMF, three times with DCM and three times with MeOH and dried over P2O5 for at least 3 hours. A cleavage mixture (TFA:H2O:TIPS (95%:2.5%:2.5%, v:v:v)) was cooled to −20°C for one hour and added to the dry resin and shaken vigorously once per 10 minutes in 60 minutes period (room temperature). The first filtrate was collected, the resin was washed with cleavage mixture, filtrates were combined, and peptide substrates were precipitated in cold Et2O, centrifuged for 5 minutes and the precipitate was washed with an additional portion of Et2O, spun down for 5 minutes and dried overnight at room temperature, followed by HPLC purification (Waters, column: Spherisorb, 5 μm particle size, L × I.D. 25 cm × 4.6 mm) using an H2O:MeOH gradient, and lyophilized. Each product was dissolved in dry DMSO to a concentration of 10mM and stored at −20°C until use. Compounds were analyzed by analytical HPLC and HRMS.
2.4. Fluorogenic substrates kinetic analysis
Kinetic parameters of selected NE, PR3 and NSP4 substrates and activity-based probes were determined in 50 mM Hepes, 0.1 M NaCl, 0.1%TRITON X-100, pH 7.45 assay buffer, while for CATG assays were carried out in Tris/HCl (0.1 mM, pH 7.5), NaCl (0.5 M). Buffers were prepared at room temperature prior to analysis.
Assays were carried out as previously described21–22. Briefly, stock substrates in DMSO were serially diluted in the assay buffer and 20μL was added to the wells of a 96 well plate prior to addition of 80μL of enzyme (preincubated for 20 minutes at 37°C in buffer). Measurements were carried out using a plate reader (Molecular Device) operating in the kinetic mode for a minimum of 20 minutes with the optimal for ACC excitation/emission wavelengths 355 nm/460 nm, and cutoff of at 455 nm. Km, kcat and kcat/Km parameters were calculated using GraphPad Prism and Excel software. Each experiment was repeated three times, and the results are presented as an average ± S.D.
2.5. Fluorescent activity-based probe synthesis
General method for the hydrobromide salt of H2N-PheP(OPh)2, H2N-AbuP(OPh)2 and H2N-NvaP(OPh)2 synthesis (9)
Electrophilic covalent diphenyl phosphonate warheads were synthesized using previously described methodology26. In a round bottom flask, 1 eq of triphenyl phosphate, 1.1 eq of adequate aldehyde (propionaldehyde for Cbz-AbuP(OPh)2, butyraldehyde for Cbz-NvaP(OPh)2 and phenylacetaldehyde for Cbz-PheP(OPh)2) and 1.1 eq of benzylcarbamate (in house) were dissolved in MeOH. The reaction mixture was cooled in an ice bath, mixed using a magnetic stirrer and HBF4 (10% in Et2O, (v/v)) was slowly added. After half an hour, the ice bath was removed and the reaction was stirred at room temperature for 4 h, followed by the evaporation of volatile components. Crude product as an oil was left for precipitation in cold MeOH at −20°C for 2–7 days. The white precipitate was filtered under reduced pressure, washed twice with MeOH, dried at room temperature, and used in the next step without additional purification.
The Cbz protecting group was removed with 30% HBr/AcOH (v:v) (1 h, room temperature). The solvent was evaporated using a rotary evaporator and the crude product was precipitated in Et2O for 1–3 days at 4°C. the white precipitate was filtered, washed twice with a small portion of Et2O, dried at room temperature, and used in the next step without additional purification. A general method for the H2N-ArgP(OPh)2 synthesis has been described previously21.
General method for Boc-linker-P4-P3-P2-OH peptide synthesis (8)
400 mg of 2-chlorotrityl chloride resin (loading 1.6 mmol/g) was placed in a glass peptide synthesis vessel and activated for 1 hour with dry DCM by shaking for 10 minutes, followed by washing (three times with dry DCM), and addition of 3 eq of Fmoc-P2-OH (1.92 mmol) (5) activated with 6 eq of DIPEA (3.84 mmol) in dry DCM. The resin was agitated for 6 hours under argon atmosphere, followed by filtration and washing with DMF (3 times) (6). After the Fmoc-group removal with 20% PIP/DMF for 5 minutes, 5 minutes and 25 minutes, the resin was washed 6 times with dry DMF. Next, 2.5 eq of Fmoc-P3-OH (1.6 mmol) was preactivated for three minutes with 2.5 eq of HOBt (1.6 mmol) and 2.5 eq of DICI (1.6 mmol) in DMF and poured onto the resin. After Fmoc group removal (as above), the peptide chain was elongated in the same manner using 2.5 eq of Fmoc-P4-OH (1.6 mmol), 2.5 eq of HOBt (1.6 mmol) and 2.5 eq of DICI (1.6 mmol) (for P4 coupling), followed by N-terminal amine group deprotection and coupling of 2.5eq of Boc-linker-OH (1.6 mmol) with 2.5 eq of HOBt (1.6 mmol) and 2.5 eq of DICI (1.6 mmol) (7).
The resin was washed 6 times with DMF, 3 times with DCM and 3 times with MeOH, dried over P2O5 for at least 3 hours and peptide derivative (Boc-linker-P4-P3-P2-OH) was cleaved from the resin using solution of TFE:AcOH:DCM (1:1:4; v/v/v). Reaction was carried out for 1.5 hour at room temperature. Filtrate was collected and the resin was washed with the cleaving mixture and filtrates were combined and the volatiles were removed on a rotary evaporator under reduced pressure. Peptide product was dissolved in MeOH:H2O (1:1, v/v), lyophilized and used for the next experiment without further purification.
General method for H2N-linker-P4-P3-P2-P1P(OPh)2 (10) synthesis
Crude peptide product (8) was dissolved in DMF and activated with 1 eq of HATU and 5 eq of 2,4,6-trimethylpiridine for three minutes and added to a pear-shaped flask with 1.2 eq of H2N-P1P(OPh)2 (9) under an argon atmosphere, constantly stirring with magnetic stirrer and monitored by analytical HPLC for 2 hours. After the reaction was complete, crude Boc-linker-P4-P3-P2-NH2-P1P(OPh)2 was extracted to ethyl acetate, washed twice with 5% NaHCO3, twice with 5% citric acid and twice with brine. The organic layer was dried over MgSO4 for half an hour. Solvent was removed on a rotary evaporator under reduced pressure and all Boc protecting groups (from Ahx and Phe(guan)) were removed with TFA:DCM (1:1, v/v) for 30 minutes, while the reaction progress was monitored by analytical HPLC. The volatiles were removed on a rotary evaporator under reduced pressure. H2N-linker-P4-P3-P2-P1P(OPh)2 (10) was purified by HPLC (Discovery BIO Wide Pore C8–10, semi-preparative column), lyophilized and its molecular weight was confirmed by HRMS.
