

Abstract
Although G protein-coupled receptors (GPCRs) are recognized as pivotal drug targets involved in multiple physiological and pathological processes, the majority of GPCRs including orphan GPCRs (oGPCRs) are unexploited. GPR88, a brain-specific oGPCR with particularly robust expression in the striatum, regulates diverse brain and behavioral functions, including cognition, mood, movement control, and reward-based learning, and is thus emerging as a novel drug target for central nervous system disorders including schizophrenia, Parkinson’s disease, anxiety, and addiction. Nevertheless, no effective GPR88 synthetic ligands have yet entered into clinical trials, and GPR88 endogenous ligands remain unknown. Despite the recent discovery and early stage study of several GPR88 agonists, such as 2-PCCA, RTI-13951–33, and phenylglycinol derivatives, further research into GPR88 pharmacology, medicinal chemistry, and chemical biology is urgently needed to yield structurally diversified GPR88-specific ligands. Drug-like pharmacological tool function and relevant signaling elucidation will also accelerate the evaluation of this receptor as a viable neurotherapeutic target.
Keywords: Orphan G protein-coupled receptors (oGPCRs), GPR88, agonists, striatum, CNS disorders, Neurotherapeutics, drug discovery
Graphical Abstract

1. INTRODUCTION
G protein-coupled receptors (GPCRs), also known as seven-transmembrane domain receptors, are the largest group of signaling proteins in the human genome, with at least 800 members.1 GPCRs mediate a myriad of important physiological and pathological processes ranging from blood pressure regulation, allergic response, renal function, hormonal disorders, psychiatric disorders, and neurodegenerative diseases to the progression of human cancers. GPCRs regulate numerous cellular and physiological functions by transducing diverse extracellular signals (e.g., light, Ca2+, odorants, amino acids, hormones, neurotransmitters, nucleotides, peptides, fatty acid derivatives, and various polypeptide ligands) into intracellular biochemical responses via the activation of heterotrimeric G proteins and other effector pathways.2–4 Owing to their unique structures with binding pockets amenable to chemical modulation, GPCRs have emerged as essential targets in both biomedical research and drug development, with approximately 40% of FDA-approved drugs targeting human GPCRs.5–9 These GPCR ligands also comprise a large number of best-selling drugs,10 including Eli Lilly’s Olanzapine, GlaxoSmithKline’s Ranitidine, Merck’s Losartan, and Otsuka Pharmaceutical’s Aripiprazole, to name a few.
Although there are a large number of marketed GPCR drugs, these ligands target ∼80 of the 350 nonsensory receptors, meaning only ∼20% of all potentially druggable GPCRs are currently targeted. In addition, roughly ∼140 of these potentially druggable receptors are orphan GPCRs (oGPCRs) whose endogenous ligands are unknown, and most oGPCR functions are poorly understood.11–13 In addition, roughly 80% of the oGPCRs are expressed in the brain, including several that are linked to diseases of the central nervous system (CNS).14 Most GPCRs started as orphan receptors, and matching them to known neuromodulators led to the elucidation of the broad diversity of the neuroreceptor families. Despite significant efforts to accelerate progress for treating CNS disorders, developing novel neurotherapeutics remains an enormous unmet medical need.15,16 To this end, the unexploited brain oGPCRs may provide valuable new therapeutic targets for future drug discovery while fueling innovation and continued interest in GPCR-based therapeutics.
The large number of druggable GPCRs includes GPR88, a class A rhodopsin family orphan receptor, that is curiously expressed almost exclusively in the striatum of the brain in rodents, primates, and humans.17 GPR88 plays an important role in the regulation of various brain and behavioral functions, including cognition, mood, movement control, and cue-based reward learning, and is emerging as a promising drug target for the treatment of basal ganglia associated disorders. Currently, GPR88 endogenous ligands remain unknown; however, several GPR88 agonists such as 2-PCCA, RTI-13951–33, and phenylglycinol derivatives have been examined in the early stage of preclinical studies. Given the therapeutic potential of GPR88 and the recent progress in exploiting the chemical space of GPR88 agonists, this concise review is structured to provide the readers with a summary of GPR88 structure, signaling, and potential functional roles in various CNS diseases, as well as a detailed description of GPR88 agonists and hit-to-lead optimizations from a medicinal chemistry and chemical biology perspective.
2. GPR88 AND ITS SIGNALING PATHWAY
The Gpr88 gene was first discovered in 2000 by the Ito lab18 using differential display screening for region-specific transcripts in rat brain. The Gpr88 gene encodes a seven transmembrane spanning receptor protein (GPR88), an orphan GPCR of the class A rhodopsin family of receptors. GPR88 was originally identified as a striatum-specific receptor (designated Strg/GPR88) in both human and rodents,18,19 though it is also expressed at low levels in the cerebral cortex, central extended amygdala, lateral, cortical, hypothalamus, and other brain regions.17,19–22
Owing to the lack of endogenous or highly selective synthetic ligands such as high affinity antagonists, there are currently no X-ray cocrystal structures of GPR88 with ligands. The human GPR88 primary sequences encoded by its full length cDNA (UniProtKB ID: Q9GZN0) clone were putatively identified as a 384-amino acid protein (molecular weight of 40.246 kDa) with seven putative transmembrane domains (TM1–TM7, Figure 1), an N-glycosylation site (amino acid residues 3–6), as well as five potential protein kinase C phosphorylation sites (amino acid residues 58–60, 111–113, 222–224, 337–339, and 346–348). Moreover, the GPR88 protein sequences are highly evolutionarily conserved between humans and rodents (95% identity).18 Using the PSI-BLAST searching method in Discovery Studio 3.1,23 we compared the sequence similarities of human GPR88 and PDB_nr95 database (nonredundant 37879 protein sequences from RCSB Protein Data Bank)24 and found that the human GPR88 do not share high sequence similarity with any protein sequences deposited in PDB_nr95 database (entire sequence identity <30%). The highest sequence similarity can be achieved between human GPR88 and the chimeric protein of 5-HT1B-BRIL (entire sequence identity: 18%, Figure 1). However, GPR88 lacks several critical features conserved in known biogenic amine receptors, such as the tripeptide motif DRY considered to be critical for receptor activation and cysteines involved in the formation of the extracellular loops.25 The genomic organization of the human Gpr88 gene, which contains a single protein coding exon, is similar to many other mammalian GPCRs, including several muscarinic acetylcholine (M1, M2, M3, M4, M5), histamine (H1), dopamine (D1, D5), cannabinoid, and angiotensin II receptors.18,26
Figure 1.

Entire amino acid sequence of the human GPR88 protein. The seven putative transmembrane spanning domains (TM1–TM7) are indicated as underlined. Identically conserved amino acids derived from sequence alignment between human h5-HT1B and GPR88 are highlighted in blue; strongly similar amino acids are marked in green; weakly similar amino acids are marked in light blue.
The specific brain signaling pathways and physiologic functions of GPR88 are largely unknown because the endogenous hormone or transmitter for the receptor has not yet been identified. However, several lines of evidence suggest that GPR88 may provide an important role regulating the excitability of glutamatergic and GABAergic neurons as well as modulating responses to dopaminergic neurons, based on the particularly robust expression of GPR88 in medium spiny neurons (MSNs) in the striatum (more details in section 3.1).17,27,28 Conversely, glutamatergic and dopaminergic depletion differentially alters Gpr88 mRNA levels in distinct MSN subpopulations, indicating neurotransmission regulates Gpr88 gene expression.17,28 Consequently, GPR88 is implicated in many behavioral functions and responses, including cognition, mood, movement control, and cued response reward learning.17,19,21,29
The molecular signaling pathway of GPR88 was initially described by the work from Bristol Myers Squibb (BMS) and Lexicon Pharmaceuticals30,31 using small molecule agonists for the receptor. These studies indicated GPR88 couples to Gαi/o G proteins, allowing GPR88 to inhibit adenylyl cyclase and reduce cAMP production and signaling, as outlined in Figure 2. GPR88 knockout (KO) mice also have provided important hints about the receptor’s potential signaling in the brain. For example, using striatal membranes from Gpr88 KO mice revealed loss of GPR88 receptor expression inhibited the function of opioid (δ/μ) and muscarinic acetylcholine receptors (M1/M4) coupling to Gi/Go proteins and possibly the signaling of other GPCRs at the cellular level.32 Further in vivo studies revealed functional antagonism between GPR88 and δ-opioid receptor (DOR) activities.32 Also, electrophysiological studies in Gpr88 KO mice demonstrated that the absence of GPR88 increased glutamatergic excitation and reduced GABAergic inhibition in striatal MSNs. This work further suggests GPR88 is inhibitory in the striatum, and when knocked out, striatal neuron firing rates increase in vivo.27
Figure 2.
