

Abstract
Vaccines and antivirals to combat dengue, Zika, and other flavivirus pathogens present a major, unmet medical need. Vaccine development has been severely challenged by the antigenic diversity of these viruses and the propensity of non-neutralizing, cross-reactive antibodies to facilitate cellular infection and increase disease severity. As an alternative, direct-acting antivirals targeting the flavivirus envelope protein, E, have the potential to act via an analogous mode of action without the risk of antibody-dependent enhancement of infection and disease. We previously discovered that structurally diverse small molecule inhibitors of the dengue virus E protein exhibit varying levels of antiviral activity against other flaviviruses in cell culture. Here we demonstrate that the broad-spectrum activity of several cyanohydrazones against dengue, Zika, and Japanese encephalitis viruses is due to specific inhibition of E-mediated membrane fusion during viral entry and provide proof of concept for pharmacological inhibition of E as an antiviral strategy in vivo.
Keywords: flavivirus, antiviral, envelope protein inhibitor, viral entry inhibitor, fusion inhibitor, dengue virus
Graphical Abstract
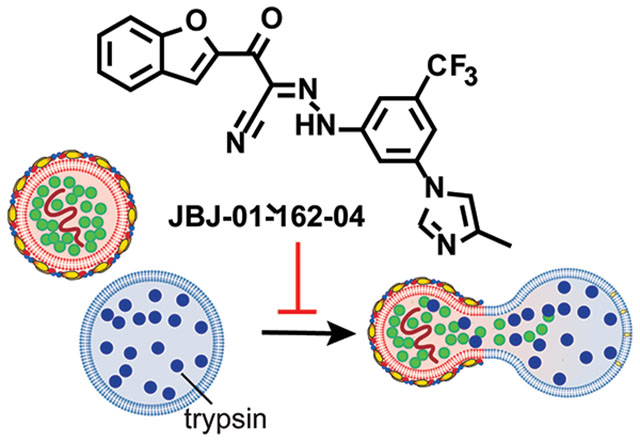
Dengue virus (DENV), Zika virus, and other flaviviruses (e.g., West Nile, St. Louis encephalitis, etc.) are significant human pathogens against which we lack effective countermeasures. Over 390 million DENV infections occur annually resulting in approximately 500,000 hospitalizations each year due to severe dengue and an estimated 2.5% fatality rate for those with severe dengue1. As evidenced during the explosive spread of Zika virus in the Western Hemisphere earlier this decade, introduction of pathogenic flaviviruses into geographic locations where prior immunity does not exist can have devastating public health consequences. Vaccine development has been challenging due to the diversity of the four DENV serotypes (DENV1-4) and the propensity of non-neutralizing antibodies to enhance viral infection2 and disease severity3. The only marketed DENV vaccine, Dengvaxia, is effective in boosting natural immunity in those who have been previously infected but significantly increases the risk of severe disease for immune-naïve vacinees who are later infected4–9. As an alternative countermeasure, we have pursued small molecules that bind to the viral envelope protein, E, on the surface of the virion and prevent viral entry. Such agents have the potential to block infection by engaging their target, E, extracellularly in a way analogous to antibodies but without the risk of antibody-dependent enhancement of infection and disease.
E exists on the surface of mature dengue virions as ninety prefusion dimers and performs multiple functions in viral entry. Entry is initiated upon attachment of the virion to the host cell through interaction of E’s domain III with any of a number of different attachment factors on the plasma surface (e.g., DC-SIGN, Tim/Tam phosphatidylserine receptors)10–12. This leads to internalization of the virion via a clathrin-dependent process13–15. Acidification of the endosomal compartment triggers fusion of the viral and endosomal membranes driven by reorganization and refolding of E as a postfusion trimeric species16–18. This critical event creates a fusion pore that enables the viral genome to escape into the cytoplasm where it can be expressed and where viral replication can ensue.
We previously used both phenotypic and target-based approaches to discover diverse small molecules that bind to the DENV prefusion E dimer (E2) on the virion surface and block infection by preventing E-mediated membrane fusion19–21. Both photocrosslinking and loss-of-binding site-directed mutagenesis experiments performed with recombinant, soluble E indicate that these compounds target a pocket between domains I and II, and sequence alignments suggest that this site is at least partially conserved across other flaviviruses22. Consistent with this idea, we previously found that representative compounds from multiple chemical series of DENV E inhibitors have variable levels of activity against West Nile, Japanese encephalitis, and/or Zika viruses (WNV, JEV, ZIKV, respectively) in cell culture experiments monitoring viral infectivity21–22; however, the biochemical mechanism of this broad-spectrum activity was uncharacterized. Interestingly, while multiple compounds from two pyrimidine series were observed to inhibit WNV, JEV, and ZIKV, they appear more selective for DENV over these other flaviviruses. In contrast, cyanohydrazone 3-110-22 uniquely exhibits comparable inhibition of DENV, WNV, JEV, and ZIKV22. A potential explanation for 3-110-22’s unusual breadth of antiviral activity is that non-specific, E-independent mechanisms, including colloidal compound aggregation or other pan-assay interference (PAINS) properties, contribute significantly to its antiviral mechanism. Alternatively, 3-110-22 and related cyanohydrazones might truly inhibit multiple flaviviruses by making conserved interactions with the same pocket of DENV, WNV, JEV, and ZIKV E proteins. Distinguishing between these possibilities is critical for establishing whether specific pharmacological inhibition of multiple flavivirus E proteins is possible.
Countermeasures against DENV, ZIKV, and other flavivirus pathogens represent a major unmet medical need. Agents with broad-spectrum activity against multiple flavivirus species are of particular interest due to the potential for the emergence or reemergence of new flavivirus species and the limited resources currently available for flavivirus drug development efforts. Towards this goal, here, we demonstrate that 3-110-22 and other related cyanohydrazones inhibit multiple flaviviruses by specific inhibition of E-mediated membrane fusion. While some cyanohydrazones exhibit activity in PAINS assays, we show that this activity is separable from the ability to inhibit multiple flaviviruses. We further report development of a cyanohydrazone, JBJ-01-162-04, with improved in vivo properties and use of this compound to establish proof-of-concept for this new class of antivirals in a murine model of DENV infection.
RESULTS AND DISCUSSION
The cyanohydrazone class of DENV E inhibitors have activity against multiple flaviviruses
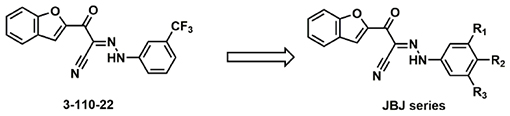
DENV E inhibitor 3-110-22 was developed through a medicinal chemistry effort guided by a cell-based assay (“viral infectivity assay”; Fig. 1A) in which single-cycle viral yield was quantified as a readout of productive viral entry. Analogous virological assays were used to demonstrate 3-110-22’s activity against WNV, JEV, and ZIKV22. To ensure that the antiviral effects we observed is due to inhibition of E’s function in viral entry and not due to other on- or off-target effects, we limit inhibitor treatment to pre-incubation with the viral inoculum and the first hour of cellular infection, then wash the cells to remove extracellular virus and inhibitor prior to the addition of fresh medium. At twenty-four hours post-infection, corresponding to a single replication cycle, supernatants are collected and the yield of infectious virions is quantified as a metric of productive viral infection. To more broadly examine the spectrum of antiviral activity of the parent cyanohydrazone scaffold, we selected a subset of compounds from our prior study19, changing either the hydrophobic head group (3-110-series) or maintaining the furan head group and changing the substituent on the benzene (3-149-series) (Fig. 1B). We quantified activity of these cyanohydrazones against DENV2, ZIKV, and JEV in the viral infectivity assay (Fig. 1A) to determine antiviral IC90 values – defined as the inhibitor concentration required to reduce the single cycle viral yield by 90% relative to the DMSO-treated control in this assay – for each virus-inhibitor pair.