Coupling of fluorogenic dyes to the peptide sequences
1 eq of ester of fluorogenic dye (BODIPY FL-NHS, Cy3-NHS, Cy5-NHS or Cy7-NHS) was dissolved in DMSO and 2 eq of DIPEA was added. After 3 minutes of preincubation, the mixture was added to a pear-shaped flask with 1.2 eq of (10) H2N-P4-P3-P2-P1P(OPh)2 (dissolved in DMSO). The reaction was carried out for one hour and monitored by analytical HPLC (Discovery BIO Wide Pore C8, analytical column). Crude product was then purified by HPLC (Discovery BIO Wide Pore C8–10, semi-preparative column), lyophilized, dissolved to a final concentration of 10mM and stored at −80°C until use. Purity of the compounds (12) was confirmed by analytical HPLC and HRMS (for HPLC and MS analysis spectra see Supporting Information, Compounds Analysis).
2.6. Determination of inhibition rate constant (kobs/I) of activity-based probes
Neutrophils contain four structurally-related serine proteases NE, PR3, CatG and NSP4 that are stored in azurophilic granules2, 27. To estimate the degree of eventual cross reactivity of the optimal probe designed for targeted enzyme, we tested each champion activity-based probe against each NSPs member. Serial probe dilutions in assay buffer were added in 96 well format in an opaque plate (Corning®) and incubated with enzyme for 5 minutes. Substrate was added and the increase of fluorescence was monitored for 15 minutes. Probes exhibiting inhibitory activity were further characterized to determine second order inhibition rates (kobs/I).
NSPs were incubated with increasing concentrations of probes in at least a two-fold excess over enzyme in appropriate assay buffers (for NE, PR3 and NSP4; 50 mM Hepes, 0.1 M NaCl, 0.1% TRITON X-100, pH 7.45, for CATG 0.1mM Tris/HCl, 0.5M NaCl, pH 7.5) in the presence of champion substrates. Fluorophore release (Rfu) was monitored over time using a BMG ClarioStar®. The pseudo-first order rate constants kobs were determined using GraphPad Prism and the second order rate constants (kobs/I) were calculated by adjusting for the substrate competition factor (1 + [S]/Km), where S is the concentration of substrate in the assay and Km is the Michaelis constant for that substrate/enzyme reaction.
2.7. Selectivity factor determination for individual fluorescent NSP probes
Analysis using purified enzymes.
NE, CATG and PR3 and NSP4 were diluted to a final concentration of 100nM. Probes from PKX05 series were diluted to final concentration of 100nM, with the first dilution in DMSO and subsequent ones in appropriate buffer (see kinetic assay buffers). To test specificity of the PK105 probe NE (100nM) was diluted in assay buffer with 100nM PK105 in a microfuge tube. In parallel, the remaining NSPs (100nM) were incubated for 10 minutes at 37°C with 100nM of PK105. Reactions were terminated for 5 minutes in 95°C in SDS buffer, and run in SDS PAGE (Gradient gels 4–12%, Bolt buffer system, Invitrogen), and visualized using Li-Cor Odyssey Infrared Imaging System Model 9120.
Analysis using neutrophil exocytosis extracts.
Preparation of exocytosis extracts. Exocytosis of azurophil granule proteins (extracts) was induced by treating 2.5 × 107neutrophils/mL with diphenyleneiodonium chloride (10μM) and cytochalasin B for 10 minutes at 37°C, followed by fMLP addition (100nM) for additional 20 minutes at 37°C and centrifuged for 5 minutes, 200 × g22. The supernatant was harvested and stored at −80°C until use. In parallel, Protein A/G-Plus agarose resin, (Santa Cruz Biotechnology) and NSP antibodies were mixed and rocked for 5 hours at 4°C.
Activity-based probe assisted immunocapture of NSPs. To each of 4 microfuge tubes 600μL portions of neutrophil extracts (2.5 × 107cells/mL) was added. The first portion of extracts was treated with Cy5 NE probe (PK105, final concentration 100nM), the second portion was treated with Cy5 CatG probe (PK205, final concentration 100nM), the third portion was treated with Cy5 PR3 probe (PK305, final concentration 100nM), and the fourth portion was treated with Cy5 NSP4 probe (PK405, final concentration 100nM). The treated extracts were mixed gently and incubated for 20 minutes at room temperature. A protease inhibitor cocktail28 (1,10-phenanthroline - 1mM final concentration, 3,4-dichloroisocoumarin - 50μM final concentration, and E-64 – 10μM final concentration) was added and mixed gently to prevent adventitious proteolysis. Each portion was divided into four microfuge tubes and anti-NSP coated protein A/G beads were added as appropriate, followed by incubation at 4°C overnight.
Mixtures were centrifuged at 4°C, the supernatant was gently removed with a needle and beads were washed four times in lysis buffer and three times with TE buffer, followed by addition of SDS buffer, captured proteins were released at 95°C for 5 minutes and run in SDS PAGE (Gradient gels 4–12%, Bolt buffer system, Invitrogen), and visualized using Li-Cor Odyssey Infrared Imaging System Model 9120.
Specific distinction between NE and PR3.
50μL of PK307 (50nM final) probe was incubated with 50μL of neutrophil extracts for 10 minutes at room temperature, followed by addition of 50μL PK105 (50nM final) probe and additional 10 minutes incubation. Parallel, 50μL of PK305 (50nM final) probe was incubated with 50μL of extracts for 10 minutes at room temperature, followed by addition of 50μL PK107 (50nM final) probe and additional 10 minutes of incubation. 50μL of PK107 (50nM final) probe was incubated with 50μL of extracts for 10 minutes at room temperature, followed by addition of 50μL of Hank’s Buffered Salt Solution (HBSS) buffer. 50μL of PK307 probe (50nM final) was incubated with 50μL of extracts for 10 minutes at room temperature, followed by addition of 50μL of HBSS buffer. In the same manner, NSP4 and CATG probes were investigated. Reaction mixtures were boiled in SDS-page buffer containing DTT for 5 minutes, before being resolved by 4–12% SDS-page (Bolt buffer system, Invitrogen). In-gel detection of fluorescently labeled enzymes was carried out directly in the wet gel using Li-Cor Odyssey Infrared Imaging System Model 9120 (700nm and 800nm).