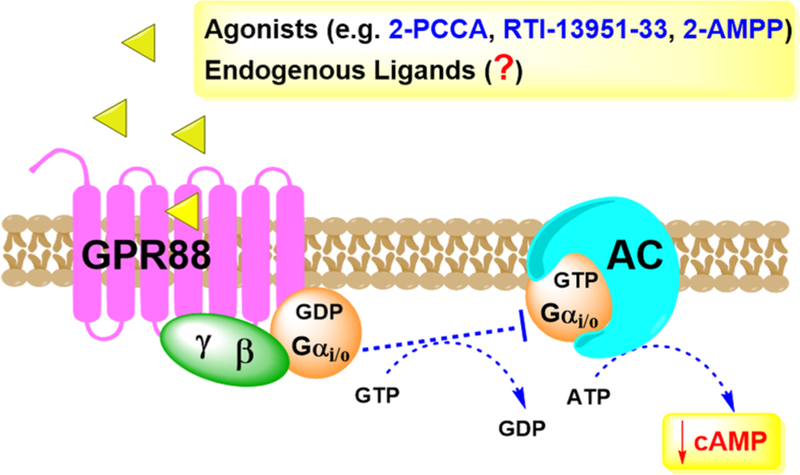
GPR88 signaling pathway. GPR88 couples to Gi/o proteins to inhibit adenylyl cyclase (AC) and reduce cAMP signaling and is likely an inhibitory receptor in the central nervous system.
3. GPR88 AS A POTENTIAL THERAPEUTIC TARGET FOR HUMAN CNS DISEASES
Because GPR88 is an orphan receptor with highly restricted localization to the brain and particularly robust expression in the dorsal and ventral striatum, GPR88 has recently attracted considerable interest for investigating its neurobiological functions and pathophysiological relevance to brain disorders. Evidence of GPR88 function, including knockout studies and transcriptional profiling studies, indicates GPR88 may provide a druggable target for human CNS diseases involving the striatum, such as schizophrenia (SZ),33–35 Huntington’s Disease (HD), Parkinson’s disease (PD),17 anxiety,32 bipolar disorder (BD),36 depression,20 and drug addiction.28,37,38
3.1. Movement Disorders.
GPR88 is highly expressed in both direct and indirect pathway populations of striatal MSNs, which represent the input circuits of the basal ganglia neuronal system. This MSN neuron network controls many basal ganglia functions, including voluntary movement, motor learning, motor planning, and decision making. One of the fascinating things about GPR88 is that it is positioned to inhibit both indirect and direct pathway MSNs, which would do opposite things to basal ganglia outputs, including modulation of motor control.27 These striatal MSNs also receive dense dopaminergic and glutamatergic signal inputs,39,40 and this neural circuitry is strongly implicated in the etiology and treatment of Parkinson’s disease, Huntington’s disease, and also neuropsychiatric diseases.41–46
Several studies relevant to PD and HD have suggested changes in GPR88 expression, and notably Gpr88 KO mice have altered motor functions. Using the unilateral 6-hydroxydopamine (6-OHDA)-lesioned rat model of PD, dopamine depletion in the striatum resulted in decreased GPR88 mRNA expression in indirect pathway MSNs and increased GPR88 expression in direct pathway MSNs.17 Notably, dopamine replacement with L-DOPA treatment in the rats normalized GPR88 expression levels.17 Gpr88 genetic knockdown negatively regulated the expression of DARPP-32, a multifunctional protein predominantly expressed in medium spiny neurons that controls the physiological characteristics of dopamine receptive neurons.47,48 Brain immunohistochemical analyses also confirmed a significant elevation of phosphorylated DARPP-32 Thr-34 in Gpr88 KO mice, indicating deletion of Gpr88 changes striatal function via DARPP-32.28 Gpr88 KO mice also show increased spontaneous locomotor activity,27 increased sensitivity to drug-induced catalepsy, and baseline motor incoordination.28 These results indicate GPR88 modulation of motor function.
Although a direct link of Gpr88 genetic mutations with human PD is lacking, sporadic cases of chorea in humans are associated with deleterious mutations in GPR88.49 Because the pathology of HD involves loss of function and/or cell death of striatal neurons leading to involuntary movement (chorea), GPR88 modulators may correct motoric abnormalities arising in HD. Studies of BACHD mice, an HD model expressing the full-length human mutant Huntington gene, show that GPR88 expression is greatly reduced in striatal neurons of aged HD mice, which is accompanied by hyper-excitability of striatal MSNs.50 Therefore, GPR88 agonists might reverse this HD pathology by increasing GPR88 function and reducing MSN hyper-excitability. Taken together, a growing body of evidence implicates GPR88 in basal ganglia motor function, and pharmacological modulation of the receptor may prove useful for normalizing abnormal motor function in PD, HD, and other related movement disorders.
3.2. Schizophrenia.
Gpr88 is, in fact, a susceptibility risk gene for sporadic cases of schizophrenia.51,52 Genetic association analyses performed by the Meloni group51 showed a positive association between Gpr88 and SZ from the Xhosa human population in South Africa. Using the neonatal treatment with the phencyclidine (PCP, a noncompetitive NMDA receptor antagonist) rat model, which induces SZ-like behavior in animals, Meloni et al.53 determined knocking down Gpr88 in the nucleus accumbens (NAcc, the ventral striatum) inhibited amphetamine-induced hyperlocomotion and reduced the impairment of social novelty discrimination in this SZ-like model. Moreover, Gpr88 KO mice behaviorally demonstrated disrupted prepulse inhibition of the startle response (PPI) and increased sensitivity to stimulants, including elevated apomorphine-induced climbing and stereotypy (AICS) and increasing locomotion and striatal dopamine release by amphetamine.28 These abnormal behaviors are general models of the positive symptoms (psychosis) arising in SZ and suggest Gpr88 KO enhances these abnormal SZ-like behaviors. Conversely, antipsychotic administration with D2 receptor antagonists (e.g., haloperidol or risperidone) to Gpr88 KO animals normalized stimulant hypersensitivity back to control levels, with improved PPI deficits and blocked AICS.28
Additionally, inactivation of striatal Gpr88 gene expression increased striatal glutamate receptor phosphorylation and altered GABAA receptor composition, resulting in enhanced MSN firing rates in vivo.27 Gpr88 genetic inactivation in these mice also resulted in abnormal behaviors, including hyper-ctivity, poor motor coordination, impaired cue-based reward learning, and deficits in acquisition of visual or auditory cues, which could be restored by striatal Gpr88 re-expression.27 Consistent with the impairment in Gpr88 KO mice, a homozygous deleterious mutation (p.C291X) in human GPR88 is also associated with marked speech delay and learning disabilities which manifest at 8–9 years old.49
All the current in vitro and in vivo results demonstrate a key role for the GPR88 in modulating the striatal dopaminergic system and glutamatergic systems, and GPR88 function may control several behaviors observed in neuropsychiatric diseases, including SZ.27
3.3. Anxiety.
Previous studies have focused on GPR88 function in striatal physiology; however, GPR88 is also expressed densely in the central nucleus (CeA) of amygdala and detected in additional brain regions including ventral striatum (NAcc and olfactory tubercles), anterior bed nucleus of the stria terminalis, and lateral septum,54 all of which are involved in regulating anxiety. Gpr88 KO mice have significantly altered gene transcriptional activity, neuronal morphology, and dopamine levels in the CeA region,32 suggesting potential effects on amygdala-dependent function and behavior.55–58 Moreover, Meirsman et al.32 confirmed that Gpr88 KO mice have markedly reduced levels of anxiety in various models, including the elevated plus maze, marble burying, and novelty suppressed feeding. Further behavioral tests in A2AR-Gpr88 KO mice (a conditional knockout of Gpr88 only in indirect pathway MSNs) showed no change in novelty preference and novelty-suppressed feeding, indicating GPR88 expressed in these neurons does not regulate anxiety-like behaviors. Future work is required to determine which GPR88 expressing neurons are involved in these anxiety-related behaviors.59 Hence, GPR88 inhibition may serve as a promising therapeutic strategy to treat anxiety-related disorders.
