

Abstract
Objective:
Interest in candidate gene and candidate gene-by-environment interaction hypotheses regarding major depressive disorder remains strong despite controversy surrounding the validity of previous findings. In response to this controversy, the present investigation empirically identified eighteen candidate genes for depression studied ten or more times and examined evidence for their relevance to depression phenotypes.
Method:
Utilizing data from large population-based and case-control samples (n ranging from 62,138 to 443,264 across subsamples), we conducted a series of preregistered analyses examining polymorphism main effects, polymorphism × environmental moderator interactions, and gene-level effects across a number of operational definitions of depression (e.g., lifetime diagnosis, current severity, episode recurrence) and environmental moderators (e.g., sexual or physical abuse during childhood, socioeconomic adversity).
Results.
There was no clear evidence for any candidate gene polymorphism associations with depression phenotypes or any polymorphism × environmental moderator effects. As a set, depression candidate genes were no more associated with depression phenotypes than noncandidate genes. We demonstrate that phenotypic measurement error is unlikely to account for these null findings.
Conclusions:
Our results do not support previous depression candidate gene findings, wherein large genetic effects are frequently reported in samples orders of magnitude smaller than those examined here. Instead, our results suggest that early hypotheses about depression candidate genes were incorrect and that the large number of associations reported in the depression candidate gene literature are likely to be false positives.
Introduction
Major Depressive Disorder (hereafter referred to as “depression”) is moderately heritable (twin-based heritability « 37%) (1), but its genetic architecture is complex, and identifying specific polymorphisms underlying depression susceptibility has been challenging. With the ability to genotype particular genetic variants and optimism about the potential public health impact of identifying reliable biomarkers for depression (2), early research focused on the effects of specific candidate polymorphisms in genes hypothesized to underlie depression liability. These genes were chosen based on hypotheses regarding the biological underpinnings of depression. The 5-HTTLPR variable number tandem repeat (VNTR) polymorphism in the promoter region of the serotonin transporter gene SLC6A4, the most commonly studied polymorphism in relation to depression (Figure 1, Table S1.1), serves as a prototypical example: given the theorized importance of the serotonergic system in the etiology of depression, a logical target for early association studies was a common, large (and hence relatively easy to genotype), and potentially functional repeat polymorphism in a serotonergic gene (3–5). Early investigations, though focused on a small number of variants by necessity (low cost genomewide arrays were not yet available), reported promising positive associations. However, replication attempts led to inconsistent results (6–8).
Figure 1.
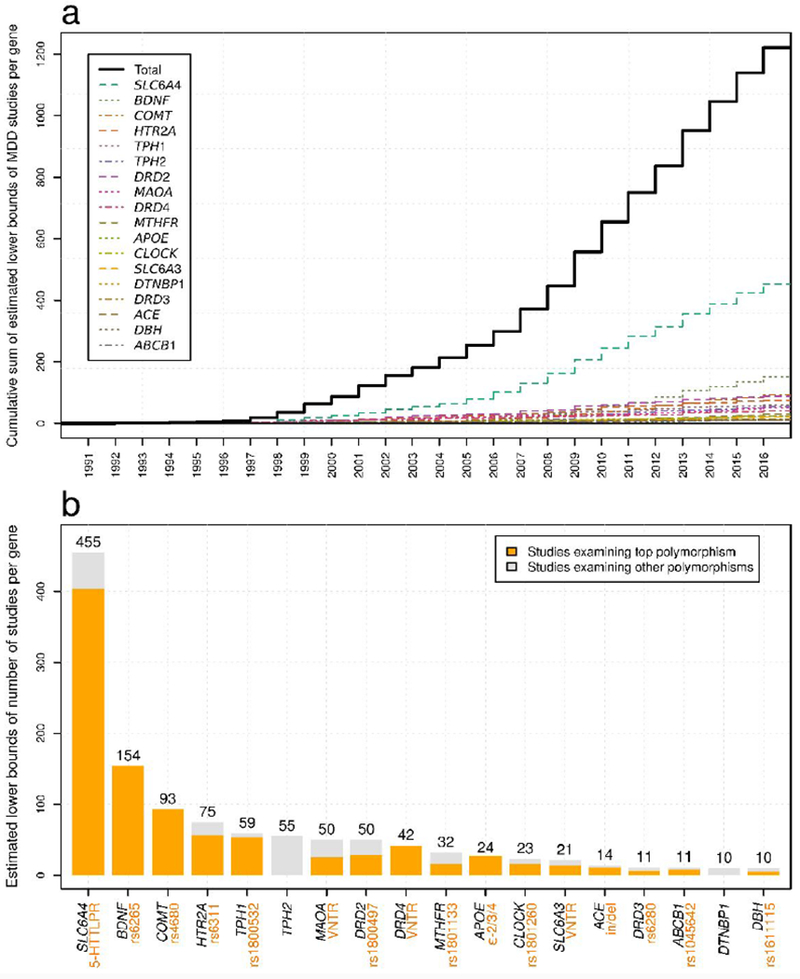
Estimated lower bounds of studies-per-candidate gene.
a. Cumulative sums of the estimated number of depression candidate gene studies identified by our algorithm per year per gene from 1991 through 2016. Estimates reflect the number of correctly classified studies among identified studies, excluding studies not detected by our protocol, and thus comprise lower bounds for the true number of studies-per-gene.
b. Eighteen candidate genes studied ≥ 10 times between 1991 and 2016. The estimated number of studies focused on the top polymorphism (Table S1.1) is displayed relative to the other identified studies within each gene. No top polymorphisms were identified for DTNBP1 or TPH2 (supplement section S1).
To critics of candidate gene findings, replication failures suggested that initial reports were artifactual (9–11). However, at least two alternative explanations could account for inabilities to replicate early reports and inconsistent results across studies. First, in the early 2000s, Caspi et al. (12) posited that previous inconsistencies might reflect the effects of candidate polymorphisms that were dependent on environment exposures (gene-by-environment interaction [G × E] effects). In what would become one of the most highly-cited (> 8000 citations as of July, 2018) and influential papers in psychiatric genetics, Caspi et al. reported that the impact of the 5-HTTLPR repeat polymorphism in SLC6A4 on depression was moderated by exposure to stressful life events, such that the positive association between stressful life events and depression was stronger in individuals carrying the “short” allele (14). This early work led many researchers to shift their attention to G × E hypotheses, focusing on the same polymorphisms first investigated for main effects (8). Second, in an alternative but complementary line of reasoning, other researchers suggested that polymorphisms other than those studied previously in the same candidate genes were likely to explain depression risk, given the genes’ putative biological relevance (15). All three lines of inquiry are well represented in the published literature of the past twenty-five years: thousands of investigations of depression or depression endophenotypes have examined the direct effects of 1. the most studied polymorphisms within candidate genes, 2. the moderation of their effects by environmental stressors, or 3. the effects of alternative polymorphisms within the same candidate genes. The popularity of these lines of inquiry has not diminished over time (Figure 1, supplement sections S1.4, S1.5), with many studies reporting statistically significant associations.