2.8. Titration of neutrophil lysates with activity-based probes
Freshly isolated neutrophils were washed with Dulbecco’s Phosphate Buffered Saline (DPBS) buffer. 5 × 106 neutrophils/mL (for NE, CATG and PR3) or 2 × 107 neutrophils/mL for NSP4 were spun down, the pellet was resuspended in cold lysis buffer (50mM Tris, 1M NaCl and 0.5% Triton X-100), and cells were lysed for three freeze/thaw cycles. 12μL lysate portions were added and treated with 12μL of appropriate inhibitor (PK101, PK201, PK301, PK401) in the rage 46–3000nM and incubated for 30 minutes. Next 12μL of appropriate probe (PK105, PK205, PK305 and PK405 respectively, 200nM final) was added to the samples and incubated for additional 30 minutes. Reaction mixtures were boiled in SDS-page buffer containing DTT for 5 minutes, before being resolved by 4–12% SDS-page (Bolt buffer system, Invitrogen), followed by immunobloting, using rabbit anti-NE antibody (ab21595, abcam, 1:300), rabbit anti-CATG antibody (ab131407, abcam, 1:300), rabbit anti-PR3 antibody (ab21592, abcam, 1:300)., followed by secondary antibody donkey anti-rabbit IRDye®8—CW (926–32213, LI-COR, 1:10000). In-gel detection of fluorescently labeled enzymes was carried out directly in the wet gel using Li-Cor Odyssey Infrared Imaging System Model 9120 (700nm).
2.9. NSP imaging in live neutrophils with fluorescent activity-based probes
Parallel imaging of individual NSPs in neutrophils: Freshly isolated neutrophils (5 × 105/mL) were incubated on cover slips 2.5%FBS/RPMI at 37°C, 5% CO2 with or without 50nM PMA for 2 hours. PK203 was added and incubated for an additional 20 minutes followed by PK302 for an additional 10 minutes, followed by addition of PK407 and PK105. All probes were added to a final concentration of 50nM. Cells were incubated for additional 30 minutes. Thus, PK203 was in contact with the neutrophils for 1 hour, PK302 was in contact with the neutrophils for 40 minutes, and PK407 and PK105 probes were in contact with the neutrophils for 30 minutes. The solution was aspirated and cells were fixed with 4% paraformaldehyde (Sigma) for 40 minutes, mounted with Fluoromount G and slides were imaged using a Leica confocal microscope with 488nm laser (for BODIPY FL), 552nm laser (for Cy3), 648nm laser (for Cy5, Cy7).
Selective labeling with activity-based probes with different fluorescent dyes coupled to the same specific sequence
Slides were prepared in a similar manner as described above, however two probes (100nM of PK105 and 100nM of PK102, 100nM of PK205 and 100nM of PK202, 100nM of PK305 and 100nM of PK302, 100nM of PK405 and 100nM of PK402) were mixed prior to addition to the neutrophils. Probes were incubated with neutrophils for 30 minutes, and the procedure described above was followed.
2.10. Immunolabeling of NSPs
Freshly isolated neutrophils (5 × 105 cells/mL) were allowed to settle on poly-D-lysine treated cover slips 30 minutes, while PMA (50nM) stimulated neutrophils were allowed to settle for 3 hours at 37°C, 5% CO2. Neutrophils were fixed with 4% PFA for 40 minutes and blocked with 10% BSA in HBSS for one hour at room temperature, followed by washing with DPBS. 1:200 dilution of antibodies (sheep anti-NE (ab80043), chicken anti-CATG (ab47787) and mouse anti-PR3 (ab91181), Abcam) in 3% BSA/0.5% TRITON X-100 in DPBS were added to the cover slips and incubated overnight at 4°C. Coverslips were washed three times with 0.5%TRITON X-100 in DPBS and secondary antibodies were added for 1 hour (AF488 goat anti-chicken, AF568 goat anti-mouse and AF680 donkey anti-sheep). Cover slips were washed twice with 3% BSA/0.5%TRITON X-100 in DPBS and mounted with Fluoromount G (for mounting details see Supporting Information Methods). Labeled neutrophils were imaged using a Leica confocal microscope
2.11. Quantitation of NSPs activity-based probes colocalization
Freshly isolated neutrophils (1 × 106/mL) were treated with mixture of probes (100nM of each: PK107, PK205, PK303 and PK40BD) or with only one probe as a control (100nM) and incubated for 30 minutes at 37°C with constant gentle agitation. Cells were centrifuged for 5 minutes at 200 × g and the supernatant was aspirated. 250μL of 4% parafolmaldehyde was added to the pellet and incubated for 40 minutes at room temperature. Cells were centrifuged, the supernatant was removed, and cells were washed in 0.5%TRITON X-100/DPBS buffer. The concentration of the cells was equilibrated to 2 × 107/mL and sorted with ImageStream, mk II, AMNIS, filters 405nm (violet), 488nm (blue) and 642nm (red).
3. RESULTS AND DISCUSSION
3.1. NSPs Substrate Specificity Matrix Using the HyCoSuL Approach
HyCoSuL is a platform for selection of optimally active and selective protease peptide-based substrates (Figure 1), consisting of a large group (over 100) of unnatural amino acids in addition to natural amino acids, and yields a substrate preference matrix of virtually any protease whose specificity determinants reside on the N-terminal side (the non-prime side) of the scissile bond. This technology explores a large number of possible structural variations including large and small aromatic, large and small aliphatic, branched, basic, acidic, hydrophilic and hydrophobic groups, D-amino acids, and amino acids with posttranslational modifications. Previous studies revealed that HyCoSuL screening resulted in highly active substrates and probes for NE, PR3, and NSP421–22. To profile the fourth member of the NSP family (CATG) we synthesized a library with Phe fixed at P1 using the same scaffold as previous HyCoSuL screens (Supporting Information Figure S1). We deployed this approach to select optimal substrates that would distinguish the NSPs from each other and carried out detailed kinetic analyses of each champion substrate (Figure 1). The champion sequence obtained from the CATG screen was synthesized as a substrate with a kcat/Km value of 8.8 (±0.8) × 103 M−1s−1.
Figure 1. Screening methodology and structures of selected champion sequences for individual NSPs.