Other CNS Disorders.
Recent studies also have focused on the regulation of Gpr88 gene expression in rat models of other CNS disorders such as bipolar disorder, depression, and drug addiction. Alterations in expression levels of Gpr88 have been observed in several brain regions after the treatment of addictive drugs,20 antidepressants,28,36,60 or mood stabilizer treatments (e.g., down-regulation by fluoxetine;36 up-regulation by lithium in the cortex,33 morphine in the central extended amygdala,20 and methamphetamine and valproate in the prefrontal cortex34) in both rodent models and humans. A recent Gpr88 KO study37 demonstrated that the GPR88-deficient mice showed increased voluntary alcohol drinking in both moderate and excessive drinking models, suggesting GPR88 may be a risk factor involved in alcohol use disorder (AUD). Later, an in vivo study further verified that administration of GPR88 agonists can significantly reduce alcohol self-administration and alcohol intake in a dose-dependent manner (more details in section 4.1).61
Moreover, Gpr88, located at 1p22-p21 chromosomal region, not only is a positive functional candidate gene for SZ in the Xhosa population mentioned before but also shows strong linkage to BD in Sardinian and Palestinian triad human populations.51,52 The association analysis further revealed a genetic association between BD and rs2030048, a single nucleotide polymorphism (SNP) marker in GPR88, in the Sardinian population. Interestingly, a positive association signal was also found with another marker of GPR88, rs2809817, in a similar BD sample of Palestinian families.
In summary, accumulating studies support that the expression, and likely function, of the Gpr88 gene is associated with a variety of CNS disorders, suggesting that pharmacological modulation of GPR88 may also provide a promising therapeutic approach for bipolar disorder, depression, and addiction.
4. GPR88 AGONISTS
Because of the emerging pharmacological implications of GPR88 and lack of endogenous ligands, considerable attention has been focused on development of potent and selective synthetic ligands as pharmacological probes to elucidate the physiological roles and therapeutic potential of GPR88 in human diseases. To the best of our knowledge, only GPR88 agonists have been identified and reported, representing two major chemotypes: 2-PCCA and phenylglycinol derivatives. Herein, we summarize current research advances in the development of chemical entities as GPR88 ligands.
4.1. 2-PCCA Analogues.
2-PCCA (1, (1R,2R)-2-pyridin-2-yl-cyclopropanecarboxylic acid ((2S,3S)-2-amino-3-methylpentyl)-(4′-propylbiphenyl-4-yl)-amide dihydrochloride, Figure 3) is the first potent small molecule GPR88 agonist with an EC50 value of 3.1 nM in a cAMP HTRF functional assay reported by BMS and Lexicon Pharmaceuticals, and its bioactivity was later confirmed in a GTPγS binding assay.30,31 To facilitate their drug discovery campaign, Jin and colleagues62 developed another cell-based assay system initially by transiently cotransfecting HEK293T cells with the human GPR88 cDNA and a luminescent cAMP biosensor. The racemate of 1 ((±)-1) produced no measurable increases in cAMP levels even at concentrations up to 30 μM, while the positive control, isoproterenol (ISO), greatly stimulated cAMP production in a concentration-dependent manner. However, compound (±)-1 inhibited ISO-induced cAMP formation with an EC50 value of 877 nM in HEK293T/GPR88 cells, indicating this ligand activates GPR88-mediated Gαi/o signaling. In addition, 1 ((1R,2R)-isomer; EC50 = 373 nM) is approximately 5-fold more potent than its (1S,2S)-isomer. Furthermore, (±)-1 did not induce calcium mobilization measured by the fluorescent imaging plate reader (FLIPR) calcium assay in HEK293T/GPR88 cells, indicating that GPR88 is likely not coupled to Gαq proteins.62 Of note, although the relative efficacy of agonists for GPCRs (e.g., partial vs full agonist) is often a key and defining parameter of small molecule ligand activity, for the orphan receptors that lack an endogenous reference agonist, defining a true measurement of relative efficacy is complex. Therefore, we focus this review of GPR88 ligand activities primarily around ligand potency (EC50).
Figure 3.

Diversified structural modifications of 1.
Further in vivo studies demonstrated that (±)-1 at 0.1–3.2 mg/kg dose-dependently decreased locomotor activity and food-maintained operant responding in rats but failed to block the behavioral effects of methamphetamine, including altering methamphetamine-induced hyperactivity and discriminative stimulus effects.63 Unfortunately, ADMET study shows 1 is a P-glycoprotein (P-gp) substrate,31 likely because of its high lipophilicity (clogP = 6.19), suggesting that 1 may have the potential risk of P-gp-mediated drug–drug interactions. More-over, 1 was recently reported to have significant off-target activity based on its obvious [35S]-GTPγS binding in Gpr88 KO mouse striatal membranes. All these data suggest that it is imperative to pursue further structural optimizations.
To further characterize GPR88 in vitro pharmacology and initially explore structure–activity relationship (SAR) studies of 2-PCCA analogues, Jin and colleagues62 established another cAMP functional assay by using HEK293 cells stably expressing the human GPR88 receptor and the GloSensor-22F cAMP construct. (±)-1 and its (1R,2R)-isomer 1 exhibit EC50 values of 911 and 603 nM, respectively. Substitution effects on the biphenyl moiety, the pyridyl cyclopropane fragment, and the substituted ethylamine moiety in (±)-1, as shown in Figure 3, were first investigated. All the tested newly synthetic 2-PCCA analogues were 1:1 diastereomeric mixtures of (1R,2R)- and (1S,2S)-isomers determined by 1H NMR and HPLC analyses. The 4-position of the biphenyl moiety tolerated the small to medium size of alkyl substitutions with the cyclohexyl analogue (±)-2 (EC50 = 746 nM, Table 1) as the most potent compound in the series. A limited examination of the pyridyl cyclopropane fragment observed that replacing the pyridyl group with a phenyl moiety exemplified by (±)-4 led to only a slight decrease in potency, while replacement of the cyclopropane moiety with a methylene group exemplified by (±)-5 resulted in a loss of activity, indicating that the central linker of the aromatic and carbonyl moieties is critical for GPR88 recognition. Investigation of the upper ethylamine moiety showed that hydrophobic substitutions were well tolerated, as shown in compounds (±)-6–(±)-8, suggesting that a hydrophobic pocket (P1) may be present in the GPR88 receptor.62
Table 1.