Perhaps surprisingly given the continued interest in studying these historic depression candidate genes and the large number of associations documented in the candidate gene literature, many researchers have expressed extreme skepticism about the validity of such findings (11,16–18). There are several reasons for this. First, genome-wide association studies (GWAS), which agnostically examine associations at millions of common single nucleotide polymorphisms (SNPs) across the genome in large samples, have consistently found that individual SNPs exert small effects on genetically-complex traits such as depression (19–21). For example, in the most recent GWAS of depression, which utilized a sample of 135,458 cases and 344,901 controls, the strongest individual signal detected (rs12552; odds ratio = 1.044; p = 6.07e-19) would require a sample of approximately 34,100 individuals to be detected with 80% power at α = .05, assuming a balanced case-control design (19). In contrast, the median study sample size in a review of 103 candidate G × E studies published during 2000-2009 was 345, with 65% of studies reporting positive results (16). Thus, given the small sample sizes typically employed, candidate gene research has likely been severely underpowered (22,23). This, in turn, may suggest that the false discovery rate for the many positive reports in the candidate gene literature is high. Consistent with this possibility, targeted, well-powered genetic association studies of depression and other psychiatric phenotypes in large samples have not supported candidate gene hypotheses (19,24–28). For example, a preregistered, collaborative meta-analysis of the stressful life event × 5-HTTLPR interaction in a sample of 38,802 individuals failed to support the original finding of Caspi et al. (29), though we note that this variant and several other candidate VNTRs have not been previously examined in a GWAS context (30,31). The absence of previous large-sample investigations of VNTR hypotheses is noteworthy as VNTRs comprise several of the earliest candidate polymorphisms to be examined in the context behavioral research; concerns about variability in VNTR genotyping procedures and analysis methods over time have further complicated the interpretation of the existing literature (32). Additionally, a number of researchers have suggested that incorrect analytic methods and inadequate control for population stratification characterize the majority of published candidate gene studies (22,33–35), and other researchers have questioned the clinical utility of focusing on individual polymorphisms or polymorphism-by-environment interactions (36). Finally, there is evidence of systematic publication bias in the candidate gene literature; in the aforementioned review of all candidate G × E studies published between 2000 and 2009, 96% percent of novel findings were significant compared to only 27% of replication attempts, and replication attempts reporting null findings had larger sample sizes than those presenting positive findings (16). In response to such skepticism, candidate gene proponents have argued that lack of replication of candidate gene associations in large sample studies may reflect poor or limited phenotyping (37–39), exclusion of non-SNP polymorphisms such as VNTRs (15,31), the “multiple-testing burden” associated with genome-wide scans (37), and failure to account for environmental moderators (37,38,40).
The current study is the most comprehensive and well-powered investigation of historic candidate polymorphism and candidate gene hypotheses in depression to date. We focus on three lines of inquiry concerning how historic candidate genes may impact depression liability:
main effects of the most commonly studied candidate polymorphisms;
moderation of the effects of these polymorphisms by environmental exposures;
main effects of common SNPs across each of the candidate genes.
We first empirically identified 18 commonly studied candidate genes represented in at least ten peer-reviewed depression-focused journal articles between 1991 and 2016 from the body of publications indexed in the PubMed database (41). Within these candidate genes, we identified the most commonly studied polymorphisms, as well as their canonical risk alleles, at which point our primary analysis plan was preregistered. Using multiple large samples (n ranging from 62,138 to 443,264 across subsamples; total N = 621,214 individuals), we examined multiple measures of depression (e.g., lifetime diagnostic status, symptom severity among individuals reporting mood disturbances, lifetime number of depressive episodes; Table 1), employing multiple statistical frameworks (e.g., main effects of polymorphisms and genes, interaction effects on both the additive and multiplicative scales) and, in G × E analyses, considering multiple indices of environmental exposure (e.g., traumatic events in childhood or adulthood). Previous large sample studies of depression have largely focused on genetic main effects on depression diagnosis in the context of SNP data across the genome. In contrast, we examined several alternative depression phenotypes, analyzed both main effects and interactions with multiple potential moderators, included the most studied polymorphisms, including VNTRs (Figure 1), and employed a liberal significance threshold. Further, we quantified the extent to which phenotypic measurement error may have biased our results. The unifying question underlying this “multiverse” analytic approach (42) was the following: do the large datasets of the whole-genome data era support any previous depression candidate gene hypotheses?
Table 1.
Depression and environmental moderator phenotypes.
| Depression phenotypes† | ||
|---|---|---|
| Phenotype | Description | Sample size |
| Estimated lifetime depression diagnosis | Binary indicator of lifetime DSM-V depression diagnosis assessed in UKBB online mental health follow-up questionnaire. To meet criteria, participants had to endorse at least four of eight DSM-V depression symptoms (motor agitation/retardation symptom was not assessed), as well as duration, frequency, and impairment criteria. |
n = 115,458 85,513: controls 29,945 cases |
| Current depression severity | Sum score of all nine DSM-V depression symptom severities (using four point Likert scale to index severity of each symptom) over the two weeks leading up to assessment. Assessed in UKBB online mental health follow-up questionnaire. |
n = 115,463 sx = 3.347 |
| Conditional lifetime symptom count | Sum of symptom indicators for eight of nine lifetime DSM-V depression symptoms (motor agitation/retardation symptom was not assessed) among individuals endorsing lifetime incidence of a two+ week period characterized by anhedonia and/or depressed mood (questionnaire skip patterns necessitated this precondition). Assessed in the UKBB online mental health follow-up questionnaire. |
n = 62,138 sx = 1.745 |
| Lifetime episode count | Ordinal measure of incidence/recurrence of a two+ week period characterized by anhedonia and/or depressed mood indicating zero episodes, a single episode, or recurrent episodes. Assessed in UKBB online mental health follow-up questionnaire. |
n = 115,457 55,388: zero 30,724: single 26,345: recurrent |
| Touchscreen probable lifetime diagnosis, ordinal classification | Ordinal measure of depression diagnostic status based on a selection items of items from the Patient Health Questionnaire (62), the Structured Clinical Interview for DSM-IV Axis I Disorders-Research Version (63), and items assessing treatment seeking behavior specific to the UKBB touchscreen interview, as described in Smith et al., 2013 (64). Categories included no depression, single depressive episode, recurrent episodes (moderate), and recurrent episodes (severe), in that order. Assessed as part of the UKBB initial touchscreen interview. |
n = 91,121 66,605: controls 6,209: 1 episode 11,634: ≤ 2 moderate 6,633: ≥ 2 severe |
| Touchscreen probable lifetime diagnosis | Dichotomized coding of the touchscreen probable life diagnosis ordinal classification, contrasting no depression with the three diagnosis categories. |
n = 91,121 66,605: controls 84,516: cases |
| Severe recurrent depression | Binary indicator of case/control status for depression excluding cases and controls with mild to moderate depression symptoms. Controls were individuals who failed to endorse incidence of a two+ week period characterized by anhedonia and/or depressed mood. Cases were individuals met criteria for estimated lifetime depression diagnosis, endorsed at least five of the eight measured DSM-V symptoms, and experienced recurrent depressive episodes. Assessed in UKBB online mental health follow-up questionnaire. |
n = 64,432 53,218: controls 14,214: cases |
| PGC lifetime depression diagnosis | Binary indicator of lifetime depression diagnosis as measured in the PGC2 depression GWAS (19). The current investigation utilized data from the full expanded cohort meta-analysis, excepting UK-based cohorts (UKBB and Generation Scotland). |
n = 443,264 323,063: controls 120,201: cases |
| Moderator phenotypes‡ | ||
| Phenotype | Description | Sample size |
| Childhood trauma | Binary indicator of sexual and or physical abuse during childhood. Assessed in the UKBB online mental health follow-up questionnaire. |
n = 157,146 118,800: unexposed 38,346: exposed |
| Adult trauma | Binary indicator of any of the following traumatic events during adulthood: physical assault, sexual assault, witness to sudden/violent death, diagnosis with life threatening illness, involvement in life threatening accident, and exposure to combat or war-zone conditions. Assessed in the UKBB online mental health follow-up questionnaire. |
n = 157,223 64,286: unexposed 92,937: exposed |
| Recent trauma | Binary indicator of whether any of the above events occurred in the year leading up to assessment. |
n = 157,220 142,008: unexposed 15,212: exposed |
| Stressor-induced depression | Binary indicator of whether period of depressed mood or anhedonia was a possible consequence of a traumatic event among individuals endorsing lifetime incidence of a two+ week period characterized by anhedonia and/or depressed mood (questionnaire skip patterns necessitated this precondition). Assessed in the UKBB online mental health follow-up questionnaire. |
n = 88,585 23,746: unrelated to stressor 64,839: stressor-induced |
| Townsend deprivation index (TDI) | Measure of socioeconomic adversity (65), with higher values indicating greater adversity. Standardized to have zero mean and unit standard deviation. Assessed during the UKBB initial touchscreen interview. | n = 187,094 |
Depression phenotypes are described in further detail in supplement section S3.1 and visually summarized in Figure S3.1.