Figure 1. The cyanohydrazones inhibit the entry of multiple flaviviruses.

A.The viral infectivity assay was designed to ensure that the antiviral activity observed is due to an effect on viral entry rather than to other on- or off-target effects. Compound treatment is limited to a preincubation with the inoculum and the initial one-hour infection, after which non-adsorbed virus and compound are washed away. After twenty-four hours, which corresponds to a single-cycle of infection, the yield of infectious virus secreted to the culture supernatant is quantified as a measure of productive viral entry by plaque-forming assay (PFA) or focus-forming assay (FFA) as indicated for each experiment. BHK-21 cells were used for DENV and JEV; Vero cells were used for ZIKV.
B. Summary of antiviral IC90 values determined against dengue virus 2 (DENV2), Zika virus (ZIKV), and Japanese encephalitis virus (JEV) in the viral infectivity assay. Viral titers were determined by focus-forming assay for DENV2 and plaque-forming assays for ZIKV and JEV. The IC90 value is defined as the concentration of inhibitor required to reduce single-cycle viral yield in the infectivity assay by 10-fold. Values were determined by non-linear regression (as shown in Figs. 1C and S1) and are presented as means ± standard deviation of n ≥ 2 independent experiments.
C. IC90 determination for 3-110-2 and 3-110-22 against DENV2, ZIKV, and JEV by non-linear regression. Viral yields were normalized to the DMSO negative control. Representative data from one of n ≥ 2 independent experiments are presented, and the mean ± standard deviation of n = 2 biological replicates is presented on the graph. Representative data for 3-149-3 and 3-149-15 are provided in Fig. S1.
D. Cyanohydrazones inhibit flavivirus entry by preventing E-mediated membrane fusion. Fusion of virions with synthetic liposomes encapsulating trypsin is triggered by low pH. Formation of a fusion pore allows trypsin (blue dots) to access and digest core protein (C, green dots) on the interior of the virion while envelope protein (E, red ovals) on the exterior of the virion remains intact and serves as an internal control. The reactions depicted in Fig. 1D were performed with DENV2, ZIKV, and JEV.
E. Western blot analysis for C and E shows that compound 3-110-22 protects core from digestion upon exposure of the virion-liposome mixture to acidic pH, indicating concentration-dependent inhibition of viral fusion for all viruses tested. Note that the target:inhibitor ratio in this assay is > 10-fold that in the viral infectivity assay. Representative data are shown for n ≥ 2 independent experiments.
All four cyanohydrazones exhibit significant antiviral activity against DENV2, ZIKV, and JEV, albeit with varying potencies and selectivities (Figs. 1B, 1C, and S1). Consistent with our previous report22, 3-110-22 inhibits the infectivity of all three flaviviruses at concentrations below 10 μM (DENV2 IC90 2.3 μM, ZIKV IC90 4.2 μM, JEV IC90 8.6 μM) with a modest selectivity for DENV > ZIKV > JEV. 3-110-2 exhibits potent activity against both DENV2 (IC90 1.0 μM) and ZIKV (IC90 4.8 μM) but has significantly less activity against JEV (IC90 > 20 μM). Compounds 3-149-3 and 3-149-15 both inhibit DENV2 infectivity with IC90 values of 15.6 and 11.8 μM, respectively, but whereas 3-149-3 exhibits more narrow-spectrum activity against DENV2 and JEV over ZIKV, 3-149-15 exhibits comparable inhibition of DENV2 and JEV infectivity while not inhibiting ZIKV. Notably, all of the compounds exhibit equipotent inhibition of at least two viruses.
To test whether these compounds inhibit DENV, ZIKV, and JEV by preventing E-mediated membrane fusion, we examined their effects on fusion in a liposome-based in vitro assay. For these experiments, we chose 3-110-22 since it has the highest antiviral potency across all three viruses. Briefly, we prepared liposomes in which we encapsulated trypsin, and then mixed these liposomes with virions in the presence and absence of inhibitor at both neutral and acidic pH. E-mediated fusion of the viral and liposome membranes at acidic pH creates a fusion pore that renders the viral core protein accessible to the protease; absence of fusion due to neutral pH or small molecule inhibition of E results in protection of core from trypsin digestion (Fig. 1D). E on the external face of the virion lipid bilayer is never accessible to the protease and serves as an internal control for liposome integrity. We monitored the levels of core and E proteins by immunoblotting and showed that 3-110-22 blocks fusion of all three viruses in a concentration-dependent manner (Fig. 1E). Together, these results indicate the antiviral activity of 3-110-22 and other cyanohydrazone inhibitors against DENV, ZIKV and JEV is due to a shared mechanism of action that targets E-mediated membrane fusion.
PAINS properties do not significantly contribute to the activity of the cyanohydrazones
Pan-assay interference compounds (PAINS) are a major source of artifacts in small molecule screening. A major source of PAINS activity is the formation of colloidal aggregates between 50-1000 nm in radius that can partially denature proteins upon absorption, leading to promiscuous inhibitory activity23–25. Although the range of viral activities across the 3-110- and 3-149-compounds suggests that specific interactions with each E protein are responsible for the observed antiviral activity and argue against non-specific, target-independent mechanisms of inhibition, we previously observed that PAINS properties can contribute significantly to antiviral activity in a high-throughput screen for inhibitors of DENV2 E21 (Lian, 2018). We therefore sought to assess the cyanohydrazone inhibitors for PAINS properties explicitly. To do this, we first tested their activity against the established counter-screening enzyme malate dehydrogenase (MDH)23–24. We also evaluated compound promiscuity by counter-screening against vesicular stomatitis virus (VSV), a rhabdovirus with a structurally unrelated envelope glycoprotein and divergent mechanism of viral entry26–28.
We observed some inhibition of VSV by the cyanohydrazones at relatively high concentrations (≥ 10 μM for the 3-110 series, ≥ 25 μM for the 3-149 series) that were for the most part significantly higher than the concentrations at which we observe activity against flaviviruses (Figs. 2A, 2B, and S2A). In examining potential PAINS activity in the enzymatic assay, we observed non-specific inhibition of MDH activity by 3-110-22 and 3-110-2 with IC50 values of 47 ± 11 μM and 8 ± 4 μM, respectively (Figs. 2A, 2C, and S2B) that was abolished in the presence of detergent (IC50 >100 μM). In contrast, 3-149-3 and 3-149-15 exhibit little to no detergent-sensitive activity against MDH (IC50 70 ± 26 μM and >100 μM, respectively). While these data indicate that PAINS properties may contribute to the antiviral activity of the 3-110-series, the significantly less potent inhibition of VSV by both 3-149- and 3-110-series and the absence of promiscuous activity for the 3-149-series in the MDH assay demonstrate that the activity of the cyanohydrazones against multiple flaviviruses is independent of compound aggregation and nonspecific interactions.
Figure 2. The activity of the cyanohydrazones against multiple flaviviruses is specific.