The first step in the procedure is to screen HyCoSuL libraries where the P1 residue (X) is fixed according to the known primary specificity of each NSP – Phe, Arg or Ala, and the P2, P3, P4 are randomized with natural and unnatural amino acids. In the second step a selection of optimal sequences are synthesized as individual substrates and scanned against all NSPs to discover the most selective sequence. These optimal and selective substrates are called champions, characterized for kcat/Km values and selectivity ratios, and constitute the sequence basis for further analysis. kcat/Km values were calculated using GraphPad Prism software.
The comprehensive HyCoSuL procedure for all four NSPs followed by synthesis of optimal sequences allowed us to generate a substrate matrix (Figure 1) for all four NSPs in terms of optimal activity and, most importantly, selectivity: Nle(O-Bzl)-Met(O)2-Oic-Abu for NE, hCha-Phe(guan)-Oic-Arg for NSP4, His(Bzl)-Val-Pro-Phe for CATG and Glu(O-Bzl)-Lys(Ac)-hPro(Bzl)-Nva for PR3 (Figure 1). We were able to develop favorably selective sequences for CATG and NSP4, while the sequence for NE was slightly cross-reacting with PR3 (ratio around 1:900) and the champion sequence for PR3 also recognized NE (ratio around 1:14) (Figure 1).
3.2. Development and Validation of Highly Selective Enzyme Activity-Based Probes for NSPs
We sought to develop imaging probes that would interact with only the active form of NSPs, allowing us to evaluate their individual activity status during neutrophil functions and disease. We converted the champion substrate sequences to activity-based probes that would allow selective labeling of each NSP in a complex mixture. All activity-based probes were synthesized using the mixed solid phase and solution phase strategy outlined in Figure 2a. We equipped each probe with N-terminal biotin, or with one of four fluorophores (BODIPYFL, Cy3, Cy5 or Cy7) that have minimal wavelength overlap29–31 to allow for simultaneous imaging by fluorescence microscopy. As an electrophilic covalent warhead selective for serine proteases we employed diphenyl phosphonate derivatives related to the P1 preference of each NSPs (Figure 2b)32–33. To avoid interaction of the detection tag with the recognition sequence we incorporated either PEG or Ahx linkers (For structures, see Supporting Information Figure S2). We determined second order inhibition rates for each probe, and to simplify the descriptions throughout the paper we gave code names to each probe (Table 1).
Figure 2. (a) Scheme of activity-based probe synthesis using PK305 as an example.

The respective P1 amino acid was loaded on the Chlorotrityl resin using DIPEA in dry DCM, followed by coupling of the P2–P4 residues by standard solid phase elongation. Following deprotection each tetrapeptide was cleaved from the resin with and attached to the hydrobromide salt of NvaP(OPh)2. The Boc protecting group was removed, followed by lyophilization and attachment of Cyanine5-NHS. The crude product was purified by HPLC, lyophilized and stored at −80°C until use. (b) Structures of the components of each activity-based probe used in this study. We utilized 16 fluorescent probes, and they are coded throughout the manuscript according to the N-terminal fluorescent tag (X) and specificity sequence as indicated in the panel. Thus, NE series probes are always PK10X, where X is one of the four fluorophores in the panel, CATG are PK20X, etc. for the other enzyme probes. (c) Second order rate constants for each Cy5 coded champion probe (PKX05 series) with each NSP. The log scale compresses the S.D. error bars from biological replicates, which were all within 10% of the mean. (d) The overall principle of activity-based probes.
Table 1.
Activity-based probes equipped with different N-terminal fluorescent tags.
| fluorophore | PKcode | kobs/I (M−1 s−1) | ||||
|---|---|---|---|---|---|---|
| NE | CATG | PR3 | NSP4 | |||
| NE probes | BODIPYFL | 102 | 2.7 ± 0.20 × 106 | NI | 4.2 ± 0.48 × 105 | NI |
| Cy3 | 103 | 2.3 ± 0.21 × 106 | NI | 4.6 ± 0.51 × 105 | NI | |
| Cy5 | 105 | 8.1 ± 0.68 × 106 | NI | 1.9 ± 0.10 × 105 | NI | |
| Cy7 | 107 | 7.6 ± 0.89 ×106 | NI | 3.6 ± 0.80 ×105 | NI | |
| CATG probes | BODIPYFL | 202 | NI | 4.3 ± 0.3 × 103 | NI | NI |
| Cy3 | 203 | NI | 3.5 ± 20 × 103 | NI | NI | |
| Cy5 | 205 | NI | 1.6 ± 15 × 103 | NI | NI | |
| Cy7 | 207 | NI | 1.4 ± 0.23 × 103 | NI | NI | |
| PR3 probes | BODIPYFL | 302 | 300 ± 50 | NI | 1.4 ± 11 × 106 | NI |
| Cy3 | 303 | 1200 ± 30 | NI | 4.7 ± 0.45 × 105 | NI | |
| Cy5 | 305 | 80 ± 20 | NI | 7.2 ± 0.12 × 105 | NI | |
| Cy7 | 307 | 600 ± 200 | NI | 7.8 ± 0.56 × 105 | NI | |
| NSP4 probes | BODIPYFL | 402 | NI | NI | NI | 3.8 ± 0.5 × 106 |
| Cy3 | 403 | NI | NI | NI | 4.0 ± 0.2 × 104 | |
| Cy5 | 405 | NI | NI | NI | 4.7 ± 0.3 × 104 | |
| Cy7 | 407 | NI | NI | NI | 5.2 ± 0.2 × 104 | |
Based on the large selectivity factors for champion probes with Cy5 equipped probes (PKX05 series) as examples (Table 1, Figure 2c,d) we predicted that we would be able to specifically identify PR3, CATG and NSP4 in complex mixtures. However, cross-reactivity of the optimal NE probe with PR3 requires an alternative approach to allow distinction between these two closely related proteases. To validate this we incubated each Cy5 probe with its targeted isolated enzyme or a mixture of the remaining three NSPs (Figure 3a). CATG, PR3 and NSP4 were all uniquely labeled by their respective Cy5 probes, but the NE probe PK105 also labeled a component in the NE-deficient mixture, likely PR3 based on our kinetic results. As a further step of validation we induced exocytosis of azurophil granule proteins to obtain a mixture of NSPs, incubated these extracts with individual probes and captured each enzyme with specific antibodies (Figure 3b). Using these extracts, obtained from fresh peripheral neutrophils isolated from healthy donors, we confirmed that PK205 and PK305 probes bind exclusively to the targeted enzymes, while PR3 binds PK305 and PK105 probes. On the basis of these specificity checks we conclude that series CATG, PR3 and NSP4 probes are highly selective, and that the NE probes are optimal, but require additional considerations to be selective.