Bioactivity of 2-PCCA Derivatives
| compd | EC50 (nM, cAMP HTRF assay) | EC50 (nM, GloSensor cAMP assay) | EC50 (nM, Lance cAMP assay) |
|---|---|---|---|
| (±)-1 | 911 | 116 | |
| 1 | 3.1 | 603 | 56 |
| (±)-2 | 746 | 176 | |
| (±)-3 | 77 | ||
| 3 | 3.5 | ||
| (±)-4 | 1250 | ||
| (±)-5 | >10000 | ||
| (±)-6 | 5330 | ||
| (±)-7 | 994 | ||
| (±)-8 | 898 | ||
| (±)-9 | 2760 | ||
| (±)-10 | 356 | ||
| (±)-11 | 439 | ||
| (±)-12 | 666 | ||
| (±)-13 | 880 | ||
| 13 | 230 | ||
| 14 | 44 | ||
| 15 | 35 | ||
| 16 | 1100 | ||
| 17 | >3000 | ||
| (±)-18 | 800 | ||
| (±)-19 | 330 | ||
| (±)-20 | 1100 | ||
| (±)-21 | 3200 | ||
| (±)-22 | 1200 | ||
| (±)-23 | >100000 | ||
| (±)-24 | 660 | ||
| (±)-25 | 570 | ||
| (±)-26 | 63 | ||
| (±)-27 | 100 | ||
| 27 (RTI-13951–33) | 25 |
To set up a more scalable assay, Jin et al.64,65 initially attempted a modified G protein coupling assay enabling GPR88/Gi/o proteins to activate calcium release and mobilization and later developed a sensitive Lance cAMP assay using Chinese Hamster Ovary (CHO) cells stably expressing PPLS-HA-GPR88. (±)-1 with its pure (1R,2R)-enantiomer display EC50 values of 116 and 56 nM, respectively. Considering that previous SAR suggests the biphenyl aniline moiety of (±)-1 as a suitable site for diverse modifications,62 systematical optimizations of substituents at the phenyl ring were investigated to get higher-potency ligands with possibly improved drug-like properties (e.g., clogP = 2–4;66 TPSA < 76 Å2;67 logBB > −168). Among these compounds, (±)-3 (clogP = 5.40, TPSA = 68.45 Å2, logBB = −0.05) was identified as the most potent compound with an EC50 value of 77 nM, which is slightly more potent than (±)-1.65 Meanwhile, in the cAMP HTRF assay, compound 3 ((1R,2R)-enantiomer) showed single-digit nanomolar potency with an EC50 value of 3.5 nM, which was as potent as 1 reported by Bi et al.30,31 Unfortunately, 3 is also a P-glycoprotein substrate with high lipophilicity. However, removal of a phenyl ring on biphenyl aniline moiety resulted in a significant loss of activity as in (±)-9 (Figure 3, EC50 = 2,760 nM).65 Replacement with a cyclohexyl group ((±)-10) led to only a slightly decreased potency (EC50 = 356 nM), while inserting 1–2 CH2 or O-linker into the biphenyl group resulted in more deteriorated activity ((±)-11–(±)-12; EC50 ≈ 500 nM; Table 1). All these results suggest that a π–π stacking interaction may be present between the biphenyl moiety and the second hydrophobic pocket of GPR88 (P2). A following computational QSAR and molecular docking study of 1 were also consistent with the SAR conclusions, indicating the positioning of the 4′-substituted biphenyl encased in a largely hydrophobic GPR88 homology model binding site.65
On the other hand, Bi et al.31 identified another two similar compounds, (±)-13 and (±)-18 (Figure 4), as hits with attractive agonist activity for GPR88 (EC50 = 800 and 880 nM, respectively) via a high-throughput screening (HTS) campaign based on cAMP HTRF assays. R,R-cyclopropyl system 13 displayed an EC50 value of 230 nM, while its S,S-cyclopropyl enantiomer was completely inactive, suggesting that the trans-cyclopropyl group appeared to be a critical feature (Table 1). However, opening the cyclopropyl ring from the distal position to give the 2-methyl-3-phenylpropyl amide (±)-22 (Figure 4) resulted in only minimal loss of activity, with an EC50 value of 1200 nM. Considering that their biphenyl group as that of 1 likely extends into a hydrophobic pocket P2, various substitutions on 13 and (±)-18 were investigated. Analogues of 13 with smaller alkyl or alkoxyl substitutions (e.g., 14 and 15; EC50 44 nM and 35, respecitively) exhibit the best potency, while compounds with substituents containing a hydrogen bond donor (e.g., hydroxyl 16 and amine 17) significantly lost their activities. Moreover, the biaryl groups of (±)-18 bearing substituents such as Me, OMe, F, and CN at the 2- or 3-position resulted in loss of activity, while the substituents at the 4-position were well tolerated (e.g., (±)-19–(±)-21). However, the SAR of the upper ethylamine moiety on 13 and (±)-18 indicated that there was little tolerance for modifications. The primary amino group was important for potency, as shown in compound (±)-23, which was completely inactive. Only the amine moiety of (±)-24 and (±)-25 with hydrophobic volume similar to that of the original hit (±)-18 maintained the potency. In addition, all attempts to constrain the scaffold (e.g., amine or the amide carbonyl to the aryl ring) unfortunately led to inactive compounds, with at least a 10-fold loss of activity.
Figure 4.

Structural modifications of 2-PCCA analogues (±)-13 and (±)-18.
To continue to lower lipophilicity of 1 for brain penetration, Jin et al.61 most recently further optimized the amino alkyl side chain of 1 by inserting an oxygen, giving (±)-26 (Figure 4) with an EC50 value of 63 nM (clogP = 4.87 vs 6.19). To further improve the drug-like properties for CNS penetration (clogP = 2–466) while retaining the potency, combination of (±)-26 with the optimized biphenyl substitution (MeOCH2 as in compound 15), yielded agonist (±)-27, and the corresponding pure (1R,2R)-diastereomer 27 (RTI-13951–33, clogP = 3.34). 27 was highly potent in the GPR88 cAMP functional assay (EC50 = 25 nM) and had no significant off-target activity when tested against 38 GPCRs, ion channels, and neurotransmitter transporters as well as no agonist signaling activity in the GTPγS binding assay using Gpr88 KO mouse striatal membranes.61 27 also displayed good biopharmaceutical properties with decent aqueous solubility (984 ± 9.4 μg/mL in phosphate-buffered saline solution) and brain permeability (13% from apical A to basal B in MDCK assays), leading to favorable pharmacokinetic properties with sufficient brain exposure (brain to plasma (B/P) ratio: 0.5) for GPR88 modulation. 27 subsequently dosed at 10 and 20 mg/kg by intraperitoneal injection significantly reduced alcohol self-administration and alcohol intake in a dose-dependent manner and exhibited no effects on locomotion and sucrose self-administration in rats,61 indicating that it can be a potential drug candidate for the treatment of alcohol use disorder.