A. Summary of assays used to evaluate non-specific activities of cyanohydrazones. Non-specific activity of the cyanohydrazones was assessed using a VSV-eGFP infectivity assay in which inhibitor treatment was limited as for the infectivity assay depicted in Figure 1A but using eGFP fluorescence at 6 hours as a readout of infection. VSV-eGFP fluorescence for each sample was normalized to the DMSO negative control and then subtracted from 100 to calculate percent VSV inhibition. Data are presented as means ± standard deviation from n = 2 independent experiments. IC50 values for inhibition of malate dehydrogenase (MDH) in the presence or absence of Triton X-100 were determined by non-linear regression (see Fig. 2C and S2) and are presented as means ± standard deviation of n = 2 independent experiments.
B. Inhibition of VSV-eGFP by 3-110-22 and 3-149-15. eGFP fluorescence at 6 hours postinfection was measured and plotted as a percentage normalized to the DMSO negative control. Assays were performed using BHK-21 cells. Representative data from one of n = 2 independent experiments are presented as means normalized to DMSO ± standard deviation of n = 2 replicates within the experiment. Graphs showing representative data for 3-110-2 and 3-149-3 are provided in Fig. S2.
C. Inhibition of MDH by 3-110-22 and 3-149-15. MDH activity measured at various cyanohydrazone concentrations was normalized to the activity of the DMSO negative control to derive the percent enzyme activity. Representative data from one of n = 2 independent experiments are presented as means normalized to DMSO ± standard deviation of n = 2 experimental replicates. Graphs showing representative data for 3-110-2 and 3-149-3 are provided in Fig. S2.
Medicinal chemistry optimization to improve in vivo properties.
Towards the goal of evaluating the efficacy of cyanohydrazone inhibitors of flavivirus E proteins in vivo, we performed preliminary studies with 3-110-22 and observed that this compound causes acute toxicity in mice (data not shown) and exhibits poor stability in the presence of mouse microsomes in vitro, with half-life (T1/2) of 13.1 minutes. To improve the metabolic stability and toxicity profile of 3-110-22, several functional groups (small halogen, cyclic aliphatic amines, morpholine and imidazole) were individually added to the trifluoromethyl-benzene ring to generate a series of JBJ analogs (Table 1). Analogs JBJ-01-162-02, -03, and -04 exhibit significantly improved half-lives of 73.0, >120, and 64.8 minutes, respectively.
Table 1. Medicinal chemistry optimization of JBJ series.
Compound stability in mouse microsomes was assessed in vitro and is reported as the half-life of the small molecule (T1/2). Antiviral activity was measured in the viral infectivity assay as depicted in Fig. 1A, limiting inhibitor treatment to preincubation with the viral inoculum and the initial one hour infection and using single-cycle viral yield as a readout for productive viral entry. Virus titers were determined by plaque-forming assay. The antiviral IC90 value refers to the concentration of inhibitor required to reduce single-cycle DENV2 viral yield by ten-fold. The CC50 value corresponds to the concentration of inhibitor causing 50% loss of BHK-21 cell viability. IC90 (Figs. 3 and S3) and CC50 values were determined by non-linear regression analysis of the data and are presented as means ± standard error of n = 2 independent experiments. Since our primary objective was to improve the microsome stability, we initially screened for T1/2 and advanced compounds with improved T1/2 to antiviral and cytotoxicity testing. “n.d.” indicates value not determined.
 | ||||||
|---|---|---|---|---|---|---|
| ID | R1 | R2 | R3 | T1/2 (min) | IC90 (μM) [DENV2 NGC) |
CC5 (μM) |
| 3-110-22 | CF3 | H | H | 13.1 | 2.5 ± 0.1 | 37.5 ± 5.6 |
| JBJ-01-162-01 | CF3 | H | 35.1 | n.d. | n.d. | |
| JBJ-01-162-02 | CF3 | H | 73.0 | 9.0 ± 2.8 | 24.5 ± 7.2 | |
| JBJ-01-162-03 | H | 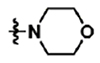 |
CF3 | > 120 | 1.6 ± 0.7 | 32.5 ± 2.2 |
| JBJ-01-162-04 | CF3 | H | 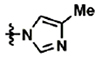 |
64.8 | 1.5 ± 0.4 | 49.1 ± 2.6 |
| JBJ-01-162-05 | CF3 | H | 29.9 | n.d. | n.d. | |
| JBJ-03-019-01 | CF3 | F | H | 7.6 | n.d. | n.d. |
| JBJ-03-019-02 | CF3 | H | F | 8.6 | n.d. | n.d. |
We verified that both JBJ-01-162-03 and -04 retain 3-110-22’s activity against DENV in the infectivity assay (Fig. 1A) (IC90 values 1.6 ± 0.6 μM and 1.5 ± 0.4 μM versus 2.5 ± 0.1 μM, respectively; Table 1 and Figs. 3A and S3A) and found that JBJ-01-162-04 exhibits reduced cellular cytotoxicity compared to the parental compound (Table 1 and Fig. S3B). For JBJ-01-162-03 and -04, CC50 values are 20-fold or more greater than antiviral IC90 values, indicating that the antiviral activity of these compounds is not due to general cytotoxicity. Additionally, we tested the JBJ series of compounds for PAINS properties and observed that whereas both JBJ-01-162-02 and -03 exhibit detergent-sensitive inhibition of malate dehydrogenase comparable to that of 3-110-22 (Fig. S3B), JBJ-01-162-04 lacks this non-specific activity (Fig. 3B).
Figure 3. JBJ-01-162-04 is a specific inhibitor of E-mediated DENV fusion.

A. Antiviral activity of JBJ-01-162-04. The IC90 value of JBJ-01-162-04 against DENV2 was determined by nonlinear regression analysis of data generated using the viral infectivity assay depicted in Fig. 1A. Assays were performed using BHK-21 cells. JBJ-01-162-04 exhibits an IC90 value against DENV2 of 1.5 ± 0.4 μM. Representative data from one of n = 2 independent experiments are shown with error bars indicating the standard deviation for 2 replicates within that experiment.
B. Malate dehydrogenase activity assay to detect PAINS activity. The effect of the compounds on malate dehydrogenase (MDH) activity was assessed in the presence and absence of Triton X-100. Slight inhibition of MDH by JBJ-01-162-04 was observed; however, this effect was independent of detergent and thus does not appear to reflect nonspecific inhibition due to colloid formation by the compound. Representative data from one of n ≥ 2 independent experiments are shown.
C. Determination of dissociation constant (KD) value for JBJ-01-162-04 bound to soluble, prefusion DENV2 envelope protein (sE2) by biolayer interferometry (BLI). The equilibrium dissociation constant (KD) for JBJ-01-162-04’s interaction with the DENV2 soluble, prefusion E dimer (sE2) is 1.4 ± 0.5 μM (average of 3 independent experiments). Representative data from one of three independent experiments are shown.
D. JBJ-01-162-04 inhibits E-mediated fusion in vitro. The liposome fusion assay depicted in Fig. 1D was performed. JBJ-01-162-04 inhibits E-mediated fusion of DENV2 in a concentration-dependent manner, as evidenced by protection of core from digestion at pH 5.3. Representative data from one of n ≥ 2 independent experiments are shown. Note that the target:inhibitor ratio in this assay is ~10-fold higher than in the infectivity assay.