Moderator phenotypes are described in further detail in supplement section S3.2 and visually summarized in Figure S3.2. All moderators were only measured in the UKBB.
Materials and methods
Identification of genes and polymorphisms
We identified eighteen candidate genes studied for their associations with depression phenotypes at least ten times from within the body of peer-reviewed biomedical literature indexed in the PubMed database (41) using the Biopython bioinformatics package (43). We used regular expressions to find articles potentially corresponding to each gene and hand-verified the number of correctly classified articles for each gene in order to estimate hypergeometric confidence intervals for the true number of correctly classified studies (for additional details, see supplemental methods S1). We identified single polymorphisms comprising a large proportion of study foci for 16 of the 18 candidate genes. Figure 1 shows the most studied candidate genes and polymorphisms within them, as well as probabilistic estimates of the minimum number of times each has been studied with respect to depression and the number of studies-per-gene-per-year (confidence intervals presented in Table S1.1).
Samples
UK Biobank samples
A large portion of the data used in the present study was collected by the UK Biobank (UKBB), a population sample of 502,682 individuals collected at 22 centers across the United Kingdom between 2006 and 2010 (44). Within this group, we analyzed several depression phenotypes and moderators among 177,950 unrelated (pairwise genome-wide relatedness, , < 0.05) European ancestry individuals for whom relevant depression measures were collected. We analyzed two partially overlapping subsets of these individuals: 91,121 individuals for whom selected items from the initial touchscreen interview were available and 115,458 individuals who completed a series of online mental health questionnaires, 62,138 of whom endorsed a two-week period characterized by anhedonia or depressed mood at some point during their lives. DNA was extracted from whole blood and genotyped using the Affymetrix UK Biobank Axiom array or the Affymetrix UK BiLEVE Axiom array and imputed to the Haplotype Reference Consortium by the UKBB (45). Further details on genotyping and sampling procedures are available online (46) and in section S2 of the supplement. Because VNTRs were not genotyped in the UKBB dataset, we used two independent whole-genome SNP datasets (the Family Transition Project (47) and the Genetics of Antisocial Drug Dependence (48,49)) that also measured these repeat polymorphisms as reference panels in order to impute highly studied VNTRs within DRD4, MAOA, SLC6A3, and SLC6A4 in the UKBB. The estimated out-of-sample imputed genotype match rates were ≥ 0.919 for all four VNTRs (complete details are provided in (30)).
Psychiatric Genetics Consortium sample
To investigate candidate gene polymorphism main effect hypotheses, we also used data from the most recent GWAS on depression conducted by the Major Depressive Disorder Working Group of the Psychiatric Genetics Consortium (PGC), which is described in detail in Wray et al., 2018 (19). Lack of access to raw genotypes for a large number of the PGC cohorts precluded imputation of VNTRs in the PGC sample. To minimize sample overlap with UKBB, UK-based cohorts were excluded from the PGC dataset, resulting in GWAS summary statistics for a total of 443,264 individuals (120,201 cases; 323,063 controls); see S2 for further details.
Phenotypes
Table 1 describes all phenotypes examined in the present investigation, with additional information provided in S3. Correlations between depression outcomes and Cohen’s κ estimates for diagnosis phenotypes are presented in Tables S3.1 and S3.2, respectively. Marker-based heritabilities of, and genetic correlations between, depression outcomes were estimated via LD score regression (50) and are presented in Tables S3.3–S3.4 and Figure S3.3 (see S4.4 for further details).
Analyses
All analyses were pre-registered through the Open Science Framework (51) and are available at (https://osf.io/akgvz/). Statistical models are described in detail in S4 and departures from the pre-registered analyses are documented in S5.
Polymorphism-wise analyses
We analyzed associations between outcomes and each of the top 16 candidate polymorphisms using a generalized linear model framework (link functions listed in Table S4.1). For two of the genes, TPH2 and DTNBP1, no particular polymorphism was investigated in a preponderance of studies, and so these genes were not included in the polymorphism-wide analyses. Covariates included genotyping batch, testing center, sex, age, age2, and the first ten European ancestry principal components. Sixteen polymorphism × environment effects were tested on both the additive and multiplicative scales for each of the 16 polymorphisms; each model tested is listed in Table S4.1. For interaction tests, we included all covariate × polymorphism and covariate × moderator terms to control for the potential confounding influences of covariates on the interaction (52). We also tested interaction models only controlling for covariate main effects, which is incorrect but common in the candidate gene literature (34). Across all outcomes we employed a preregistered significance threshold of αpoly = .05/16 = 3.13e-03, corresponding to a Bonferroni correction across the top 16 candidate polymorphisms. This threshold is liberal because it does not account for the multiple ways each polymorphisms was analyzed or the multiple outcomes it was assessed with respect to. Further details are provided in S4.1.
Gene-wise and gene-set analyses
We used the NCBI Build 37 gene locations to annotate SNPs to genes, allowing SNPs within a 25kb window of the gene start and end points to be mapped to each gene. We used MAGMA software version 1.05b (53) to perform gene-wise and gene-set analyses for the top eighteen candidate genes separately in the UKBB and PGC datasets. Gene-wise tests summarize the degree of association between a phenotype and polymorphisms within a given gene; in contrast, gene-set tests examine the association between a phenotype and a set of genes rather than individual genes.
We conducted gene-wise association analyses for each gene and outcome using the MAGMA default gene-level association statistic (sum −log p-based statistics or principal components regression, for tests based on summary statistics or individual-level genotypes, respectively) and using a liberal significance threshold of αgene = .05/18 = 2.78e-03 to correct for multiple tests across the 18 candidate genes. We used summary statistics from the PGC2 depression GWAS (19) (excluding UK-based cohorts) as input for the PGC analyses, whereas individual-level genotypes were available for the UKBB. The gene-level association statistics were in turn used to perform “competitive” gene-set tests that compared enrichment of depressin phenotype-associated-loci between our set of 18 candidate genes and all other genes not in the gene set, controlling for potentially confounding gene characteristics. Further analyses, which compared the 18 candidate genes to negative control sets of genes involved in type 2 diabetes, height, or synaptic processes, are described in S4.2 and reported in S11.
Results
Polymorphism-level analyses
Table 2 shows the most significant result for each of the most-studied candidate gene polymorphisms for the main effect across the eight outcomes investigated (eight main effect tests per polymorphism; first column) and the interaction effect across five moderators measured in the UKBB (32 interaction tests per polymorphism [Table S4.6]; second column). Given the number of tests conducted, there was little evidence that any effect was larger than what would be expected by chance under the null hypothesis. Only for COMT rs4680 on current depression severity was there was evidence of a small main effect that surpassed our liberal threshold of significance, such that the incident rate of current depression severity scores decreased by a factor of 0.983 per copy of the G allele (odds ratio 95% CI=0.967-0.999; p = 0.002; Figure 2). Detecting an effect of this size at an alpha level of 0.05 with 80% power would require a sample of over 100,000 individuals (S4.3). Similarly, across all polymorphisms, outcomes, and exposures, on both the additive and multiplicative scales, no polymorphism-by-exposure moderation effects attained significance at αpoly. Failing to include all covariate × polymorphism and covariate × moderator terms as covariates, as is common in the published G × E literature (34), inflated product term test statistics on average but did not result in any additional significant effects (S10). Complete results for all outcomes are provided in S7–S10.