4.2. Phenylglycinol and its Derivatives (2-AMPP Analogues).
Other than compounds (±)-13 and (±)-18,31 Bi and Dzierba et al.69 also identified a novel hit (±)-28 (a diastereomeric mixture, EC50 = 6 μM, Figure 5) with new phenylglycinol scaffold by inhibition of forskolin-stimulated cAMP in HEK cells in their high-throughput screening campaign. Only (R)-amino, (S,S)-cyclopropyl isomer 28 showed moderate activity with an EC50 value of 2.5 μM, while the other three diastereomers were completely inactive (EC50 > 100 μM). Based on the SAR findings of the 2-PCCA series, early structural modifications of 28 resulted in biaryl analogues, such as 29 with a 25-fold improvement in potency in the cAMP HTRF assay (EC50 = 93 nM), and comparable potency between the cAMP inhibition assay and the GTPγS binding assay (EC50 = 240 nM). Although compound 29 had an ideal ratio of total brain (695 nM) to plasma (290 nM) concentrations of ∼2, 29 exhibits relatively high lipophilic with a clogP of 4.2, high plasma protein binding (PPB, 96.4%), and poor metabolic stability (CL = 47 mL/min/kg; T1/2 = 0.6 h) at 1 mg/kg iv in mouse. To improve clearance and half-life, as well as maintain or improve CNS exposure, analogues with high potency and low calculated lipophilicity were therefore designed. Replacement of the terminal ring of the biaryl group with an aliphatic ether resulted in ethers 30 and 31 with lower lipophilicity (clogP = 3.4–3.9). Although ether 31 was slightly more potent than 29, it showed lower B/P ratios (1.1) and high clearance (144 mL/min/kg), which limited its utility for in vivo efficacy studies. Optimization of amide cap of 31 with (S)-α-methylbenzyl moiety led to compound 32 with only a 2-fold decrease in potency (EC50 of 190 vs 95 nM). Further introduction of smaller substitutions on the central phenyl group of compound 32 was shown to be well tolerated, such as methyl (33) or fluoro (34), and methoxy group (35) at its 3-position, except incorporating a nitrogen atom. Extending the ether chain in one carbon increments from (S)-2-methylbutyl (32) to 2-methylpentyl (36) improved potency with an EC50 value of 110 nM, but incorporating an additional oxygen into the side chain led to complete loss of activity. However, optimizing site A with trans-cyclopropyl phenyl, phenyl, benzyl group, and its isomer (R)-α-methylbenzyl decreased potency (e.g., 37–39). Only α-hydroxymethylbenzyl and (S)-α-ethylbenzyl groups (40 and 41) were well tolerated (EC50 = 120 nM).69 Additionally, introduction of the geminal dimethyl group next to the hydroxyl group led to a boost in potency and lipophilicity relative to the unsubstituted alcohol (42 vs 36, EC50 = 29 vs 110 nM, clogP = 5.3 vs 4.7). Moreover, an amino group (43, 2-AMPP) was well tolerated at this position of the hydroxyl group as exemplified in compound 33. However, substitutions on the nitrogen of 43 increased the lipophilicity (clogP of 5.4 vs 4.4) with no gain in potency (44), and further incorporating them into a ring led to a modest loss in potency. 43 elicited strong [35S]-GTPγS binding (EC50 = 940 nM) in mice striatal membranes but was ineffective in the samples from the Gpr88 KO mice, demonstrating that the compound is a GPR88 striatum-specific agonist.32
Figure 5.

Structural modifications of phenylglycinol analogues.
Given that 36 and 43 can be viewed as promising leads for further optimizations to probe GPR88 in vivo functions, Jin et al.70 re-evaluated 36 and 43 in their Lance cAMP assay, with EC50 values of 194 and 414 nM, respectively, which are slightly different from previous HTRF assays. Interestingly, replacing the hydroxyl or amine group with an azide group gave the most potent compound (45, EC50 = 134 nM) in the series (Table 2). However, amide 46 and methyl ester 47 were moderately active, with EC50 values of 616 nM and 538 nM, respectively. All the substitutions on the terminal phenyl ring of site A led to detrimental activities.70 These findings suggest that the GPR88 receptor is associated with a hydrogen bonding/electrostatic interaction at site B and has limited steric tolerance at site A.
Table 2.
Bioactivity of Phenylglycinol Derivatives
| no. | EC50 (nM, cAMP HTRF assays) | EC50 (nM, Lance cAMP assays) |
|---|---|---|
| 28 | 2500 | |
| (±)-28 | 6000 | |
| 29 | 93 | |
| 30 | 270 | |
| 31 | 81 | |
| 32 | 190 | |
| 33 | 170 | |
| 34 | 260 | |
| 35 | 410 | |
| 36 | 110 | 194 |
| 37 | 810 | |
| 38 | 860 | |
| 39 | 1500 | |
| 40 | 120 | |
| 41 | 145 | |
| 42 | 29 | |
| 43 | 90 | |
| 44 | 91 | 414 |
| 45 | 134 | |
| 46 | 616 | |
| 47 | 538 |
5. CONCLUSION AND FUTURE DIRECTIONS
Since the discovery of GPR88 as an orphan GPCR in 2000, the scientific community has made progress in understanding GPR88 function and, moreover, advanced GPR88 as a promising and attractive therapeutic target for brain disorders including schizophrenia, Parkinson’s disease, anxiety, bipolar disorder, depression, and drug addiction.
Success in this new era of orphan receptor probe discovery and pharmacology can be attributed to the synergy of three approaches: loss-of-function genetics (knockout studies), immunohistochemistry, and sensitive and versatile cellular and high-throughput chemical screening assays. The development of orphan GPCR knockout studies and transcriptional profiling studies have proven to be a transformative approach for elucidating GPR88 function and biochemical signaling. Immunohistochemistry has become an integral component for assessing the anatomical distribution and expression level of Gpr88 in specific brain regions. Sensitive and versatile high-throughput screening assays, including innovative and scalable cAMP assays (GloSensor, Lance), have facilitated identification of selective GPR88 synthetic agonist ligands (e.g., 2-PCCA, RTI-13951–33 and 2-AMPP). The probes have proven to be valuable pharmacological tools for elucidating GPR88 function and for testing agonist modulation of GPR88 for neuro-therapeutic potential in a variety of CNS disorders. In addition to the existing agonist ligands, discovery and optimization of GPR88 selective antagonists would provide valuable tools to further study receptor function and therapeutic potential.
Progress in understanding and targeting this orphan GPCR has been hampered by the lack of the endogenous ligand(s) as well as cocrystal structures. Conventional deorphanization studies to discover endogenous ligands is challenging and time-consuming;71 however, future studies aimed at screening fractionated brain tissues against GPR88 may reveal the receptor’s endogenous peptide or ligand. There is also the intriguing possibility that GRP88 may dimerize with another GPCR, thereby regulating the ligand specificity and function of the latter.72,73 Identifying the endogenous ligand would provide a key discovery to elucidate the biochemical signaling of GPR88 in the brain and also provide a foundation to fully define the neurochemistry and neurobiology of this receptor. In addition, without knowledge of the endogenous GPR88 ligand, identifying and validating other types of receptor modulators (e.g., antagonists, NAMs, PAMs) will be challenging. However, GPR88 antagonists would be valuable tools to further our understanding about GPR88 structure and function. Therefore, deorphanization of GPR88 is an important goal for future studies.
The development of highly selective and druglike GPR88 synthetic ligands remains an attractive and alternative approach to identify endogenous active ligands, and cocrystal structures with synthetic ligands could provide atomic level structural information that may also help solve the puzzle of the receptor’s endogenous ligand (e.g., by revealing the structure of potential binding pockets). GPCRs are notoriously challenging for crystallography and require either significant protein engineering or nanobody stabilization, and the vast majority of current GPCR-ligand structures are with antagonists.74 Therefore, discovery and optimization of a high-affinity GPR88 antagonist could empower future structural studies of the receptor. Currently identified synthetic GPR88 agonists, represented by 2-PCCA and 2-AMPP, have limited translational and clinical development potential because of various suboptimal drug-like properties, including high lipophilicity, suboptimal target selectivity, and poor metabolic stability. Further diligent and iterative efforts on hit-to-lead optimization will be needed to improve current leads for their drug-like properties, especially their in vivo drug metabolism and pharmacokinetics (DMPK) properties.44 Although RTI-13951–33 with a favorable PK profile is effective in a rat model of alcohol self-administration, further studies of both WT and Gpr88 KO mice will be imperative for validating agonist efficacy, on-target specificity, and if GPR88 is a druggable target for alcohol use disorders.
Because of the low sequence similarities with 5-HT1B (sequence identity = 18%) and other protein receptors with solved crystallized structures from PDB, the reliable 3D structure of GPR88 may not be easily generated by using the homology modeling technique. With the rapid development of structural biology and computational methods and tools used in computer-aided drug design (CADD), the precise GPR88 homology model/crystal structure might be generated or reported in the near future. This can provide new avenues to find novel orthosteric/allosteric chemotypes7,75 of GPR88 by using structure-based drug design (SBDD), such as a virtual screening strategy.76 Fragment-based drug design (FBDD)77–80 and ligand-based drug design23,81,82 may also each provide efficient strategies to identify more effective and selective GPR88 agonists and also antagonists with improved druglikeness by taking advantage of privileged fragments and newly identified synthetic ligands. Moreover, it is of great value for structural biologists to resolve the GPR88 structure and GPR88 receptor–ligand complexes using biophysical methods to pave the way for mechanistic studies and rational drug design. Additionally, with recent discoveries of orphan GPCR signaling pathways and physiological functions83 and novel screening approaches,13 deorphanization campaigns can be redesigned to better capture the native signaling of orphan receptors. Both deorphanization and continued probe development will accelerate understanding of GPR88 biological functions, facilitate evaluation of GPR88 as a drug target, and open new avenues for potential neurotherapeutics.