Based on its superior stability, toxicity profile, lack of PAINS activity, and antiviral potency, we selected JBJ-01-162-04 for additional characterization prior to in vivo experiments. We measured an equilibrium dissociation constant (KD) for its interaction with the recombinant, soluble prefusion DENV2 E protein (sE2) by biolayer interferometry, observing a KD of 1.4 ± 0.5 μM (Fig. 3C), consistent with a direct interaction with its intended target. We also confirmed that JBJ-01-162-04 inhibits viral fusion in the liposome-based assay in a concentration-dependent manner (Fig. 3D). Together, these efforts enabled advancement of JBJ-01-162-04 as a suitable candidate to assess antiviral efficacy in a murine model.
JBJ-01-162-04 reduces DENV viral burden in vivo.
To evaluate JBJ-01-162-04’s antiviral efficacy in vivo, we utilized the AG129-DENV2 S221 model. Mouse-adapted strain DENV2 S221 strain was cloned following alternate passaging of DENV2 strain PL046 in mosquito cells and mice29–30. Infection of mice lacking interferon receptors (e.g., AG129) leads to productive viral replication and dissemination29, 31–32. The model is well-established for evaluating inhibition of DENV by small molecules and siRNAs30, 33. We first verified that JBJ-01-162-04 potently inhibits DENV2 strain S221, observing an antiviral IC90 of 1.4 ± 0.6 μM (Fig. 4A) against DENV2 S221 in the viral infectivity assay. We further confirmed this activity in a multi-cycle experiment in which we infected at a low multiplicity of infection (MOI 0.01) to allow viral spread. JBJ-01-162-04 potently reduced viral yield in supernatants collected at 48 and 72 hours post-infection, with 5 μM JBJ-01-162-04 sufficient to reduce viral yield by over 95% during the multi-cycle infection (Fig. 4B). At lower concentrations, the antiviral effect observed decreases over time, presumably due to amplification of the virus and decreased stoichiometric ratio of inhibitor:target. Notably, this is avoided at inhibitor concentrations high enough to prevent viral replication (e.g., 5 μM).
Figure 4. JBJ-01-162-04 inhibits DENV2 S221 in vitro and in vivo.

A. JBJ-01-162-04’s activity against the mouse-adapted strain DENV2 S221 was quantified in the viral infectivity assay shown in Fig. 1A using an MOI of 0.5 and the plaque-forming assay to quantify viral titers. Assays were performed using BHK-21 cells. The average IC90 value from two independent experiments was 1.4 ± 0.6 μM. Representative data from one of two independent experiments are shown with error bars indicating the standard deviation for 2 replicates within that experiment.
B. Assessment of JBJ-01-162-04’s antiviral activity in a multicycle infection. DENV2 S221 was used to infect cells a multiplicity of infection of 0.01 in the presence of varying concentrations of JBJ-01-162-04. JBJ-01-162-04 in the medium was replenished at 24 and 48 hours postinfection. Culture supernatants were harvested at 48 and 72 hours postinfection, and virus titers at these time points were quantified by plaque-forming assay. Representative data from one of n ≥ 2 independent experiments are shown with error bars indicating the standard deviation for 2 replicates within that experiment.
C. Analysis of JBJ-01-162-04 pharmacokinetics (PK) following intraperitoneal dosing. JBJ-01-162-04 was formulated in 10/10/80 DMSO/Tween80/water and administered to C57BL/6 female, 6-8 week old mice at a dosage of 20 mg/kg by intraperitoneal route twice daily for three days. Compound concentrations in plasma and liver were sampled at 4, 8, and 12 hours following dosing on days 1 and on day 3. Data presented are the average of three mice with the exception of those marked with “*” which represent the average of two animals.
D. Inhibition of DENV2 S221 in vivo. Six- to eight-week old AG129 mice were inoculated with 1 × 103 PFU DENV2 S221 by retro-orbital intravenous injection. Mice were treated with either JBJ-01-162-04 at 40 mg/kg (5 males, 5 females) or vehicle (5 males, 5 females) via intraperitoneal route twice daily starting one day before viral inoculation. Peak viremia occurs on day 3 postinfection and was quantified by plaque-forming assay using BHK-21 cells. Representative data from one of two independent experiments are shown, with statistical significance between with treated and control groups determined by paired 2-tailed t test (p = 0.0083).
E. JBJ-01-162-04’s antiviral activity is inhibited in the presence of serum. JBJ-01-162-04 was preincubated with 100, 25, or 0 percent heat-inactivated mouse serum (2 hours, 37 degrees C) and then used in viral infectivity assay as described in Fig. 1A. Viral titers were quantified by plaque-forming assay using BHK-21 cells. Antiviral activity was severely reduced in the presence of serum. Representative data from one of n ≥ 2 independent experiments are shown with error bars indicating the standard deviation for 2 replicates within that experiment.
In preliminary examination of JBJ-01-162-04’s pharmacokinetics (PK) properties in mice, we first tested oral and intravenous administration of JBJ-01-162-04 (Table S1), but the limited oral bioavailability of only 14% prompted us to examine intraperitoneal administration as an alternative. JBJ-01-162-04 was administered twice daily at 20 mg/kg for 3 days, and the concentration of compound in the plasma and liver tissue of mice at 4, 8, and 12 hours after JBJ-01-162-04 treatment was measured on days 1 and 3 (Fig. 4C). Inhibitor exposures in plasma and liver decreased over the course of 4 to 12 hours from 75.8 μM to 2.9 μM in plasma and 37.2 μM to 13.0 μM in liver on day 3. Based on these results, we chose twice daily dosing at 40 mg/kg as a regimen that would enable us to maintain a plasma concentration of JBJ-01-162-04 above the antiviral IC90 value of 1.4 μM measured in the infectivity assay (Fig. 4A).
To evaluate the antiviral activity of JBJ-01-162-04 in vivo, 6- to 8-week old AG129 mice were treated prophylactically with either JBJ-01-162-04 at 40 mg/kg or vehicle via the intraperitoneal route twice daily starting one day prior to inoculation with 1×103 plaque-forming units (PFU) of mouse-adapted DENV2-S221. Under these conditions, we previously observed that the peak of viremia occurs on day 3 post-infection and decreases on day 4 and thereafter (data not shown). Quantification of viremia on day 3 post-infection showed that JBJ-01-162-04 reduces viremia when compared to the vehicle controls (Fig. 4D). Although this effect is modest, we subsequently determined that JBJ-01-162-04 has a high level of plasma-protein binding (> 99.9%; data not shown). This reduces JBJ-01-162-04’s antiviral activity in vitro in a concentration-dependent manner (Fig. 4E), and almost certainly limits the compound’s antiviral effect in vivo. Taking this into account, the reproducible antiviral effect we observed is notable and suggests that further medicinal chemistry to reduce JBJ-01-162-04’s plasma protein-binding would enable more potent inhibition.