Table 2.
Minimum p-value effect across 8 main effect models and 32 interaction effect models per polymorphism.
| Polymorphism | MAF | Outcome: Additive effect | β | min p | Outcome: Interaction effect | Moderator | Scale | β | min p | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | SLC6A4 – 5-HTTLPR†,⁑ | 0.499 | Current depression severity | 0.008 | .138 | Lifetime episode count | TDI | primary | 0.019 | .041 |
| 2 | BDNF – rs6265 | 0.188 | Severe recurrent depression | 0.018 | .325 | Estimated lifetime depression diagnosis | TDI | alternate | 0.007 | .008 |
| 3 | COMT – rs4680 | 0.483 | Current depression severity | −0.017 | .002* | Conditional lifetime symptom count | Stressor-induced depression‡ | alternate | 0.048 | .040 |
| 4 | HTR2A – rs6311 | 0.402 | Estimated lifetime depression diagnosis | 0.020 | .045 | Estimated lifetime depression diagnosis | Childhood trauma | alternate | 0.008 | .072 |
| 5 | TPH1 – rs1800532 | 0.391 | Current depression severity | −0.012 | .036 | Conditional lifetime symptom count | Childhood trauma | primary | −0.045 | .049 |
| 6 | DRD4 – VNTR† | 0.223 | Touchscreen probable lifetime diagnosis (ord.) | 0.022 | .079 | Severe recurrent depression | TDI | primary | 0.011 | .094 |
| 7 | DRD2 – rs1800497 | 0.201 | PGC lifetime diagnosis | −0.019 | .006 | Conditional lifetime symptom count | Stressor-induced depression‡ | alternate | −0.044 | .134 |
| 8 | MAOA – VNTR† | ⁂ | Severe recurrent depression | 0.023 | .073 | Conditional lifetime symptom count | TDI | primary | −0.024 | .014 |
| 9 | APOE – rs429358/rs7412† | 0.148 | Lifetime episode count | 0.019 | .091 | Current depression severity | Recent trauma | alternate | −0.182 | .009 |
| 10 | MTHFR – rs1801133 | 0.334 | Current depression severity | −0.012 | .034 | Estimated lifetime depression diagnosis | Adult trauma | alternate | −0.007 | .054 |
| 11 | CLOCK – rs1801260 | 0.268 | Touchscreen probable lifetime diagnosis | 0.030 | .013 | Severe recurrent depression | TDI | primary | 0.014 | .012 |
| 12 | SLC6A3 – VNTR† | 0.255 | Touchscreen probable lifetime diagnosis | 0.019 | .114 | Estimated lifetime depression diagnosis | Childhood trauma | alternate | −0.008 | .099 |
| 13 | ACE – in/del | 0.474 | Touchscreen probable lifetime diagnosis | 0.016 | .143 | Lifetime episode count | TDI | primary | 0.015 | .107 |
| 14 | ABCB1 – rs1045642 | 0.456 | PGC lifetime diagnosis | −0.006 | .164 | Current depression severity | Recent trauma | alternate | −0.108 | .027 |
| 15 | DRD3 – rs6280 | 0.336 | Current depression severity | −0.010 | .078 | Current depression severity | Recent trauma | alternate | −0.111 | .031 |
| 16 | DBH – rs1611115 | 0.205 | Estimated lifetime depression diagnosis | −0.014 | .236 | Severe recurrent depression | Adult trauma | alternate | −0.005 | .087 |
Minimum p value for each polymorphism across outcomes/moderators for additive and interaction effects (on additive and multiplicative scales), respectively. Interaction tests were not conducted in the PGC sample because moderators were unavailable for the PGC sample. Only one effect was significant after a liberal correction for the number of polymorphisms (but not for outcomes or moderators; αpoly = .05/16 = 3.125×10−3). Details of each model are provided in supplement section S4, with all interaction models listed in Table S4.6. Complete results are presented in sections S7–S9.
Significant at αpoly = .05/16.
VNTRs and the triallelic APOE polymorphism were unavailable for the PGC samples, and thus these variants were examined only across the seven UKBB outcomes.
Variant × stressor-induced depression estimates reflect differences in the magnitude of variant/outcome associations between individuals reporting that their depression was induced by a stressful event and those reporting otherwise.
allele frequency reflects the low activity VNTR/rs25531 haplotype (5).
MAOA is located on the X chromosome; frequencies were 0.336, 0.341 for females, males, respectively. MAF indicates the minor allele frequency in the subset of UKBB sample for whom estimated lifetime depression diagnosis was available.
Figure 2.
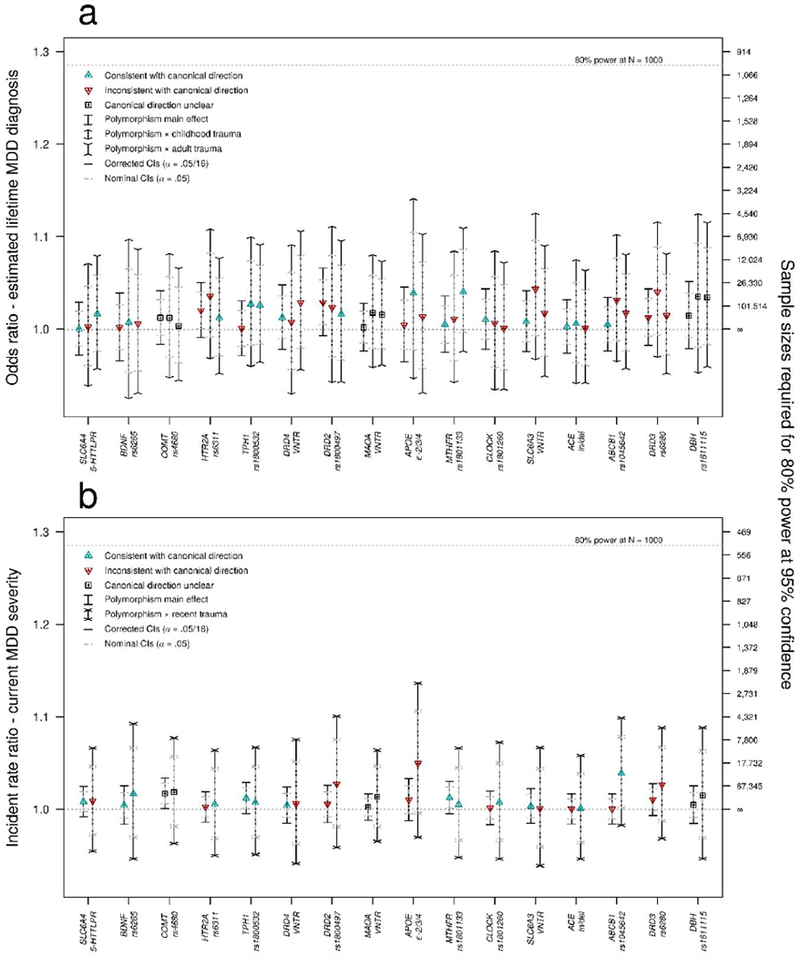
Main effects and G × E effects of 16 candidate polymorphisms on estimated lifetime depression diagnosis and current depression severity in the UK Biobank.