ACKNOWLEDGMENTS
We are grateful to Dr. Youyong Li in the Institute of Functional Nano & Soft Materials (FUNSOM) at Soochow University for providing softwares for amino acid sequence alignment.
Funding
This work was supported by the National Institutes of Health (Grants R01 DA038446 and P30 DA028821 to J.Z., T32 DA07287 and F31 DA045511 to E.A.W.), the Pharmaceutical Research and Manufacturers of America Foundation (PhRMA Research Starter Grant RSGPT18 to J.A.A.), the National Natural Science Foundation of China (81703330 to N.Y.), the Natural Science Foundation of Jiangsu Province (BK20170347 to N.Y.), and Jiangsu Key Laboratory of Neuropsychiatric Diseases (BM2013003 to N.Y.).
ABBREVIATIONS USED
- GPCRs
G protein-coupled receptors
- oGPCRs
orphan GPCRs
- HD
Huntington’s Disease
- MSNs
medium spiny neurons
- DOR
δ-opioid receptor
- PDB
RCSB Protein Data Bank
- SZ
schizophrenia
- PD
Parkinson’s Disease
- 6-OHDA 6
hydroxydopamine
- KD
knockdown
- KO
knockout
- AICS
apomorphine-induced climbing and stereotypy
- PPI disrupted
prepulse inhibition of startle response
- RGS4
G-protein signaling 4
- CeA
central nucleus
- AUD
alcohol use disorder
- BD
bipolar disorder
- SNP
single nucleotide polymorphism
- FLIPR
fluorescent imaging plate reader
- P-gp
P-glycoprotein
- PPB
plasma protein binding
- SAR
structure–activity relationship
- B/P
brain to plasma
- DMPK
drug metabolism and pharmacokinetics
- CADD
computer-aided drug design
- SBDD
structure-based drug design
- FBDD
fragment-based drug design
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Allen JA, and Roth BL (2011) Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 51, 117–144. [DOI] [PubMed] [Google Scholar]
- (2).Katritch V, Cherezov V, and Stevens RC (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci 33, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tang XL, Wang Y, Li DL, Luo J, and Liu MY (2012) Orphan G protein-coupled receptors (GPCRs): Biological functions and potential drug targets. Acta Pharmacol. Sin 33, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wacker D, Stevens RC, and Roth BL (2017) How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Rask-Andersen M, Almen MS, and Schioth HB (2011) Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov 10, 579–590. [DOI] [PubMed] [Google Scholar]
- (6).Wold EA, Chen J, Cunningham KA, and Zhou J (2018) Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem, 10.1021/acs.jmedchem.8b00875. [DOI] [PMC free article] [PubMed]
- (7).Lu S, and Zhang J Small Molecule Allosteric Modulators of G- Protein-Coupled Receptors: Drug-Target Interactions. (2018) J. Med. Chem, 10.1021/acs.jmed-chem.7b01844. [DOI] [PubMed]
- (8).Topiol S (2018) Current and Future Challenges in GPCR Drug Discovery. Methods Mol. Biol 1705, 1–21. [DOI] [PubMed] [Google Scholar]
- (9).Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, and Gloriam DE (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Filmore D (2004) It’s a GPCR world. Mod. Drug Discov 7 (24–26), 28. [Google Scholar]
- (11).Chung S, Funakoshi T, and Civelli O (2008) Orphan GPCR research. Br. J. Pharmacol 153 (S1), S339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fang Y, Kenakin T, and Liu C (2015) Editorial: Orphan GPCRs as emerging drug targets. Front. Pharmacol 6, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM, Sciaky N, and Roth BL (2015) PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Civelli O (2012) Orphan GPCRs and neuromodulation. Neuron 76, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Marder SR, Roth B, Sullivan PF, Scolnick EM, Nestler EJ, Geyer MA, Welnberger DR, Karayiorgou M, Guidotti A, Gingrich J, Akbarian S, Buchanan RW, Lieberman JA, Conn PJ, Haggarty SJ, Law AJ, Campbell B, Krystal JH, Moghaddam B, Saw A, Caron MG, George SR, Allen JA, and Solis M (2011) Advancing drug discovery for schizophrenia. Ann. N. Y. Acad. Sci 1236, 30–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Conn PJ, and Roth BL (2008) Opportunities and challenges of psychiatric drug discovery: roles for scientists in academic, industry, and government settings. Neuropsychopharmacol 33, 2048–2060. [DOI] [PubMed] [Google Scholar]
- (17).Massart R, Guilloux JP, Mignon V, Sokoloff P, and Diaz J (2009) Striatal GPR88 expression is confined to the whole projection neuron population and is regulated by dopaminergic and glutamatergic afferents. Eur. J. Neurosci 30, 397–414. [DOI] [PubMed] [Google Scholar]
- (18).Mizushima K, Miyamoto Y, Tsukahara F, Hirai M, Sakaki Y, and Ito T (2000) A Novel G-Protein-Coupled Receptor Gene Expressed in Striatum. Genomics 69, 314–321. [DOI] [PubMed] [Google Scholar]
- (19).Ghate A, Befort K, Becker JA, Filliol D, Bole-Feysot C, Demebele D, Jost B, Koch M, and Kieffer BL (2007) Identification of novel striatal genes by expression profiling in adult mouse brain. Neuroscience 146, 1182–1192. [DOI] [PubMed] [Google Scholar]
- (20).Befort K, Filliol D, Ghate A, Darcq E, Matifas A, Muller J, Lardenois A, Thibault C, Dembele D, Le Merrer J, Becker JA, Poch O, and Kieffer BL (2008) Mu-opioid receptor activation induces transcriptional plasticity in the central extended amygdala. Eur. J. Neurosci 27, 2973–2984. [DOI] [PubMed] [Google Scholar]
- (21).Waes VV, Tseng KY, and Steiner H (2011) GPR88: A putative signaling molecule predominantly expressed in the striatum: Cellular localization and developmental regulation. Basal Ganglia 1, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Massart R, Mignon V, Stanic J, Munoz-Tello P, Becker JA, Kieffer BL, Darmon M, Sokoloff P, and Diaz J (2016) Developmental and adult expression patterns of the G-protein-coupled receptor GPR88 in the rat: Establishment of a dual nuclear-cytoplasmic localization. J. Comp. Neurol 524, 2776–2802. [DOI] [PubMed] [Google Scholar]
- (23).Foti RS, Rock DA, Han X, Flowers RA, Wienkers LC, and Wahlstrom JL (2012) Ligand-based design of a potent and selective inhibitor of cytochrome P450 2C19. J. Med. Chem 55, 1205–1214. [DOI] [PubMed] [Google Scholar]
- (24).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bockaert J, and Pin JP (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J 18, 1723–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gentles AJ, and Karlin S (1999) Why are human G-protein-coupled receptors predominantly intronless? Trends Genet 15, 47–49. [DOI] [PubMed] [Google Scholar]
- (27).Quintana A, Sanz E, Wang W, Storey GP, Guler AD, Wanat MJ, Roller BA, La Torre A, Amieux PS, McKnight GS, Bamford NS, and Palmiter RD (2012) Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat. Neurosci 15, 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Logue SF, Grauer SM, Paulsen J, Graf R, Taylor N, Sung MA, Zhang L, Hughes Z, Pulito VL, Liu F, Rosenzweig-Lipson S, Brandon NJ, Marquis KL, Bates B, and Pausch M (2009) The orphan GPCR, GPR88, modulates function of the striatal dopamine system: a possible therapeutic target for psychiatric disorders? Mol. Cell. Neurosci 42, 438–447. [DOI] [PubMed] [Google Scholar]
- (29).Becker JA, Befort K, Blad C, Filliol D, Ghate A, Dembele D, Thibault C, Koch M, Muller J, Lardenois A, Poch O, and Kieffer BL (2008) Transcriptome analysis identifies genes with enriched expression in the mouse central extended amygdala. Neuroscience 156, 950–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bi Y, Dzierba CD, Bronson JJ, Fink C, Green M, Kimball D, Macor JE, Kwon S, Zhang Y, and Zipp G (2011) Modulators of G protein-Coupled Receptor 88 U.S. Patent 2011/0251204 A1.