Conclusion
Countermeasures against DENV, ZIKV, and other flavivirus pathogens represent a major unmet medical need. While effective vaccines represent the gold standard for prevention of infection, the genetic diversity of the four DENV serotypes and antibody-dependent enhancement of infection and disease have, to date, complicated efforts to develop a broadly protective vaccine. These challenges are exacerbated by recent studies indicating cross-reactivity of ZIKV with anti-DENV antibodies and vice versa and demonstrating that cross-reacting DENV antibodies enhance ZIKV infection and disease in murine models34–39. Antivirals provide an alternative approach to limit transmission and reduce disease by reducing viral burden and disease severity in those with confirmed infection or by preventing infection entirely if administered prophylactically. In the case of ZIKV, persistence of virus in body fluids for weeks to months after the onset of symptoms40–42 suggests that antivirals may be needed to accelerate elimination of the virus from immune-privileged sites. The antiviral effect sufficient to have a significant clinical impact on DENV, ZIKV, and other flavivirus infections has not been established. This is largely due to limitations of current animal models for evaluating impact on flavivirus pathogenesis but also due to the lack of antivirals against DENV, ZIKV, or other flavivirus pathogens. Indeed, despite efforts in both industry and academia43, direct-acting antivirals targeting essential viral enzymes akin to those now used successfully to treat human immunodeficiency virus (HIV) and hepatitis C virus (HCV) have not been developed for any flaviviruses.
We hypothesized that small molecules that bind to the viral envelope protein, E, can potentially engage their target extracellularly and block infection analogously to antibodies but without the risk of antibody-dependent enhancement of infection and disease. Although the genetic diversity of DENV and other flaviviruses that has confounded vaccine development efforts could potentially stymie the development of E-targeting antivirals, our discovery of small molecules that can block the entry of multiple flaviviruses in vitro has indicated otherwise. In contrast to surface epitopes that are the main targets of the antibody response, the small molecule E inhibitors target a pocket located at the interface of domains I and II. This is a hinge region for the conformational changes in E triggered by low pH, and thus, residues in this pocket may be functionally conserved. Consistent with this, mutations introduced to the analogous pocket of other flavivirus E proteins have been shown to affect the pH threshold for viral fusion44–49. We also note that we have experienced difficulties in selecting for antiviral resistance by serial passage of DENV2 in the presence of inhibitors or engineering resistance by site-directed mutagenesis of the pocket (reference 22 and unpublished data), suggesting that mutation of this pocket may be functionally constrained. Here, we have shown that multiple cyanohydrazones can block the entry of DENV, ZIKV, and JEV through inhibition of E-mediated fusion. This antiviral activity cannot be attributed to non-specific or PAINS-like mechanisms but, rather, appears to reflect conserved interactions of these compounds with the E protein. We further demonstrate that JBJ-01-162-04 inhibits DENV in an in vivo model and thus provide the first demonstration of antiviral activity for inhibition of a flavivirus by this mechanism in vivo. Collectively, these findings provide proof of concept for the development of direct-acting antivirals that can block E-mediated membrane fusion of multiple flavivirus pathogens.
Experimental Section
Compound synthesis and characterization
Starting materials, reagents, and solvents were purchased from commercial suppliers and were used without further purification unless otherwise noted. All reactions were monitored using a Waters Acquity UPLC/MS system (Waters PDA eλ Detector, QDa Detector, Sample manager - FL, Binary Solvent Manager) using Acquity UPLC® BEH C18 column (2.1 × 50 mm, 1.7 μm particle size): solvent gradient = 85 % A at 0 min, 1 % A at 1.7 min; solvent A = 0.1 % formic acid in Water; solvent B = 0.1 % formic acid in acetonitrile; flow rate : 0.6 mL/min. Reaction products were purified by flash column chromatography using CombiFlash®Rf with Teledyne Isco RediSep® normal-phase silica flash columns (4 g, 12 g, 24 g, 40 g or 80 g) and Waters HPLC system using SunFire™ Prep C18 column (19 × 100 mm, 5 μm particle size): solvent gradient = 80 % A at 0 min, 10 % A at 25 min; solvent A = 0.035 % TFA in Water; solvent B = 0.035 % TFA in MeOH; flow rate : 25 mL/min. 1H NMR spectra were recorded on 500 MHz Bruker Avance III spectrometers and 13C NMR spectra were recorded on 125 MHz Bruker Avance III spectrometer. Chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as br (broad), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet).
1. Representative Procedure: Synthesis of JBJ-01-162-01 [(E)-2-(benzofuran-2-yl)-N-(4-(4-methylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide].
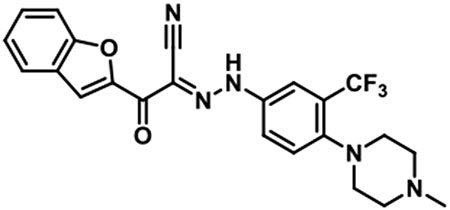
To a solution of 4-(4-methylpiperazin-1-yl)-3-(trifluoromethyl)aniline (154 mg, 0.51 mmol) in 6 N hydrochloric acid (1 mL) was added dropwise a solution of sodium nitrite (41 mg, 0.59 mmol) in water (1 mL) at 0 °C under icebath. After stirring at room temperature for 30 min, a mixture of sodium acetate (49 mg) and 3-(benzofuran-2-yl)-3-oxopropanenitrile (100 mg, 0.54 mmol) in 1,4-dioxane (2 ml) was added to the reaction mixture. After stirring for 6 hr, the reaction mixture was filtered through celite, and the filtrate was diluted with EtOAc and washed with sat. NaHCO3 and brine. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by prepHPLC to give JBJ-01-162-01 as a yellowish solid (103 mg, 42 %).
1H NMR (500 MHz, DMSO-d6) δ 9.79 (br s, 1H), 7.97 (s, 1H), 7.95 - 7.93 (m, 1H), 7.90 - 7.83 (m, 2H), 7.75 (d, J = 8.5 Hz, 1H), 7.70 (d, J = 8.9 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H), 3.56 - 3.40 (m, 4H), 3.20 - 3.07 (m, 4H), 2.90 (s, 3H); LC/MS (ESI) calcd. C23H21F3N5O2 [M+H]+ m/z 456.16, found 456.22; retention time 0.99 min.
JBJ-01-162-02 [(E)-2-(benzofuran-2-yl)-N-(3-(4-methylpiperazin-1-yl)-5-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide].
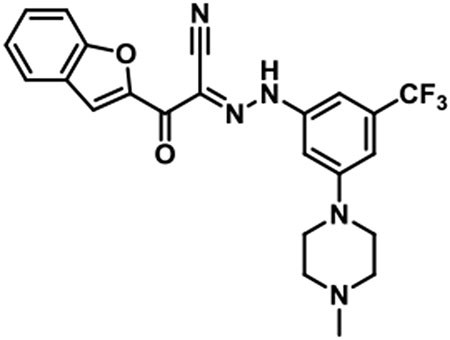
1H NMR (500 MHz, DMSO-d6) δ 9.85 (br s, 1H), 7.97 (s, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.36 (br s, 2H), 7.19 (s, 1H), 4.00 (br s, 2H), 3.53 (br s, 2H), 3.27 - 3.03 (m, 4H), 2.89 (s, 3H); LC/MS (ESI) calcd. C23H21F3N5O2 [M+H]+ m/z 456.16, found 456.28; retention time 1.1 min.
JBJ-01-162-03 [(E)-2-(benzofuran-2-yl)-N-(4-morpholino-3-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide].
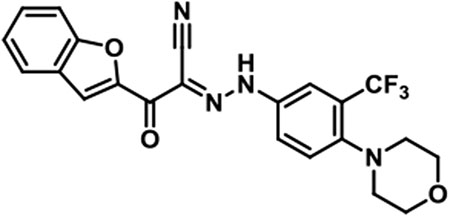
1H NMR (500 MHz, DMSO-d6) δ 9.85 (br s, 1H), 7.98 (s, 1H), 7.92 (d, J = 1.8 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.85 - 7.81 (m, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.70 (d, J = 8.9 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 3.74 - 3.68 (m, 4H), 2.89 - 2.82 (m, 4H); LC/MS (ESI) calcd. C22H18F3N4O3 [M+H]+ m/z 443.13, found 443.19; retention time 1.6 min.