Effect size estimates for 16 candidate polymorphisms (in order of estimated number of tops from left to right, descending) on a. estimated lifetime depression diagnosis and b. past two-week depression symptom severity from the online mental health follow-up assessment in the UKBB sample (n = 115,257). Both polymorphism main effects and polymorphism × environmental moderator interaction effects are presented for each outcomes. Detailed descriptions of the variables, and of the association and power analysis models are provided in S3 and S4, respectively.
Despite the lack of evidence for G × E effects, all moderators exhibited large significant effects on all outcomes in the expected directions (S6). For example, experiencing childhood trauma increased odds for estimated lifetime depression diagnosis by a factor of 1.655 (z = 32.048, p = 2.33e-225) and experiencing a traumatic event in the past two years increased incidence rate of current depression severity index by a factor of 1.431 (z = 27.004, p = 1.32e-160).
Gene-level analyses
Across all candidate genes and outcomes, only DRD2 showed a significant gene-wise effect (αgene =.05/18=2.78e-03) and only on PGC lifetime depression diagnosis using both the sum −log p statistic (p = 5.14e-07) as well as using the minimum p-value statistic (p = 2.74e-03; see Tables S11.1 and S11.2 for full results and section S4.2 for comparison of methods). The former estimate, based on the sum −log p statistic, was also significant at the more stringent genome-wide level (αGW = .05/19,165 = 2.61e-6). DRD2 did not exhibit a significant effect on any of the UKBB outcomes despite its high genetic correlations with the UKBB depression phenotypes (Table S3.3, Figure S3.3). Investigating the effects of the 18 genes together as a set revealed no associations with depression above what would be expected by chance under the null; the set of 18 depression candidate genes did not show stronger associations with any depression phenotype compared to all other genes at α = .05 (S11.2.1).
Attempted replication of top 16 loci implicated by PGC GWAS results
In order to contextualize the lack of replication of the of 16 candidate genetic polymorphisms, we sought to replicate the top 16 independent genome-wide significant loci implicated for PGC lifetime diagnosis by examining their associations with estimated lifetime diagnosis in the independent UKBB sample (see S4.5 for details). Three loci attained significance at –poly = .05/16 (rs12552, rs12658032, rs11135349; S12), which is consistent with the low power to detect small associations; median power for the 16 loci was 0.143 and the 95% CI for number of replications we’d expect given power estimates was 2 – 7 (Figure S4.6).
Sensitivity of results to measurement error
One reason why candidate gene polymorphism associations detected in small samples are not replicated in large GWAS datasets is the potentially worse phenotyping and higher measurement error in predictor or outcome variables in the GWAS datasets. To investigate this possibility, we used a Monte Carlo procedure to quantify the extent to which measurement error may have impacted statistical power of our tests. As a lower bound on a candidate gene polymorphism study effect sizes, we used the minimally detectable log odds ratio for both main and interaction effects that had 50% power at α = .05 in a balanced case/control study of 1000 individuals and where the risk allele frequency was 0.5 (e.g., for main effects, genomic relative risk = 1.16). Simulations demonstrated that we had ≈100% power to detect such effects under multiple severe measurement error scenarios in a sample of size typical of that in our UKBB analyses (≈ 30,000 cases and ≈ 85,000 controls; see S4.3.3). This was true even in the extreme scenario wherein half of diagnoses and half of traumatic exposures were determined via coin toss (Figure S4.5).
Discussion
The present study examined multiple types of associations between 18 highly studied candidate genes for depression and multiple depression phenotypes. The study was very well powered compared to previous candidate gene studies, with n ranging from 62,138 to 443,264 across subsamples. Despite the high statistical power, none of the most highly studied polymorphisms within these genes demonstrated substantial contributions to depression liability. Furthermore, we found no evidence to support moderation of polymorphism effects by exposure to traumatic events or socioeconomic adversity. We also found little evidence to support contributions of other common polymorphisms within these genes to depression liability excepting DRD2, which showed a genome-wide significant gene-wise effect on depression diagnosis in the PGC sample, though not on any outcomes in the UKBB sample. Reasons for the failure of DRD2 to replicate in the UKBB are unclear, but could be due to sampling variability, lower statistical power in the UKBB, or false positive or negative findings. Phenotypic heterogeneity, however, is an unlikely explanation as genetic correlation estimates between depression phenotypes across samples were high (Table S3.3, Figure S3.3)—for example, PGC lifetime depression diagnosis was strongly associated with estimated lifetime depression diagnosis from the UKBB online follow-up questionnaire (, , respectively; , p = 2.08e-57), which was in turn strongly associated with probable lifetime diagnosis from the UKBB initial touchscreen interview (; , p = 2.83e-30). Finally, as a set, depression candidate genes were no more related to depression phenotypes than non-candidate genes. Our results stand in stark contrast to the published candidate gene literature, where large, statistically significant effects are commonly reported for the specific polymorphisms in the 18 candidate genes we investigated here.
There are several features of the current investigation that set it apart from previous candidate gene replication attempts, meta-analyses of candidate gene studies, and genome-wide studies that failed to support roles for depression candidate polymorphisms. First, this is the only study to have imputed and examined the effects of several highly-studied VNTR polymorphisms in a large GWAS dataset, including 5-HTTLPR in SLC6A4, which was examined in 38.14% of the depression candidate gene studies we identified (see (30) for imputation details). Second, we thoroughly examined several distinct depression phenotypes (e.g., diagnosis, depressive episode recurrence, symptom count among depressed individuals) to ensure that our results did not reflect a single operationalization of depression. Some researchers have attributed the poor replicability of candidate gene findings to specificity of effects with respect to particular types of depression or stressors (e.g., prior versus subsequent depression onset with respect to stress exposure (39), recurrent versus single episode depression (54), financial versus other stress exposure (55)). As such, we examined all available depression and exposure phenotypes reflecting constructs of interest in the candidate gene literature. Results for all measures and modeling choices (e.g., multiplicative versus additive interactions), presented in detail in the supplement (S7–S11), were consistently null with respect to candidate gene hypotheses. Third, we employed exceedingly liberal significance thresholds (e.g., for polymorphism-wise analyses αpoly = 3.13e-03 as opposed to the standard αgwas = 5e-08 utilized in GWAS) across all outcomes to ensure no possible effect was missed, correcting only for the number of polymorphisms we examined. As such, our results suggest that the zero or near-zero effect sizes of these candidate polymorphisms, rather than the multiple-testing burden induced by genome-wide scans, account for the previous failures of large GWAS to detect candidate polymorphisms effects. Finally, and perhaps most importantly, unlike meta-analyses that use previously published candidate gene findings, our results cannot be affected by selective publication or reporting practices that can inflate type-I errors and lead to biased representations of evidence for candidate gene hypotheses.