- (31).Bi Y, Dzierba CD, Fink C, Garcia Y, Green M, Han J, Kwon S, Kumi G, Liang Z, Liu Y, Qiao Y, Zhang Y, Zipp G, Burford N, Ferrante M, Bertekap R, Lewis M, Cacace A, Westphal RS, Kimball D, Bronson JJ, and Macor JE (2015) The discovery of potent agonists for GPR88, an orphan GPCR, for the potential treatment of CNS disorders. Bioorg. Med. Chem. Lett 25, 1443–1447. [DOI] [PubMed] [Google Scholar]
- (32).Meirsman AC, Le Merrer J, Pellissier LP, Diaz J, Clesse D, Kieffer BL, and Becker JA (2016) Mice lacking GPR88 show motor deficit, improved spatial learning, and low anxiety reversed by delta opioid antagonist. Biol. Psychiatry 79, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Brandish PE, Su M, Holder DJ, Hodor P, Szumiloski J, Kleinhanz RR, Forbes JE, McWhorter ME, Duenwald SJ, Parrish ML, Na S, Liu Y, Phillips RL, Renger JJ, Sankaranarayanan S, Simon AJ, and Scolnick EM (2005) Regulation of gene expression by lithium and depletion of inositol in slices of adult rat cortex. Neuron 45, 861–872. [DOI] [PubMed] [Google Scholar]
- (34).Ogden CA, Rich ME, Schork NJ, Paulus MP, Geyer MA, Lohr JB, Kuczenski R, and Niculescu AB (2004) Candidate genes, pathways and mechanisms for bipolar (manic-depressive) and related disorders: an expanded convergent functional genomics approach. Mol. Psychiatry 9, 1007–1029. [DOI] [PubMed] [Google Scholar]
- (35).Le-Niculescu H, Balaraman Y, Patel S, Tan J, Sidhu K, Jerome RE, Edenberg HJ, Kuczenski R, Geyer MA, Nurnberger JI Jr., Faraone SV, Tsuang MT, and Niculescu AB (2007) Towards understanding the schizophrenia code: an expanded convergent functional genomics approach. Am. J. Med. Genet., Part B 144B, 129–158. [DOI] [PubMed] [Google Scholar]
- (36).Conti B, Maier R, Barr AM, Morale MC, Lu X, Sanna PP, Bilbe G, Hoyer D, and Bartfai T (2007) Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol. Psychiatry 12, 167–189. [DOI] [PubMed] [Google Scholar]
- (37).Ben Hamida S, Mendonca-Netto S, Arefin TM, Nasseef MT, Boulos LJ, McNicholas M, Ehrlich AT, Clarke E, Moquin L, Gratton A, Darcq E, Harsan LA, Maldonado R, and Kieffer BL (2018) Increased alcohol seeking in mice lacking Gpr88 involves dysfunctional mesocorticolimbic networks. Biol. Psychiatry 84, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rainwater A, Sanz E, Palmiter RD, and Quintana A (2017) Striatal GPR88 Modulates Foraging Efficiency. J. Neurosci 37, 7939–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lobo MK (2009) Molecular profiling of striatonigral and striatopallidal medium spiny neurons past, present, and future. Int. Rev. Neurobiol 89, 1–35. [DOI] [PubMed] [Google Scholar]
- (40).Sesack SR, and Grace AA (2010) Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacol 35, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Stone JM, Morrison PD, and Pilowsky LS (2007) Glutamate and dopamine dysregulation in schizophrenia–a synthesis and selective review. J. Psychopharmacol 21, 440–452. [DOI] [PubMed] [Google Scholar]
- (42).Ye N, Neumeyer JL, Baldessarini RJ, Zhen X, and Zhang A (2013) Update 1 of: Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev 113, PR123–178. [DOI] [PubMed] [Google Scholar]
- (43).Zhang H, Ye N, Zhou S, Guo L, Zheng L, Liu Z, Gao B, Zhen X, and Zhang A (2011) Identification of N-propylnoraporphin- 11-yl 5-(1,2-dithiolan-3-yl)pentanoate as a new anti-Parkinson’s agent possessing a dopamine D2 and serotonin 5-HT1A dual-agonist profile. J. Med. Chem 54, 4324–4338. [DOI] [PubMed] [Google Scholar]
- (44).Ye N, Song Z, and Zhang A (2013) Dual ligands targeting dopamine D2 and serotonin 5-HT1A receptors as new antipsychotical or anti-Parkinsonian agents. Curr. Med. Chem 21, 437–457. [DOI] [PubMed] [Google Scholar]
- (45).Zhao R, Lu W, Fang X, Guo L, Yang Z, Ye N, Zhao J, Liu Z, Jia J, Zheng L, Zhao B, Zhang A, and Zhen X (2014) (6aR)-11- amino-N-propyl-noraporphine, a new dopamine D2 and serotonin 5- HT1A dual agonist, elicits potent antiparkinsonian action and attenuates levodopa-induced dyskinesia in a 6-OHDA-lesioned rat model of Parkinson’s disease. Pharmacol., Biochem. Behav 124, 204–210. [DOI] [PubMed] [Google Scholar]
- (46).Ye N, Wu Q, Zhu L, Zheng L, Gao B, Zhen X, and Zhang A (2011) Further SAR study on 11-O-substituted aporphine analogues: identification of highly potent dopamine D3 receptor ligands. Bioorg. Med. Chem 19, 1999–2008. [DOI] [PubMed] [Google Scholar]
- (47).Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, and Greengard P (2004) DARPP-32: an integrator of neuro- transmission. Annu. Rev. Pharmacol. Toxicol 44, 269–296. [DOI] [PubMed] [Google Scholar]
- (48).Walaas SI, Hemmings HC Jr., Greengard P, and Nairn A (2011) Beyond the dopamine receptor: regulation and roles of serine/threonine protein phosphatases. Front. Neuroanat 5, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Alkufri F, Shaag A, Abu-Libdeh B, and Elpeleg O (2016) Deleterious mutation in GPR88 is associated with chorea, speech delay, and learning disabilities. Neurol. Genet 2, No. e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Rocher AB, Gubellini P, Merienne N, Boussicault L, Petit F, Gipchtein P, Jan C, Hantraye P, Brouillet E, and Bonvento G (2016) Synaptic scaling up in medium spiny neurons of aged BACHD mice: A slow-progression model of Huntington’s disease. Neurobiol. Dis 86, 131–139. [DOI] [PubMed] [Google Scholar]
- (51).Del Zompo M, Deleuze JF, Chillotti C, Cousin E, Niehaus D, Ebstein RP, Ardau R, Mace S, Warnich L, Mujahed M, Severino G, Dib C, Jordaan E, Murad I, Soubigou S, Koen L, Bannoura I, Rocher C, Laurent C, Derock M, Faucon Biguet N, Mallet J, and Meloni R (2014) Association study in three different populations between the GPR88 gene and major psychoses. Mol. Genet. Genomic Med 2, 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Del Zompo M, Severino G, Ardau R, Chillotti C, Piccardi M, Dib C, Muzard G, Soubigou S, Derock M, Fournel R, Vaubien Y, Roche S, Bowen-Squires L, Genin E, Cousin E, Deleuze JF, Biguet NF, Mallet J, and Meloni R (2010) Genome-scan for bipolar disorder with sib-pair families in the Sardinian population: a new susceptibility locus on chromosome 1p22-p21? Am. J. Med. Genet., Part B 153B, 1200–1208. [DOI] [PubMed] [Google Scholar]
- (53).Ingallinesi M, Le Bouil L, Biguet NF, Thi AD, Mannoury la Cour C, Millan MJ, Ravassard P, Mallet J, and Meloni R (2015) Local inactivation of Gpr88 in the nucleus accumbens attenuates behavioral deficits elicited by the neonatal administration of phencyclidine in rats. Mol. Psychiatry 20, 951–958. [DOI] [PubMed] [Google Scholar]
- (54).Aupperle RL, and Paulus MP (2010) Neural systems underlying approach and avoidance in anxiety disorders. Dialogues Clin. Neurosci 12, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).de la Mora MP, Gallegos-Cari A, Arizmendi-Garcia Y, Marcellino D, and Fuxe K (2010) Role of dopamine receptor mechanisms in the amygdaloid modulation of fear and anxiety: Structural and functional analysis. Prog. Neurobiol 90, 198–216. [DOI] [PubMed] [Google Scholar]