JBJ-01-162-04 [(E)-2-(benzofuran-2-yl)-N-(3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide].
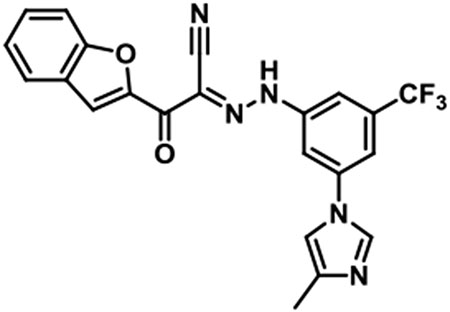
1H NMR (500 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.83 (br s, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.63 (s, 1H), 7.62 - 7.59 (m, 3H), 7.44 (t, J = 7.6 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 2.18 (s, 3H); LC/MS (ESI) calcd. C22H15F3N5O2 [M+H]+ m/z 438.12, found 438.17; retention time 1.1 min.
JBJ-01-162-05 [(E)-2-(benzofuran-2-yl)-N-(4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide].
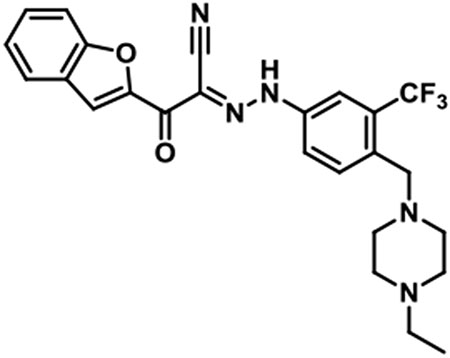
1H NMR (500 MHz, DMSO-d6) δ 9.38 (br s, 1H), 8.01 (s, 1H), 7.97 (s, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.85 (s, 2H), 7.77 (d, J = 8.2 Hz, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 3.72 (s, 2H), 3.15 (q, J = 7.3 Hz, 2H), 3.05 - 2.90 (m, 4H), 2.42 (t, J = 11.9 Hz, 2H), 1.22 (t, J = 11.9 Hz, 3H); LC/MS (ESI) calcd. C25H25F3N5O2 [M+H]+ m/z 484.20, found 484.29; retention time 0.98.
JBJ-03-019-01 [(E)-2-(benzofuran-2-yl)-N-(4-fluoro-3-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide].
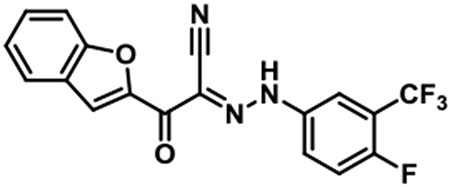
1H NMR (500 MHz, DMSO-d6) δ 7.97 (s, 1H), 7.95 - 7.90 (m, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.63 (t, J = 9.6 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H); LC/MS (ESI) calcd. C18H10F4N3O2 [M+H]+ m/z 376.07, found 376.07; retention time 1.6 min.
JBJ-03-019-02 [(E)-2-(benzofuran-2-yl)-N-(3-fluoro-5-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide].
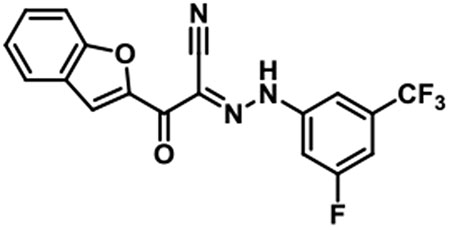
1H NMR (500 MHz, DMSO-d6) δ 8.00 (s, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.76 (s,1H), 7.73 (d, J = 8.5 Hz, 1H), 7.62 - 7.57 (m, 2H), 7.50 (t, J = 8.2 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H); LC/MS (ESI) calcd. C18H10F4N3O2 [M+H]+ m/z 376.07, found 376.07; retention time 1.6 min.
Synthesis of 3-110-22, 3-110-2, 3-149-3, and 3-149-15 were previously described19–20.
Cell lines
Vero cells (ATCC, RRID:CVCL_0059), baby hamster kidney-derived BHK-21 cells (laboratory of Eva Harris, University of California, Berkeley), C6/36 cells derived from Aedes albopictus (Diptera: Culicidae) embryonic tissue (ATCC, RRID:CVCL_Z230), Spodoptera frugiperda cells (Sf9) (laboratory of Stephen C. Harrison, Cat # B825-01 Invitrogen/Thermo Fisher Scientific), and Trichoplusia ni cells (High Five™, laboratory of Stephen C. Harrison, Invitrogen/Thermo Fisher Scientific) were maintained as previously described21–22.
Viruses
All work with infectious virus was performed in a biosafety level 2 (BSL2) laboratory using additional safety practices as approved by the Harvard Committee on Microbiological Safety. Dengue virus serotype 2 strain New Guinea C (DENV2 NGC; Lee Gehrke, MIT), dengue virus serotype 2 strain S221 (DENV2 S221; Sujan Shresta, La Jolla Institute for Allergy and Immunology), and Zika virus (ZIKV) strain PF-251013-18 (Didier Musso, Institut Louis Malardé, Tahiti Island, French Polynesia) were propagated in C6/36 cells as previously described21–22. The Japanese encephalitis virus (JEV) vaccine strain SA14-14-2 was kindly provided by Gregory Gromowski (Walter Reed Army Institute of Research). The viral stocks used in the current study were produced in C6/36 cells, and were not propagated more than twice.
Antibodies
Monoclonal antibody 4G2 against the DENV E protein was produced from culture supernatants of hybridoma D1-4G2-4-15(ATCC HB-112, RRID:CVCL_J890). Mouse hybridoma producing monoclonal antibody 6F3.1 against DENV2 core protein was generously provided by John Aaskov (Queensland University of Technology)50. Rabbit polyclonal antibodies against the ZIKV core protein, rabbit polyclonal antibodies against the JEV E protein, and mouse monoclonal antibodies against the JEV core protein were purchased from GeneTex (GTX133317, GTX125867 and GTX634153, respectively). Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and anti-rabbit IgG antibodies were obtained from Bio-Rad Laboratories (170-6516 and 170-6515, respectively).
Cytotoxicity assay
Cytotoxicity was evaluated against BHK-21 cells using a commercially available kit (CellTiter-Glo, Promega) following the manufacturer’s instructions. Values for the concentrations that cause 50% cytotoxicity (CC50) were calculated using the nonlinear fit variable slope model (GraphPad Software).
Plaque-forming assay (PFA) and focus-forming assay
DENV2 and JEV titers were quantified by plaque-formation assay (PFA) on BHK-21 cells, and ZIKV titers were quantified by PFA on Vero cells following published procedures21–22. Incubation times for plaque formation at 37 °C were 2.5 days (ZIKV), 3 days (JEV), or 4.5 days (DENV2). Where indicated, DENV2 titers were quantified by focus-formation assay (FFA) on BHK-21 cells. The procedure was as described above for PFA except that after incubation at 37°C for 3 days, the cells were fixed with methanol for 15 min at −20°C and then immunostaining was performed using monoclonal antibody 6F3.1 against the DENV2 core protein.