There are several limitations to the present investigation. First, it is possible that we failed to identify a small number of candidate gene publications and that these failures resulted in the omission of some depression candidate genes examined in ten or more publications. Nevertheless, the top nine of the eighteen identified genes accounted for 86.59% of the estimated number of studies, and it is unlikely that we omitted any depression candidate genes with popularity approaching that of, for example, SLC6A4 or COMT. Second, a subset of the UKBB sample were ascertained for smoking behaviors (the BiLEVE study (56)), and controlling for genotyping batch (which differentiates the two subsamples) has the potential to induce collider bias (57). However, only one of the sixteen candidate gene polymorphisms demonstrated allele frequency differences across these two subsamples (rs6311; χ2(2)=12.558, p = .002; MAF = .402 in the BiLEVE sample, MAF = .405 otherwise) and it is unlikely that ascertainment in the BiLEVE subsample unduly influenced association statistics. However, the potential influence of ascertainment in the BiLEVE subsample on interaction effect estimates, as well as other possible sources of selection-induced bias, remains unclear. Third, whereas some of phenotypes we examined closely matched standard diagnostic instruments (e.g., current depression severity was based on the widely used PHQ-9 questionnaire (58)), others were of undetermined reliability. For example, one of the nine DSM-V depression symptoms (motor agitation/retardation) was omitted from the UKBB online mental health follow-up questionnaire, and our estimated lifetime depression diagnosis phenotype required ≥ 4 of 8 symptoms rather than the standard ≥ 5 of 9 symptoms (in addition to episode duration and impairment criteria; S3.1). However, enforcing stricter case/control criteria (i.e., comparing individuals who endorsed no two-week period of either anhedonia or depressed mood throughout their lifetimes to individuals reporting recurrent episodes, endorsing ≥ 5 of 8 symptoms, and meeting duration and impairment criteria) failed to alter results (S7, S8, S9), despite the fact that even this diminished sample size (n = 67,304) was much larger than any previous candidate gene study we are aware of. Fourth, some of the phenotypes we examined were possibly measured with greater error than is typical in smaller candidate gene studies, an issue for which large studies are often criticized. For example, the prevalence of our measure of traumatic exposure in adulthood was uncommonly high (59.11%) and most of our retrospective measurements were likely corrupted by recall bias. However, as demonstrated in S4.3.3, even extreme measurement error cannot explain our failure to detect the relatively large effects necessary for detection in smaller samples. Further, follow-up analyses demonstrated strong effects of all environmental moderators across all outcomes (S6), suggesting that both moderators and depression phenotypes were measured with sufficient accuracy to detect known environmental effects. It is exceedingly difficult to construct a plausible measurement error model that could, for example, comfortably reconcile the large effect estimate of childhood trauma on estimated lifetime diagnosis (odds ratio = 1.655, p = 2.96e-225) and the negligible estimate for the 5-HTTLPR × childhood trauma interaction effect (odds ratio = 0.988, p = .914) with the existence of a substantial G × E interaction effect.
The genetic underpinnings of common complex traits such as depression appear to be far more complicated than originally hoped (59,60), and large collaborative efforts have not supported the existence of common genetic variants with large effects on depression liability (19). In the context of our understanding of psychiatric genetics in the 1990s and early 2000s, the most studied candidate genes and the polymorphisms within them were defensible targets for association studies. However, our results demonstrate that historic depression candidate gene polymorphisms do not have detectable effects on depression phenotypes. Further, the candidate genes themselves (with the possible exception of DRD2) were no more associated with depression phenotypes than genes chosen at random. The present study had > 99.99% power at αgwas = 5e-08 to detect a main effect of the magnitude commonly reported in candidate gene studies, even allowing for extreme measurement error in both outcome and moderator phenotypes (S4.3). Thus, it is extremely unlikely that we failed to detect any true associations between depression phenotypes and these candidate genes. The implication of our study, therefore, is that previous positive main effect or interaction effect findings for these 18 candidate genes with respect to depression were false positives. Our results mirror those of well-powered investigations of candidate gene hypotheses for other complex traits including those of schizophrenia (17,26) and white matter microstructure (20). The potential for self-correction is an essential strength of the scientific enterprise; it is with this mechanism in mind that we present these findings. In agreement with the recent recommendations of the National Institute of Mental Health Council Workgroup on Genomics (61), we conclude that it is time for depression research to abandon historic candidate gene and candidate gene-by-environment interaction hypotheses.
Supplementary Material
Figure 3.
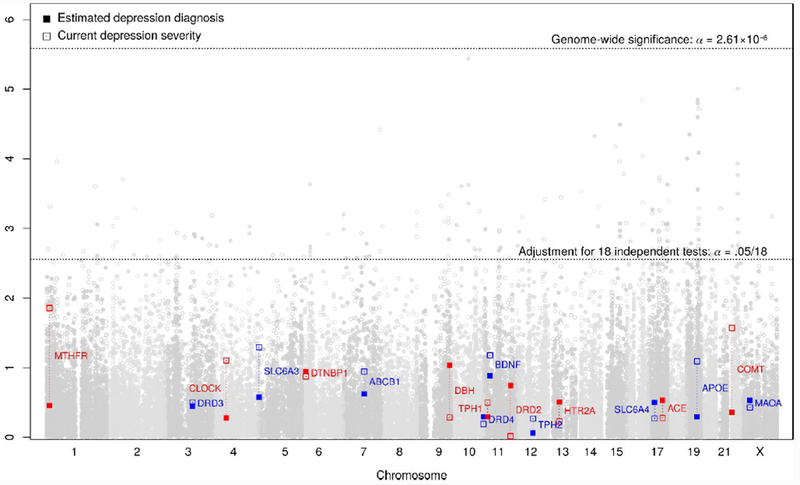
Gene-wise statistics for effects of 18 candidate genes on primary depression outcomes in the UK Biobank.
Gene-wise p-values across the genome, highlighting the 18 candidate polymorphisms’ effects on estimated depression diagnosis (filled points) and past two-week depression symptom severity (hollow points) from the online mental health follow-up assessment in the UKBB sample (n = 115,257). Detailed descriptions of the variables, and of the association and power analysis models are provided in S3 and S4, respectively.
Disclosures and acknowledgments:
RB was supported by the National Institute of Mental Health (NIMH; T32 MH016880). PFS was supported by the NIMH (U01 MH109528) and the Swedish Research Council (D0886501). LME and MCK were supported by the NIMH (R01 MH00141) and the Institute for Behavioral Genetics. This research has been conducted using the UK Biobank Resource under application numbers 1665, 16651, and 24795. This work utilized the RMACC Summit supercomputer, which is supported by the National Science Foundation (awards ACI-1532235 and ACI-1532236), the University of Colorado Boulder, and Colorado State University. The Summit supercomputer is a joint effort of the University of Colorado Boulder and Colorado State University. We thank SURFsara (www.surfsara.nl) for the support in using the Lisa Compute Cluster. We thank the research participants of the PGC, UK Biobank, and employees of 23andMe, Inc. for their contribution to this study.
Footnotes
The authors report no conflicts of interest.