Figure 3. Selectivity of champion NSP probes.

(a) Champion probes (100nM) were incubated with their target NSP or with a mixture of the other NSPs for 10 minutes followed by SDS PAGE and fluorescence imaging of the gel. (b) Neutrophil extracts were treated with probes (100nM), followed by a protease inhibitor cocktail (1mM 1,10-phenanthroline, 50μM DCI, 10μM E-6) and the indicated specific antibodies, captured by Protein A/G-Plus agarose beads, followed by SDS PAGE of the captured proteins, and fluorescence imaging of the gels. No bands were detected with the NSP4 antibody capture, likely because the antibody is poor at recognizing the native protein. (c) PK10X series probes preferentially label NE in the presence of PR3. PR3 probes were added to neutrophil extracts at time = 0, followed in lanes 3–4 by a second NE probe after 10 min as indicated above the gel. Each sample was incubated for a total of 20 min before SDS page, and gels were analyzed for Cy5 and Cy7 fluorescence. All probes were used at 50nM final concentration. PK107 labels NE glycoforms and a small amount of PR3, whereas PK305 labels PR3 exclusively. The small amount of cross reactivity of PK10X series probes (see the faint overlaid band in lane 3) represents off target labeling of PR3 by PK107. Images describe the individual channels that form the basis of the overlay. (d) Lysates were treated with PKX01 (biotin-tagged) activity-based probes to act as titration inhibitors (I) in the indicated concentration range for 45 minutes at 37°C, followed by 200nM Cy5 probes (PK X05 series) and an additional 30 minutes incubation. For NE, CATG and PR3, western blots were performed and membranes were stained with appropriate antibodies. The membranes were imaged for the Cy5 probes (red) and IRDye800W® (green).
Inhibition rate constants (kobs/I) were calculated under pseudo first order conditions34. All calculations were repeated in triplicate represent (± SD). Code numbers are given to simplify discussion of the probes throughout the paper. NI – no inhibition observed.
There are essentially two ways that the activity-based probes can be used to address neutrophil protease function. One would require complete inhibition of each enzyme by saturating with probe, and the other would be to add trace amounts of probe to follow the location and disposition of each enzyme without affecting their overall function. Because the NE probe is so efficient (kobs/I > 107 M−1s−1) it also reacts with the closely related PR3, and so we needed to develop a reliable protocol that would diminish labeling of PR3 by NE probes (10X series). We found that the most efficient way to do this was by incubating the PR3 probe with neutrophils before adding the NE probe, and providing the NE probe at trace-labeling concentrations. With this caveat, all probes are now stringently selective for their individual target enzymes (Figure 3c, Supporting Information Figure S3). To date the total amount of active NSPs has not been reported, due to lack of available reagents. We titrated neutrophil lysates with different concentrations of biotinylated probes (PK101, PK201, PK301 and PK401) and after 45 minutes incubation we added Cy5 probes (PK105, PK205, PK305 and PK405 respectively). The total amount of targeted enzyme is thus defined by the concentration of covalent PKX01 inhibitors that prevent PKX05 binding. NSP antibodies were used to ensure that all lanes contain equivalent amount of protein (Figure 3d, for full length gels, please see Supporting Information Figure S4). The fluorescent probes signals were quantitated and plotted against inhibitor concentration to derive IC50 values from at least two biological replicates (individual blood donors). Interestingly, we observed quiet of broad range and we attribute this to the donor effect, since each of the biological replicates was from separate donors. Active NSP concentrations (measured in 107 cells/mL) ranged from 230–340 nM for NE, 150–530 nM for PR3, 75–188 nM for CATG and 17–34 nM for NSP4 (Figure 3d), in agreement with literature data that concluded NSP4 is present at about 5–10 % the amount of NE2. The large amounts of active NSPs in neutrophils mitigated against saturating with probe, so we conducted experiments where trace amounts of NE and PR3 selective probes were added sequentially (50–100nM). As demonstrated in Figure 3c, the trace labeling experiments provided good selectivity of PK10X series probes for NE, even in the presence of PR3.
3.3. Localization of active NSPs in neutrophils
NSPs are processed to their active forms in the trans-Golgi prior to targeting to, and storage in azurophil granules: first removal of a short N-terminal dipeptide and second by removal of a C-terminal peptide35–36. To test whether NSPs are sorted and localize to the same azurophil granule we coded each specific probe with a different fluorophore and incubated them with freshly prepared unstimulated and PMA-stimulated neutrophils. After fixing, washing and mounting, we imaged neutrophils with confocal microscopy, reducing the emission wavelengths to diminish potential fluorophore signal overlap.
The probes revealed that individual NSPs appeared to have a non-overlapping punctate distribution (Figure 4a). Since this observation was unexpected, we hypothesized that some property of the probes was leading toward an uneven distribution. To test this possibility we chose three strategies. In the first we incubated two different fluorophore-tagged probes (Cy5 and BODIPY FL) for each individual NSP asking whether the different tags may alter localization (Figure 4c). In the second we used antibodies specific for the individual enzymes combined with three secondary antibodies equipped with different fluorescent tags (Figure 4d). In the third we employed selective NSP probes followed by fixation and co-staining with specific antibodies (Figure 4e). In each case these experiments demonstrated that probes with the same selectivity sequence, regardless of visualization tag, are targeted to the same localization within the neutrophil confirming, by complementary methods, the dislocated distribution of NSPs within single neutrophils (See Supporting Information Figure S4).
Figure 4. Localization of active NSPs in neutrophil granules.