Viral infectivity assays for IC90 determination
Virus inocula were diluted in EBSS to achieve an MOI of 1, and were pre-incubated with the given small molecule at varying concentrations for 45 min at 37 °C. The mixture was then added to cells for 1 hour at 37 °C to allow infection, after which the inoculum was remved, and the cells were washed with 1× PBS to remove unbound virus and compound. Cells were overlaid with medium lacking inhibitor and incubated at 37 °C for 24 hours. Unless stated otherwise, culture supernatants were harvested at this time, stored at −80 °C, and the yield of infectious particles produced was quantified by PFA or FFA. The values for the concentrations that lead to 90% inhibition (IC90) were calculated using the nonlinear fit variable slope model (GraphPad Software).
Antiviral activity assays were performed with the following cell lines: DENV infections, PFA, and FFA performed in BHK-21; JEV infections and PFA performed in BHK-21; ZIKV infections and PFA performed in Vero; VSV-eGFP infections performed in BHK-21.
Vesicular stomatitis virus-eGFP (VSV-eGFP) counter screen
Virus inocula were diluted in EBSS to achieve a MOI of 1 and were preincubated with the given small molecule at varying concentrations for 45 min at 37 °C. The virus-inhibitor mixture was then added to cells for 1 h at 37 °C to allow infection, after which the inoculum was removed, and the cells were washed with 1X PBS to remove unbound virus and compound. Cells were overlaid with medium lacking inhibitor and incubated at 37 °C for 6 h, corresponding to a single cycle of infection. Following removal of the supernatants, the cells were washed with 1X PBS and overlaid with PBS and then imaged. Fluorescence (excitation 473 nm, emission 650 nm) was measured using a Typhoon FLA 9500 (GE Healthcare Life Sciences) and quantified using ImageJ software (http://imagej.nih.gov/ij/).
Capsid protection assay to monitor fusion of virions with liposomes
The composition of liposomes was designed to mimic late endosomal membranes as previously described19–20, 51–52. Liposomes were made with (1,2-dioleoyl-sn-glycero-3-phosphocholine) (DOPC) (Avanti Polar Lipids), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (Avanti Polar Lipids), L-α-phosphatidylinositol, Soy (PI), bis(monooleoylglycero)phosphate, S,R Isomer (BMP) and cholesterol (Avanti Polar Lipids) at 4/1/1/2/4 molar ratio in TAN buffer (20 mM triethanolamine, 100 mM NaCl, pH 8.0). Lipids were dried down and resuspended thoroughly by vortexing and sonication. Liposomes were prepared by extrusion through a 0.2 micron filter after five freeze/thaw cycles. For trypsin-containing liposomes, 3.8 mg of trypsin was added to 0.38 mL of lipids (2.8 mg each) after the third freeze/thaw cycle, prior to extrusion. Liposomes were separated from unincorporated trypsin by size-exclusion chromatography using a 10/300 Increase Superdex 200 column (GE Healthcare) on an Akta fast performance liquid chromatography (FPLC) system.
50 μL of DENV2, strain NGC (5×105 PFU); ZIKV, strain PF13 (9.5×105 PFU); and JEV, strain SA14–14-2 (9.5×105 PFU) were incubated with various concentrations of compounds (0, 10 μM, 50 μM) for 45 min at 37 °C in TAN buffer (pH 8.0) prior to addition of 20 μL liposomes for 5 minutes. This represents > 10-fold increase in virus and hence target abundance relative to the viral infectivity assays. After incubation with liposomes, 2.5 μL 1M sodium acetate (pH 5.0) was added to drop the pH to 5.3 for 5 minutes. Samples were back-neutralized with 2.5 μL 2M triethanolamine (pH 8.1) and incubated for 20 minutes at 37°C to allow trypsin digestion. SDS sample buffer was added, and samples were boiled for 20 minutes before separation by SDS-PAGE on a 10% gel. Proteins were transferred to a PVDF membrane using a semi-dry transfer apparatus. The following antibodies were used for detection of envelope and capsid protein respectively: 4G2 (1:100) and 6F3.1 antibodies for DENV, 4G2 (1:100) and ZIKV capsid protein antibodies (1:100) for ZIKV, and JEV envelope protein antibodies (1:1000), and JEV core antibodies (1:500) for JEV. The membrane was developed with enhanced chemiluminescence reagents (Pierce), and the signal was captured using the Amersham Imager 600.
Malate dehydrogenase (MDH) assay
Small molecule inhibitors were serially diluted (2-fold dilution series from 100 μM) and were mixed with 200 μM oxaloacetic acid (VWR) and 200 μM NADH (VWR) in working buffer (100 mM potassium phosphate, pH 7.0). Malate dehydrogenase (EMD Millipore) was added to a final concentration of 0.7 nM, and absorbance was immediately monitored at 340 nm for 5 min. The final concentration of DMSO was 1% for all samples. All assays were repeated in the presence of 0.01% Triton X-100. The values for the concentrations that lead to 50% inhibition (IC50) were calculated using the nonlinear fit variable slope model (GraphPad Software).
Expression, purification, and biotinylation of recombinant soluble, prefusion DENV2 envelope protein (sE2) and determination of dissociation constant (KD) values by bio-layer interferometry (BLI)
The soluble DENV2 prefusion E dimer (sE2) bearing a 6His-N-terminal purification tag and AviTagTdeM4 C-terminal tag as previously described21–22. Equilibrium dissociation constant for binding of inhibitor to sE2 was measured by biolayer interferometry using an Octet RED384 system (FortéBio) using superstreptavidin (SSA) biosensors. Association with small molecules was monitored for 120 seconds with inhibitor concentrations that ranged from 50 nM to 20 μM; dissociation was performed in reaction buffer and monitored for 120 seconds. Equilibrium dissociation constants (KD) values were determined by plotting the local fit maximum response (nm) as a function of small molecule concentrations (μM) using Octet Data Analaysis Software (FortéBio) and Prism (GraphPad Software). Titration curves were fit to the following steady-state analysis equation:
where Rmax is the local fit response maximum; Conc is the concentration of small molecule; and KD is the equilibrium dissociation constant.
Pharmacokinetic study in mice
Pharmacokinetic studies in vivo were performed by the Drug Metabolism and Pharmacokinetics Core at Scripps Florida following protocols reviewed and approved by the Institutional Animal Care and Use Committee. Briefly, C57BL/6 mice were administered JBJ-01-162-04 formulated in 0.1 and 1 mg/mL solution in DMSO:Tween-80:water (10:10:80) at dosages of 1 mg/kg by intravenous (i.v.) and 10 mg/kg by oral gavage (n = 3 mice per group), respectively. Blood samples were collected at 9 time points over 500 min post-dosing for determination of PK parameters’ calculation. Due to limited oral bioavailability and clearance issue by the i.v. route, we further tested intraperitoneal (i.p.) administration of JBJ-01-162-04 with the same formulation at 20 mg/kg twice daily for 3 days. Blood and liver samples were collected and compound concentrations were quantified at 4, 8, 12 h on days 1 and day 3.