References
- 1.Sullivan PF, Neale MC, Kendler KS. Genetic Epidemiology of Major Depression: Review and Meta-Analysis. AJP [Internet]. 2000. October 1 [cited 2017 Aug 2];157(10):1552–62. Available from: http://ajp.psychiatryonline.org/doi/abs/10.1176/appi.ajp.157.10.1552 [DOI] [PubMed] [Google Scholar]
- 2.McInnes LA, Freimer NB. Mapping genes for psychiatric disorders and behavioral traits. Curr Opin Genet Dev. 1995. June;5(3):376–81. [DOI] [PubMed] [Google Scholar]
- 3.Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, et al. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA. 1993. March 15;90(6):2542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owens MJ, Nemeroff CB. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem. 1994. February;40(2):288–95. [PubMed] [Google Scholar]
- 5.Heils A, Teufel A, Petri S, Stöber G, Riederer P, Bengel D, et al. Allelic variation of human serotonin transporter gene expression. J Neurochem. 1996. June;66(6):2621–4. [DOI] [PubMed] [Google Scholar]
- 6.Stoltenberg SF, Burmeister M. Recent progress in psychiatric genetics-some hope but no hype. Hum Mol Genet. 2000. April 12;9(6):927–35. [DOI] [PubMed] [Google Scholar]
- 7.Buckland PR. Genetic association studies of alcoholism--problems with the candidate gene approach. Alcohol Alcohol. 2001. April;36(2):99–103. [DOI] [PubMed] [Google Scholar]
- 8.Munafò MR. Candidate gene studies in the 21st century: meta-analysis, mediation, moderation. Genes, Brain and Behavior [Internet]. 2006. February 1 [cited 2018 Feb 20];5(S1):3–8. Available from: http://onlinelibrary.wiley.com/doi/10.1111/j.1601-183X.2006.00188.x/abstract [DOI] [PubMed] [Google Scholar]
- 9.Lander ES, Schork NJ. Genetic dissection of complex traits. Science [Internet]. 1994. September 30 [cited 2018 Jul 26];265(5181):2037–48. Available from: http://science.sciencemag.org/content/265/5181/2037 [DOI] [PubMed] [Google Scholar]
- 10.Terwilliger JD, Weiss KM. Linkage disequilibrium mapping of complex disease: fantasy or reality? Current Opinion in Biotechnology [Internet]. 1998. December 1 [cited 2018 Jul 26];9(6):578–94. Available from: http://www.sciencedirect.com/science/article/pii/S0958166998801353 [DOI] [PubMed] [Google Scholar]
- 11.Colhoun HM, McKeigue PM, Smith GD. Problems of reporting genetic associations with complex outcomes. The Lancet [Internet]. 2003. March 8 [cited 2018 Jul 26];361(9360):865–72. Available from: http://www.sciencedirect.com/science/article/pii/S0140673603127158 [DOI] [PubMed] [Google Scholar]
- 12.Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002. August 2;297(5582):851–4. [DOI] [PubMed] [Google Scholar]
- 13.Rutter M, Moffitt TE, Caspi A. Gene-environment interplay and psychopathology: multiple varieties but real effects. J Child Psychol Psychiatry. 2006. Apr;47(3–4):226–61. [DOI] [PubMed] [Google Scholar]
- 14.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003. July 18;301(5631):386–9. [DOI] [PubMed] [Google Scholar]
- 15.Niculescu AB. DISCovery in psychiatric genetics. Mol Psychiatry. 2014. February;19(2):145. [DOI] [PubMed] [Google Scholar]
- 16.Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry. 2011. Oct;168(10):1041–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrell MS, Werge T, Sklar P, Owen MJ, Ophoff RA, O’Donovan MC, et al. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry. 2015. May;20(5):555–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munafò MR. Reliability and replicability of genetic association studies. Addiction [Internet]. 2009. September 1 [cited 2016 Jul 30];104(9):1439–40. Available from: http://onlinelibrary.wiley.com/doi/10.1111/j.1360-0443.2009.02662.x/abstract [DOI] [PubMed] [Google Scholar]
- 19.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature Genetics [Internet]. 2018. May [cited 2018 May 21];50(5):668–81. Available from: https://www.nature.com/articles/s41588-018-0090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson PM, Stein JL, Medland SE, Hibar DP, Vasquez AA, Renteria ME, et al. The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav [Internet]. 2014. [cited 2018 Jul 15];8(2):153–82. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4008818/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014. July 24;511(7510):421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munafò MR, Gage SH. Improving the reliability and reporting of genetic association studies. Drug and Alcohol Dependence [Internet]. 2013. October 1 [cited 2016 Jul 30];132(3):411–3. Available from: http://www.sciencedirect.com/science/article/pii/S037687161300135X [DOI] [PubMed] [Google Scholar]
- 23.Burton PR, Hansell AL, Fortier I, Manolio TA, Khoury MJ, Little J, et al. Size matters: just how big is BIG?: Quantifying realistic sample size requirements for human genome epidemiology. Int J Epidemiol. 2009. February;38(1):263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosker FJ, Hartman CA, Nolte IM, Prins BP, Terpstra P, Posthuma D, et al. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol Psychiatry [Internet]. 2011. May [cited 2016 Jul 30];16(5):516–32. Available from: http://www.nature.com/mp/journal/v16/n5/full/mp201038a.html [DOI] [PubMed] [Google Scholar]
- 25.Coleman JRI, Consortium MDDWG of the PG, Consortium UBMH, Eley TC, Breen G. Genome-wide gene-environment analyses of depression and reported lifetime traumatic experiences in UK Biobank. bioRxiv [Internet]. 2018. January 12 [cited 2018 Feb 20];247353 Available from: https://www.biorxiv.org/content/early/2018/01/12/247353 [Google Scholar]
- 26.Johnson EC, Border R, Melroy-Greif WE, et al. : No evidence that schizophrenia candidate genes are more associated with schizophrenia than noncandidate genes. Biol Psychiatry. 2017; 82:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jahanshad N, Ganjgahi H, Bralten J, Braber A den, Faskowitz J, Knodt A, et al. Do Candidate Genes Affect the Brain’s White Matter Microstructure? Large-Scale Evaluation of 6,165 Diffusion MRI Scans. bioRxiv [Internet]. 2017. February 20 [cited 2018 Jun 30];107987 Available from: https://www.biorxiv.org/content/early/2017/02/20/107987 [Google Scholar]
- 28.Van der Auwera S, Peyrot WJ, Milaneschi Y, Hertel J, Baune B, Breen G, et al. Genomewide gene-environment interaction in depression: A systematic evaluation of candidate genes. Am J Med Genet [Internet]. 2018. January 1 [cited 2018 Feb 20];177(1):40–9. Available from: http://onlinelibrary.wiley.com/doi/10.1002/ajmg.b.32593/abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culverhouse RC, Saccone NL, Horton AC, Ma Y, Anstey KJ, Banaschewski T, et al. Collaborative meta-analysis finds no evidence of a strong interaction between stress and 5-HTTLPR genotype contributing to the development of depression. Molecular Psychiatry [Internet]. 2018. January [cited 2018 Jun 5];23(1):133–42. Available from: https://www.nature.com/articles/mp201744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Border R, Smolen A, Corley R, Stallings M, Brown S, Conger R, et al. Imputation of Behavioral Candidate Gene Repeat Polymorphisms in 486,551 Publicly-Available UK Biobank Individuals. bioRxiv [Internet]. 2018. June 29 [cited 2018 Jun 30];358267 Available from: https://www.biorxiv.org/content/early/2018/06/29/358267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brookes KJ. The VNTR in complex disorders: The forgotten polymorphisms? A functional way forward? Genomics [Internet]. 2013. May 1 [cited 2018 May 25];101(5):273–81. Available from: http://www.sciencedirect.com/science/article/pii/S0888754313000451 [DOI] [PubMed] [Google Scholar]
- 32.Wendland JR, Martin BJ, Kruse MR, Lesch K-P, Murphy DL. Simultaneous genotyping of four functional loci of human SLC6A4, with a reappraisal of 5-HTTLPR and rs25531. Molecular Psychiatry [Internet]. 2006. March [cited 2018 Oct 24];11(3):224–6. Available from: https://www.nature.com/articles/4001789 [DOI] [PubMed] [Google Scholar]