(a) Freshly isolated neutrophils were allowed to settle on cover slips, treated or untreated with PMA for 3 hours, incubated with the indicated probes (50nM) for 30 min and fixed (b) scheme of experimental flow (c) Neutrophils labeled with two probes (100nM each) with the same sequence and two different fluorescent dyes and fixed. Each signal overlays well, demonstrating specific probe labeling of the targeted enzyme. For separate channels see Supporting Information Figure S6. (d) Neutrophil staining with antibodies for three individual NSPs. Neutrophils were prepared as in (a) without probe labeling, stained with anti-NE, anti-PR3 and anti-CATG followed by secondary antibodies with the fluorophores indicated in the figure. (e) Co-staining with activity-based probes and antibodies. Neutrophils were allowed to settle on cover slips, treated with 50nM of the indicated probes, fixed and stained with anti-NE, anti-PR3, anti-CATG and anti-NSP4 respectively, followed by a secondary antibodies with the indicated fluorophores. (For separate channels see Supporting Information Figure S7). All slides were mounted and imaged by confocal microscopy with 488nm, 552nm and 648nm lasers. Images are representative of 3 separate donors (For more examples and z-stack movies see Supporting Information Figure S5–S7 and Supporting Information movies S1–S4)
These observations imply that NSPs are localized separately in distinctive azurophilic neutrophil granules, and by way of quantifying this finding we sorted cells according to their contents/loading using imaging flow cytometry (Figure 5a). To quantitate at a population level the degree of colocalization of NSPs revealed by the probes we utilized flow cytometry (Amnis ImageStream), which provides qualitative and quantitative microscopic image data of every event acquired in flow in bright field and all fluorophore channels utilized by our probes (Figure 5). The level of magnification does not allow for quantitation of individual puncta (azurophil granules), however we were able to assess the degree of colocalization between probes on a per cell basis using IDEAS image cytometry analysis software. The Bright Detail Similarity R3 feature (BDS) calculates the log transformed Pearson’s correlation coefficient of the localized bright spots in two input images, which removes diffuse signals and compares the spatial distribution of the bright puncta from two NSP probes37. BDS was calculated in a pairwise fashion for every combination of the four NSP probes (Figure 5b).
Figure 5. Quantitation of NSP probe colocalization and potential sorting signals encoded within NSPs.

Freshly isolated neutrophils from a healthy donor were treated with PK107, PK205, PK303 and PK402, followed by washing, fixation, and analysis by imaging flow cytometry with 60X objective and EDF. Single in-focus neutrophils were selected in IDEAS software with the Brightfield Gradient RMS, Brightfield Area, Brightfield Aspect Ratio, and Side Scatter Intensity features prior to NSP colocalization analysis. (a) Bright detail similarity (BDS) was calculated for each pair of probes according to fluorescence intensity from specifically labeled NSPs in neutrophils. Cells were considered colocalized for a probe pair if BDS ≥ 1.5. (b) Compilation of colocalized BDS events – final colocalization of all four BDSs is 0.2%. The analysis is a representative of experiments from 2 healthy donors. (c) An example of 5 of the 9,394 events used for quantitation. Each row represents images from one neutrophil. (d) Alignment of the C-terminal tails of HeSPs with the archetype of the family chymotrypsin. Red text indicates C-terminal extensions that are known (PR3, CATG, NE) or suspected (NSP4) to be cleaved during protease maturation.
With this experiment we calculated only 0.2% of the total amount of neutrophils have granules containing all four enzymes, while the majority of the total amount of neutrophils demonstrate individual loading of the granules (Figure 5b, c) – confirming at a population level that neutrophil enzymes are loaded separately. Indeed the extent of colocalization was so low that it seemed to be that the NSPs are anti-localized (dislocated or positively segregated) by a mutual exclusion mechanism.
Azurophil granules were once considered to be neutrophil lysosomes, but later shown to have functional characteristics of a regulated secretory granule38. Normally considered to constitute a homogeneous population, there has been tentative evidence that there are two distinct subpopulations of azurophil granules based on centrifugation analysis39, later expanded to between 10 and 13 suggestive of both functional and maturational differences40. Thus there is precedence for distinct neutrophil granule heterogeneity, but the degree of non-localization of contents of the azurophil granules was unexpected. Indeed, we were not able to find any evidence in the literature for specialized protein targeting in any post Golgi vesicle system similar to that we have demonstrated for the spatial compartmentalization of the four NSPs.
Mechanisms for protein targeting to neutrophil granules from the trans-Golgi network remain unidentified, primarily because these cells are terminally differentiated and do not synthesize granule proteins41. NE and CATG undergo two processing steps during packaging. The first is the removal of an N-terminal dipeptide that generates proteolytic activity. The second is the removal of a C-terminal peptide, and it is this latter event that may pertain to packaging mechanisms35–36. Thus, the C-terminus of NE contains an adaptor protein 3 (AP-3) signal that has been suggested to provide sorting signals42. Little if anything is known about potential sorting signals in the C-terminal peptides of the other NSPs. Significantly, these C-terminal extensions seem to be unique to NSPs among the broader family of HeSPs and are distinct for each NSP (Figure 5d). They exist, but are not well conserved, in NSPs throughout mammals, and so we would predict that they represent a specialized neutrophil sorting function. As yet there is no understanding of why they are removed following packaging, although it has been suggested that the removal in NE reveals a cryptic sorting signal43. Our demonstration of dislocated packaging of NSPs is concrete visual and quantifiable evidence related to the previous finding that there may be as many as 13 varieties of azurophil granule40.
4. CONCLUSIONS
In recent years, development of activity-based probes has led to a significant improvement in the imaging of active proteases in situ44–45. A daunting challenge has been to image in parallel several enzymes in their active forms at the same time and location46, a problem hindered by the absense of sequences that can distinguish between closely related enzymes47. In addressing this challenge we set out to design a toolbox to reliably image the four known neutrophil serine proteases. Initial experiments demonstrated the feasibility of selecting highly active substrates and biotin-labeled probes for NE and NSP4 through HyCoSuL screening21–22, 48. We have now utilized this technique to develop selective probes for the other two human NSPs, CATG and PR3. NE and PR3 probes are very efficient (kobs/I>107 and 5 × 105 respectively, while NSP4 and CATG probes are less efficient (kobs/I> 4 × 104 and 1.4 × 103 respectively) yet show high discrimination between NSPs. The stringency of the probes was successfully demonstrated by labeling of active NSPs in neutrophil exocytosis extracts and lysates.
Armed with these selective fluorescent probes we were now able to address the question of the localization of active NSPs in neutrophils. The degree of colocalization is so low that it appears that a discriminatory packaging system operates at the stage of myelopoiesis when azurophil granules are populated. A number of mechanisms seem plausible, and need to be investigated in future studies. For example, neutrophils may be released from the bone marrow with an even NSP distribution that deviates as they age. Formation of individual azurophil granules may proceed in kinetic or spatial waves that tend to translate messages at different times or locations within neutrophil progenitors. Equally likely is that each NSP is sorted into pre-azurophil vesicles based on information in their distinct C-terminal peptides (Figure 5d). Importantly, all prior work has considered that NSPs are co-sorted, and the discriminatory localization based on NSP content, revealed in our work thus has no precedent.