- (56).Hebb AL, Robertson HA, and Denovan-Wright EM (2008) Phosphodiesterase 10A inhibition is associated with locomotor and cognitive deficits and increased anxiety in mice. Eur. Neuro-psychopharmacol 18, 339–363. [DOI] [PubMed] [Google Scholar]
- (57).Luuk H, Plaas M, Raud S, Innos J, Sutt S, Lasner H, Abramov U, Kurrikoff K, Koks S, and Vasar E (2009) Wfs1-deficient mice display impaired behavioural adaptation in stressful environment. Behav. Brain Res 198, 334–345. [DOI] [PubMed] [Google Scholar]
- (58).Smith KS, and Rudolph U (2012) Anxiety and depression: mouse genetics and pharmacological approaches to the role of GABA(A) receptor subtypes. Neuropharmacol 62, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Meirsman AC, Robe A, de Kerchove d’Exaerde A, and Kieffer BL (2016) GPR88 in A2AR neurons enhances anxiety-like behaviors. eNeuro 3.3 10.1523/ENEURO.0202-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Bohm C, Newrzella D, Herberger S, Schramm N, Eisenhardt G, Schenk V, Sonntag-Buck V, and Sorgenfrei O (2006) Effects of antidepressant treatment on gene expression profile in mouse brain: cell type-specific transcription profiling using laser microdissection and microarray analysis. J. Neurochem 97, 44–49. [DOI] [PubMed] [Google Scholar]
- (61).Jin C, Decker AM, Makhijani VH, Besheer J, Darcq E, Kieffer BL, and Maitra R (2018) Discovery of a potent, selective, and brain-penetrant small molecule that activates the orphan receptor GPR88 and reduces alcohol intake. J. Med. Chem 61, 6748–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jin C, Decker AM, Huang XP, Gilmour BP, Blough BE, Roth BL, Hu Y, Gill JB, and Zhang XP (2014) Synthesis, pharmacological characterization, and structure-activity relationship studies of small molecular agonists for the orphan GPR88 receptor. ACS Chem. Neurosci 5, 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li JX, Thorn DA, and Jin C (2013) The GPR88 receptor agonist 2-PCCA does not alter the behavioral effects of methamphetamine in rats. Eur. J. Pharmacol 698, 272–277. [DOI] [PubMed] [Google Scholar]
- (64).Decker AM, Gay EA, Mathews KM, Rosa TC, Langston TL, Maitra R, and Jin C (2017) Development and validation of a high-throughput calcium mobilization assay for the orphan receptor GPR88. J. Biomed. Sci 24, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Jin C, Decker AM, Harris DL, and Blough BE (2016) Effect of substitution on the aniline moiety of the GPR88 agonist 2- PCCA: Synthesis, structure-activity relationships, and molecular modeling studies. ACS Chem. Neurosci 7, 1418–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, and Jeffrey P (2007) Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J. Pharmacol. Exp. Ther 322, 205–213. [DOI] [PubMed] [Google Scholar]
- (67).Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, and Mallamo JP (2012) Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci 3, 50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Clark DE, and Pickett SD (2000) Computational methods for the prediction of ‘drug-likeness’. Drug Discov. Today 5, 49–58. [DOI] [PubMed] [Google Scholar]
- (69).Dzierba CD, Bi Y, Dasgupta B, Hartz RA, Ahuja V, Cianchetta G, Kumi G, Dong L, Aleem S, Fink C, Garcia Y, Green M, Han J, Kwon S, Qiao Y, Wang J, Zhang Y, Liu Y, Zipp G, Liang Z, Burford N, Ferrante M, Bertekap R, Lewis M, Cacace A, Grace J, Wilson A, Nouraldeen A, Westphal R, Kimball D, Carson K, Bronson JJ, and Macor JE (2015) Design, synthesis, and evaluation of phenylglycinols and phenyl amines as agonists of GPR88. Bioorg. Med. Chem. Lett 25, 1448–1452. [DOI] [PubMed] [Google Scholar]
- (70).Jin C, Decker AM, and Langston TL (2017) Design, synthesis and pharmacological evaluation of 4-hydroxyphenylglycine and 4-hydroxyphenylglycinol derivatives as GPR88 agonists. Bioorg. Med. Chem 25, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Civelli O, Reinscheid RK, Zhang Y, Wang Z, Fredriksson R, and Schioth HB (2013) G protein-coupled receptor deorphanizations. Annu. Rev. Pharmacol. Toxicol 53, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Lovinger DM (2012) New twist on orphan receptor GPR88 function. Nat. Neurosci 15, 1469–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Levoye A, Dam J, Ayoub MA, Guillaume JL, and Jockers R (2006) Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep 7, 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Xiang J, Chun E, Liu C, Jing L, Al-Sahouri Z, Zhu L, and Liu W (2016) Successful Strategies to Determine High-Resolution Structures of GPCRs. Trends Pharmacol. Sci 37, 1055–1069. [DOI] [PubMed] [Google Scholar]
- (75).Huang W, Lu S, Huang Z, Liu X, Mou L, Luo Y, Zhao Y, Liu Y, Chen Z, Hou T, and Zhang J (2013) Allosite: a method for predicting allosteric sites. Bioinformatics 29, 2357–2359. [DOI] [PubMed] [Google Scholar]
- (76).Hou T, and Xu X (2004) Recent development and application of virtual screening in drug discovery: an overview. Curr. Pharm. Des 10, 1011–1033. [DOI] [PubMed] [Google Scholar]
- (77).Rees DC, Congreve M, Murray CW, and Carr R (2004) Fragment-based lead discovery. Nat. Rev. Drug Discov 3, 660–672. [DOI] [PubMed] [Google Scholar]
- (78).Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, Liu H, Shen Q, and Zhou J (2013) Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem 62, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Chen H, Wang CZ, Ding C, Wild C, Copits B, Swanson GT, Johnson KM, and Zhou J (2013) A combined bioinformatics and chemoinformatics approach for developing asymmetric bivalent AMPA receptor positive allosteric modulators as neuroprotective agents. ChemMedChem 8, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Chen H, Zhou X, Wang A, Zheng Y, Gao Y, and Zhou J (2015) Evolutions in fragment-based drug design: the deconstruction- reconstruction approach. Drug Discov. Today 20, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Liu J, Pham PT, Skripnikova EV, Zheng S, Lovings LJ, Wang Y, Goyal N, Bellow SM, Mensah LM, Chatters AJ, Bratton MR, Wiese TE, Zhao M, Wang G, and Foroozesh M (2015) A ligand-based drug design. Discovery of 4-trifluoromethyl-7,8- pyranocoumarin as a selective inhibitor of human cytochrome P450 1A2. J. Med. Chem 58, 6481–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Lee CH, Huang HC, and Juan HF (2011) Reviewing ligand-based rational drug design: the search for an ATP synthase inhibitor. Int. J. Mol. Sci 12, 5304–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy S, Saddoris KA, Nikolova VD, Farrell MS, Wang S, Mangano J, Deshpande DA, Jiang A, Penn RB, Jin J, Koller BH, Kenakin T, Shoichet BK, and Roth BL (2015) Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]