- 33.Dick DM, Agrawal A, Keller MC, Adkins A, Aliev F, Monroe S, et al. Candidate Gene-Environment Interaction Research: Reflections and Recommendations. Perspect Psychol Sci [Internet]. 2015. January [cited 2018 Feb 20];10(1):37–59. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4302784/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller MC. Gene-by-environment interaction studies have not properly controlled for potential confounders: The problem and the (simple) solution. Biol Psychiatry [Internet]. 2014. January 1 [cited 2016 Jul 30];75(1). Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3859520/] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Border R, Keller MC. Commentary: Fundamental problems with candidate gene-byenvironment interaction studies – reflections on Moore et Thoemmes (2016). J Child Psychol Psychiatry [Internet]. 2017. March [cited 2018 Mar 19];58(3):328–30. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5312579/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munafò MR, Zammit S, Flint J. Limitations of Gene × Environment Interaction Models in Psychiatry. J Child Psychol Psychiatry [Internet]. 2014. October [cited 2018 Oct 12];55(10):1092–101. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4961234/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Assary E, Vincent JP, Keers R, Pluess M. Gene-environment interaction and psychiatric disorders: Review and future directions. Seminars in Cell & Developmental Biology [Internet]. 2018. May 1 [cited 2018 Jun 5];77:133–43. Available from: http://www.sciencedirect.com/science/article/pii/S108495211730280X [DOI] [PubMed] [Google Scholar]
- 38.Moore SR. Commentary: What is the case for candidate gene approaches in the era of highthroughput genomics? A response to Border and Keller (2017). J Child Psychol Psychiatr [Internet]. 2017. March 1 [cited 2017 Apr 3];58(3):331–4. Available from: http://onlinelibrary.wiley.com/doi/10.1111/jcpp.12697/abstract [DOI] [PubMed] [Google Scholar]
- 39.Moffitt TE. Letter to Culverhouse [Internet]. 2012. [cited 2018 Jun 26]. Available from: https://moffittcaspi.com/sites/moffittcaspi.com/files/Letter_to_Culverhouse_JUNE2012.pdf [Google Scholar]
- 40.Uher R Gene-environment interactions in severe mental illness. Front Psychiatry. 2014;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.National Center for Biotechnology Information, US National Library of Medicine, National Institutes of Health. [Internet]. [cited 2018 Jun 25]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/ [Google Scholar]
- 42.Steegen S, Tuerlinckx F, Gelman A, Vanpaemel W. Increasing Transparency Through a Multiverse Analysis. Perspect Psychol Sci [Internet]. 2016. September 1 [cited 2018 Feb 20];11(5):702–12. Available from: 10.1177/1745691616658637 [DOI] [PubMed] [Google Scholar]
- 43.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics [Internet]. 2009. June 1 [cited 2018 Jun 25];25(11):1422–3. Available from: https://academic.oup.com/bioinformatics/article/25/11/1422/330687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Medicine [Internet]. 2015. March 31 [cited 2018 Jun 5];12(3):e1001779 Available from: http://journals.plos.org/plosmedicine/article?id=10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loh P-R, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, et al. Reference-based phasing using the Haplotype Reference Consortium panel. NatureGenetics [Internet]. 2016. November [cited 2018 Jun 30];48(11):1443–8. Available from: https://www.nature.com/articles/ng.3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.UKBiobank_genotyping_QC_documentation-web.pdf [Internet]. [cited 2018 Jul 19]. Available from: http://www.ukbiobank.ac.uk/wpcontent/uploads/2014/04/UKBiobank_genotyping_QC_documentation-web.pdf
- 47.Conger RD, Schofield TJ, Neppl TK. Intergenerational Continuity and Discontinuity in Harsh Parenting. Parent Sci Pract [Internet]. 2012. January 1 [cited 2018 Oct 11];12(2–3):222–31. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3383029/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Derringer J, Corley RP, Haberstick BC, Young SE, Demmitt BA, Howrigan DP, et al. Genome-Wide Association Study of Behavioral Disinhibition in a Selected Adolescent Sample. Behav Genet. 2015. July;45(4):375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stallings MC, Corley RP, Hewitt JK, Krauter KS, Lessem JM, Mikulich SK, et al. A genome-wide search for quantitative trait loci influencing substance dependence vulnerability in adolescence. Drug and alcohol dependence [Internet]. 2003. [cited 2016 Mar 7];70(3):295–307. Available from: http://www.sciencedirect.com/science/article/pii/S0376871603000310 [DOI] [PubMed] [Google Scholar]
- 50.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. An atlas of genetic correlations across human diseases and traits. Nature Genetics [Internet]. 2015. November [cited 2018 Jul 25];47(11):1236–41. Available from: https://www.nature.com/articles/ng.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Center for Open Science. Open Science Framework [Internet]. [cited 2016 Mar 11]. Available from: https://osf.io/ [Google Scholar]
- 52.Yzerbyt VY, Muller D, Judd CM. Adjusting researchers’ approach to adjustment: On the use of covariates when testing interactions. Journal of Experimental Social Psychology [Internet]. 2004. May [cited 2016 Jul 30];40(3):424–31. Available from: http://www.sciencedirect.com/science/article/pii/S0022103103001598 [Google Scholar]
- 53.Leeuw CA de, Mooij JM, Heskes T, Posthuma D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLOS Computational Biology [Internet]. 2015. April 17 [cited 2017 Nov 30];11(4):e1004219 Available from: http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uher R, Caspi A, Houts R, Sugden K, Williams B, Poulton R, et al. Serotonin transporter gene moderates childhood maltreatment’s effects on persistent but not single-episode depression: Replications and implications for resolving inconsistent results. J Affect Disord [Internet]. 2011. December [cited 2018 Oct 10];135(0):56–65. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3752793/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonda X, Eszlari N, Kovacs D, Anderson IM, Deakin JFW, Juhasz G, et al. Financial difficulties but not other types of recent negative life events show strong interactions with 5-HTTLPR genotype in the development of depressive symptoms. Transl Psychiatry [Internet]. 2016. May [cited 2018 Oct 10];6(5):e798 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070066/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller S, Wain L, Shrine N, Ntalla I, Cook J, Sayers I, et al. The UK BiLEVE Study: The First Genetic Study in UK Biobank Identifies Novel Regions Associated with Airway Obstruction and Smoking Behaviour In: B31 INFLAMMATION AND COPD [Internet]. American Thoracic Society; 2015. [cited 2018 Oct 10]. p. A2714–A2714. (American Thoracic Society International Conference Abstracts). Available from: https://www.atsjournals.org/doi/abs/10.1164/ajrccmconference.2015.191.1_MeetingAbstracts.A2714 [Google Scholar]
- 57.Munafò MR, Tilling K, Taylor AE, Evans DM, Davey Smith G. Collider scope: when selection bias can substantially influence observed associations. Int J Epidemiol [Internet]. 2018. February [cited 2018 Oct 4];47(1):226–35. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5837306/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kroenke K, Spitzer RL, Williams JB. The phq-9. Journal of general internal medicine. 2001;16(9):606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McClellan J, King M-C. Genetic Heterogeneity in Human Disease. Cell [Internet]. 2010. April 16 [cited 2018 Jul 1];141(2):210–7. Available from: https://www.cell.com/cell/abstract/S0092-8674(10)00320-X [DOI] [PubMed] [Google Scholar]
- 60.Kapur S, Phillips AG, Insel TR. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol Psychiatry. 2012. December;17(12):1174–9. [DOI] [PubMed] [Google Scholar]
- 61.Hyman SE, Krystal JH. Report of the National Advisory Mental Health Council Workgroup on Genomics [Internet]. [cited 2018 Jul 1]. Available from https://www.nimh.nih.gov/about/advisory-boards-and-groups/namhc/reports/report-of-thenational-advisory-mental-health-council-workgroup-on-genomics.shtml#roster [Google Scholar]
- 62.Spitzer RL, Kroenke K, Williams JB, Group PHQPCS. Validation and utility of a selfreport version of PRIME-MD: the PHQ primary care study. Jama. 1999;282(18):1737–1744. [DOI] [PubMed] [Google Scholar]
- 63.First MB, Gibbon M, Spitzer RL, et al. : User’s Guide for the Structured Clinical Interview for DSM-IV Axis I Disorders–Research Version. New York, Biometrics Research Department, New York State Psychiatric Institute, 1996 [Google Scholar]
- 64.Smith DJ, Nicholl BI, Cullen B, Martin D, Ul-Haq Z, Evans J, et al. Prevalence and Characteristics of Probable Major Depression and Bipolar Disorder within UK Biobank: Cross-Sectional Study of 172,751 Participants. PLOS ONE [Internet]. 2013. November 25 [cited 2018 Jan 31];8(11):e75362 Available from: http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0075362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Townsend P, Phillimore P, Beattie A. Health and Deprivation: Inequality and the North. Croom Helm; 1988. 250 p. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.