In summary, we have developed a first in class toolbox for reliable parallel imaging of active neutrophil serine proteases and demonstrated that these enzymes are unevenly located in the granules of human neutrophils to an extent that implies a hitherto unknown discriminatory packaging system. The utility of this approach can be readily extended to other groups of proteases using HyCoSuL as a tool for selection of champion recognition sequences in combination with different fluorophores with non overlapping emission spectra.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants NIH R01GM099040 to GS, and 2014/14/M/ST5/00619 by National Science Centre in Poland to MD. PK is the beneficiary of a START scholarship from the Foundation for Polish Science. We would like to thank Daniel Kirchhofer, Ph.D. for kind gift of the NSP4 protease. We would like to thank to Scott Snipas for his technical assistance.
Footnotes
SUPPORTING INFORMATION
Supporting figures: CATG substrate specificity, structure of PKX0X activity-based probes, experimental methods and characterisation of all compounds
COMPETING INTEREST STATEMENT
The authors declare no competing financial interests
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding authors upon request.
REFERENCES
- 1.Cowland JB; Borregaard N, Granulopoiesis and granules of human neutrophils. Immunol Rev 2016, 273 (1), 11–28. [DOI] [PubMed] [Google Scholar]
- 2.Perera NC; Schilling O; Kittel H; Back W; Kremmer E; Jenne DE, NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci U S A 2012, 109 (16), 6229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brinkmann V; Reichard U; Goosmann C; Fauler B; Uhlemann Y; Weiss DS; Weinrauch Y; Zychlinsky A, Neutrophil extracellular traps kill bacteria. Science 2004, 303 (5663), 1532–5. [DOI] [PubMed] [Google Scholar]
- 4.Korkmaz B; Attucci S; Juliano MA; Kalupov T; Jourdan ML; Juliano L; Gauthier F, Measuring elastase, proteinase 3 and cathepsin G activities at the surface of human neutrophils with fluorescence resonance energy transfer substrates. Nat Protoc 2008, 3 (6), 991–1000. [DOI] [PubMed] [Google Scholar]
- 5.Belaaouaj A; Kim KS; Shapiro SD, Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science 2000, 289 (5482), 1185–8. [DOI] [PubMed] [Google Scholar]
- 6.Tkalcevic J; Novelli M; Phylactides M; Iredale JP; Segal AW; Roes J, Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity 2000, 12 (2), 201–10. [DOI] [PubMed] [Google Scholar]
- 7.Korkmaz B; Horwitz MS; Jenne DE; Gauthier F, Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010, 62 (4), 726–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horwitz M; Benson KF; Person RE; Aprikyan AG; Dale DC, Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet 1999, 23 (4), 433–6. [DOI] [PubMed] [Google Scholar]
- 9.Horwitz MS; Corey SJ; Grimes HL; Tidwell T, ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am 2013, 27 (1), 19–41, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kollner I; Sodeik B; Schreek S; Heyn H; von Neuhoff N; Germeshausen M; Zeidler C; Kruger M; Schlegelberger B; Welte K; Beger C, Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood 2006, 108 (2), 493–500. [DOI] [PubMed] [Google Scholar]
- 11.Salvesen GS; Hempel A; Coll NS, Protease signaling in animal and plant-regulated cell death. FEBS J 2016, 283 (14), 2577–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caughey GH; Schaumberg TH; Zerweck EH; Butterfield JH; Hanson RD; Silverman GA; Ley TJ, The human mast cell chymase gene (CMA1): mapping to the cathepsin G/granzyme gene cluster and lineage-restricted expression. Genomics 1993, 15 (3), 614–20. [DOI] [PubMed] [Google Scholar]
- 13.Schechter I; Berger M, On the size of the active site in proteases. Biochem. Biophys. Res. Commun 1967, 27, 157–162. [DOI] [PubMed] [Google Scholar]
- 14.Wei AZ; Mayr I; Bode W, The refined 2.3 A crystal structure of human leukocyte elastase in a complex with a valine chloromethyl ketone inhibitor. FEBS Lett 1988, 234 (2), 367–73. [DOI] [PubMed] [Google Scholar]
- 15.Rao NV; Hoidal JR, Chapter 589 - Myeloblastin In Handbook of Proteolytic Enzymes, Rawlings ND, Salvesen Guy, Ed. Academic Press: 2013; pp 2666–2675. [Google Scholar]
- 16.Harris JL; Backes BJ; Leonetti F; Mahrus S; Ellman JA; Craik CS, Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci U S A 2000, 97 (14), 7754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schilling O; Overall CM, Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol 2008, 26 (6), 685–94. [DOI] [PubMed] [Google Scholar]
- 18.O’Donoghue AJ; Jin Y; Knudsen GM; Perera NC; Jenne DE; Murphy JE; Craik CS; Hermiston TW, Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS One 2013, 8 (9), e75141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salvesen GS, Chapter 588 - Cathepsin G In Handbook of Proteolytic Enzymes, Rawlings ND, Salvesen Guy, Ed. Academic Press: 2013; pp 2661–2666. [Google Scholar]
- 20.Perera NC; Wiesmuller KH; Larsen MT; Schacher B; Eickholz P; Borregaard N; Jenne DE, NSP4 is stored in azurophil granules and released by activated neutrophils as active endoprotease with restricted specificity. J Immunol 2013, 191 (5), 2700–7. [DOI] [PubMed] [Google Scholar]
- 21.Kasperkiewicz P; Poreba M; Snipas SJ; Lin SJ; Kirchhofer D; Salvesen GS; Drag M, Design of a Selective Substrate and Activity Based Probe for Human Neutrophil Serine Protease 4. PLoS One 2015, 10 (7), e0132818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasperkiewicz P; Poreba M; Snipas SJ; Parker H; Winterbourn CC; Salvesen GS; Drag M, Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc Natl Acad Sci U S A 2014, 111 (7), 2518–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poreba M; Solberg R; Rut W; Lunde NN; Kasperkiewicz P; Snipas SJ; Mihelic M; Turk D; Turk B; Salvesen GS; Drag M, Counter Selection Substrate Library Strategy for Developing Specific Protease Substrates and Probes. Cell Chem Biol 2016, 23 (8), 1023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rut W; Zhang L; Kasperkiewicz P; Poreba M; Hilgenfeld R; Drag M, Extended substrate specificity and first potent irreversible inhibitor/activity-based probe design for Zika virus NS2B-NS3 protease. Antiviral Res 2017, 139, 88–94. [DOI] [PubMed] [Google Scholar]
- 25.Maly DJ; Leonetti F; Backes BJ; Dauber DS; Harris JL; Craik CS; Ellman JA, Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J Org Chem 2002, 67 (3), 910–5. [DOI] [PubMed] [Google Scholar]
- 26.Soroka M, G. W, Sposób wytwarzania estrów diarylowych kwasów 1-alkiloksykarbonyloaminoalkilofosfonowych. Polish Patent 2007, PL196222. [Google Scholar]
- 27.Korkmaz B; Moreau T; Gauthier F, Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 2008, 90 (2), 227–42. [DOI] [PubMed] [Google Scholar]
- 28.Salvesen G; Nagase H, Inhibition of proteolytic enzymes In Proteolytic Enzymes: A Practical Approach, Beynon RJ; Bond JS, Eds. IRL Press: Oxford, 1989; pp 83–104. [Google Scholar]
- 29.Giepmans BN; Adams SR; Ellisman MH; Tsien RY, The fluorescent toolbox for assessing protein location and function. Science 2006, 312 (5771), 217–24. [DOI] [PubMed] [Google Scholar]
- 30.Berlier JE; Rothe A; Buller G; Bradford J; Gray DR; Filanoski BJ; Telford WG; Yue S; Liu J; Cheung CY; Chang W; Hirsch JD; Beechem JM; Haugland RP; Haugland RP, Quantitative comparison of long-wavelength Alexa Fluor dyes to Cy dyes: fluorescence of the dyes and their bioconjugates. J Histochem Cytochem 2003, 51 (12), 1699–712. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi-Takanaka Y; Stasevich TJ; Kurumizaka H; Nozaki N; Kimura H, Evaluation of chemical fluorescent dyes as a protein conjugation partner for live cell imaging. PLoS One 2014, 9 (9), e106271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sienczyk M; Oleksyszyn J, Irreversible inhibition of serine proteases - design and in vivo activity of diaryl alpha-aminophosphonate derivatives. Curr Med Chem 2009, 16 (13), 1673–87. [DOI] [PubMed] [Google Scholar]
- 33.Oleksyszyn J; Powers JC, Irreversible inhibition of serine proteases by peptide derivatives of (alpha-aminoalkyl)phosphonate diphenyl esters. Biochemistry 1991, 30 (2), 485–93. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Q; Salvesen GS, Viral caspase inhibitors CrmA and p35. Methods Enzymol 2000, 322, 143–54. [DOI] [PubMed] [Google Scholar]
- 35.Salvesen G; Enghild JJ, An unusual specificity in the activation of neutrophil serine proteinase zymogens. Biochemistry 1990, 29, 5304–5308. [DOI] [PubMed] [Google Scholar]
- 36.Gullberg U; Lindmark A; Lindgren G; Persson AM; Nilsson E; Olsson I, Carboxyl-terminal prodomain-deleted human leukocyte elastase and cathepsin G are efficiently targeted to granules and enzymatically activated in the rat basophilic/mast cell line RBL. J Biol Chem 1995, 270 (21), 12912–8. [DOI] [PubMed] [Google Scholar]
- 37.Pugsley HR, Quantifying autophagy: Measuring LC3 puncta and autolysosome formation in cells using multispectral imaging flow cytometry. Methods 2017, 112, 147–156. [DOI] [PubMed] [Google Scholar]
- 38.Cieutat AM; Lobel P; August JT; Kjeldsen L; Sengelov H; Borregaard N; Bainton DF, Azurophilic granules of human neutrophilic leukocytes are deficient in lysosome-associated membrane proteins but retain the mannose 6-phosphate recognition marker. Blood 1998, 91 (3), 1044–58. [PubMed] [Google Scholar]
- 39.Kinkade JM Jr.; Pember SO; Barnes KC; Shapira R; Spitznagel JK; Martin LE, Differential distribution of distinct forms of myeloperoxidase in different azurophilic granule subpopulations from human neutrophils. Biochem Biophys Res Commun 1983, 114 (1), 296–303. [DOI] [PubMed] [Google Scholar]
- 40.Rice WG; Kinkade JM Jr.; Parmley RT, High resolution of heterogeneity among human neutrophil granules: physical, biochemical, and ultrastructural properties of isolated fractions. Blood 1986, 68 (2), 541–55. [PubMed] [Google Scholar]
- 41.Sheshachalam A; Srivastava N; Mitchell T; Lacy P; Eitzen G, Granule protein processing and regulated secretion in neutrophils. Front Immunol 2014, 5, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horwitz M; Benson KF; Duan Z; Li FQ; Person RE, Hereditary neutropenia: dogs explain human neutrophil elastase mutations. Trends Mol Med 2004, 10 (4), 163–70. [DOI] [PubMed] [Google Scholar]
- 43.Benson KF; Li FQ; Person RE; Albani D; Duan Z; Wechsler J; Meade-White K; Williams K; Acland GM; Niemeyer G; Lothrop CD; Horwitz M, Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 2003, 35 (1), 90–6. [DOI] [PubMed] [Google Scholar]
- 44.Sanman LE; Bogyo M, Activity-based profiling of proteases. Annu Rev Biochem 2014, 83, 249–73. [DOI] [PubMed] [Google Scholar]
- 45.Yang P; Liu K, Activity-based protein profiling: recent advances in probe development and applications. Chembiochem 2015, 16 (5), 712–24. [DOI] [PubMed] [Google Scholar]
- 46.Edgington-Mitchell LE; Barlow N; Aurelio L; Samha A; Szabo M; Graham B; Bunnett N, Fluorescent diphenylphosphonate-based probes for detection of serine protease activity during inflammation. Bioorg Med Chem Lett 2017, 27 (2), 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasperkiewicz P; Poreba M; Groborz K; Drag M, Emerging challenges in the design of selective substrates, inhibitors and activity-based probes for indistinguishable proteases. FEBS J 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guarino C; Legowska M; Epinette C; Kellenberger C; Dallet-Choisy S; Sienczyk M; Gabant G; Cadene M; Zoidakis J; Vlahou A; Wysocka M; Marchand-Adam S; Jenne DE; Lesner A; Gauthier F; Korkmaz B, New selective peptidyl di(chlorophenyl) phosphonate esters for visualizing and blocking neutrophil proteinase 3 in human diseases. J Biol Chem 2014, 289 (46), 31777–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.