Hepatic microsomal stability
Microsome stability assays were performed by the Drug Metabolism and Pharmacokinetics Core at Scripps Florida. 1 μM test compound was incubated with 1 mg/mL hepatic microsomes in 100 mM potassium phosphate buffer, pH 7.4. The reaction was initiated by adding NADPH (1 mM final concentration). Aliquots were removed at 0, 5, 10, 20, 40, and 60 minutes and added to acetonitrile (5X v:v) to stop the reaction and precipitate the protein. NADPH dependence of the reaction was evaluated with -NADPH samples. At the end of the assay, the samples were centrifuged through a Millipore Multiscreen Solvinter 0.45 micron low binding PTFE hydrophilic filter plate and analyzed by LC-MS/MS. Data were log-transformed and represented as half-life and intrinsic clearance.
Antiviral efficacy in vivo.
All experiments performed in mice followed protocols and procedures reviewed and approved by the Institutional Animal Care and Use Committee of Harvard University. Breeding pairs of AG129 mice, which lack type I and type II interferon receptors, were generously donated by Sujan Shresta (LJIA) and bred in the specific-pathogen-free animal facility at Harvard Medical School. 6-8 week old AG129 mice were challenged with 1 × 103 PFU of DENV-2 S221 by retro-orbital injection in 100 μL of DPBS with 0.2% BSA. For prophylaxis experiments, mice were treated with either JBJ-01-162-04 at 40 mg/kg formulated in DMSO:Tween-80:water (10:10:80) or vehicle (10 mice per group for both sexes) via the intraperitoneal route twice daily starting 1 day prior to inoculation with DENV until day 3 post-infection. Blood was collected on day 3 post-infection by the retro-orbital route, as this reproducibly coincides with the peak of viremia under these experimental conditions. The serum was separated and harvested after centrifugation in a microvette blood collection tube (Sarstedt), and then stored at −80 degrees C until processed. Serum viremia was assessed by viral plaque formation assay. The in vivo experiments were performed in two independent studies. Statistical analysis was performed by paired t test for two-tailed analysis.
Statistical Analysis
Statistical tests and the associated error bars are identified in the corresponding figure legends. ‘‘Independent’’ experiments describe the number of biological replicates, which were repeat experiments performed on different days. Statistical analysis was performed using Prism (GraphPad Software).
Supplementary Material
Scheme 1.
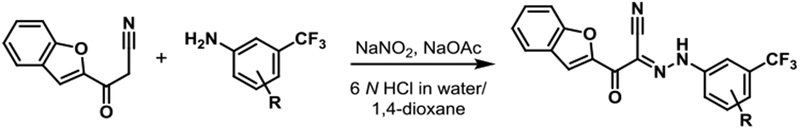
General synthetic route19.
Acknowledgement:
Stephen C. Harrison and Ian Walker are thanked for technical assistance in expression of recombinant DENV E proteins. We thank the following colleagues for sharing reagents: Didier Musso and Nathalie Pardigon for Zika PF13-251013-18; John Aaskov for the hybridoma cell line producing Mab 6F3.1; Lee Gehrke for DENV2 New Guinea C virus; Aravinda de Silva for other DENV strains; Sean Whelan for VSV-eGFP reporter virus; Eva Harris for BHK-21 cells; Stephen C. Harrison for the synthetic DENV2 sE gene and insect cell lines. Kelly Arnett and the Center for Macromolecular Interactions and Jennifer Smith and the ICCB-Longwood Screening Facility provided access to instruments and technical support. We thank Brian Shoichet for help with the PAINS assays utilizing MDH. Isabella Zafra is thanked for performing antiviral assays in a preliminary phase of this work. This work was supported by NIH awards R56AI095499, R01AI095499, and U19AI109740.
Funding Sources This work was supported by NIH awards R56AI095499, R01AI095499, and U19AI109740 and an award from the Blavatnik Biomedical Accelerator at Harvard University.
Notes Nathanael S. Gray is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, B2S and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Voronoi, Her2llc, Deerfield and Sanofi.
Footnotes
Ancillary Information
Supporting Information. Figures S1, S2, S3 and Table S1 provide additional data. Detailed descriptions of materials and methods are also provided.
REFERENCES
- 1.Bhatt S; Gething PW; Brady OJ; Messina JP; Farlow AW; Moyes CL; Drake JM; Brownstein JS; Hoen AG; Sankoh O; Myers MF; George DB; Jaenisch T; Wint GRW; Simmons CP; Scott TW; Farrar JJ; Hay SI, The global distribution and burden of dengue. Nature 2013, 496, 504–507. DOI 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halstead SB; O’Rourke EJ, Antibody-enhanced dengue virus infection in primate leukocytes. Nature 1977, 265 (5596), 739–41. [DOI] [PubMed] [Google Scholar]
- 3.Katzelnick LC; Gresh L; Halloran ME; Mercado JC; Kuan G; Gordon A; Balmaseda A; Harris E, Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358 (6365), 929–932. DOI 10.1126/science.aan6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabchareon A; Wallace D; Sirivichayakul C; Limkittikul K; Chanthavanich P; Suvannadabba S; Jiwariyavej V; Dulyachai W; Pengsaa K; Wartel TA; Moureau A; Saville M; Bouckenooghe A; Viviani S; Tornieporth NG; Lang J, Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. The Lancet 2012, 380, 1559–1567. DOI 10.1016/S0140-6736(12)61428-7. [DOI] [PubMed] [Google Scholar]
- 5.Capeding MR; Tran NH; Hadinegoro SRS; Ismail HIHM; Chotpitayasunondh T; Chua MN; Luong CQ; Rusmil K; Wirawan DN; Nallusamy R; Pitisuttithum P; Thisyakorn U; Yoon I-K; van der Vliet D; Langevin E; Laot T; Hutagalung Y; Frago C; Boaz M; Wartel TA; Tornieporth NG; Saville M; Bouckenooghe A, Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. The Lancet 2014, 384, 1358–1365. DOI 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- 6.Hadinegoro SR; Arredondo-García JL; Capeding MR; Deseda C; Chotpitayasunondh T; Dietze R; Hj Muhammad Ismail HI; Reynales H; Limkittikul K; Rivera-Medina DM; Tran HN; Bouckenooghe A; Chansinghakul D; Cortés M; Fanouillere K; Forrat R; Frago C; Gailhardou S; Jackson N; Noriega F; Plennevaux E; Wartel TA; Zambrano B; Saville M, Efficacy and Long-Term Safety of a Dengue Vaccine in Regions of Endemic Disease. New England Journal of Medicine 2015, 373, 1195–1206. DOI 10.1056/NEJMoa1506223. [DOI] [PubMed] [Google Scholar]
- 7.Villar L; Dayan GH; Arredondo-Garcia JL; Rivera DM; Cunha R; Deseda C; Reynales H; Costa MS; Morales-Ramirez JO; Carrasquilla G; Rey LC; Dietze R; Luz K; Rivas E; Miranda Montoya MC; Cortes Supelano M; Zambrano B; Langevin E; Boaz M; Tornieporth N; Saville M; Noriega F; Group CYDS, Efficacy of a tetravalent dengue vaccine in children in Latin America. N Engl J Med 2015, 372 (2), 113–23. DOI 10.1056/NEJMoa1411037. [DOI] [PubMed] [Google Scholar]
- 8.Halstead SB, Critique of World Health Organization Recommendation of a Dengue Vaccine. J Infect Dis 2016, 214 (12), 1793–1795. DOI 10.1093/infdis/jiw340. [DOI] [PubMed] [Google Scholar]
- 9.Halstead SB; Russell PK, Protective and immunological behavior of chimeric yellow fever dengue vaccine. Vaccine 2016, 34 (14), 1643–7. DOI 10.1016/j.vaccine.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Tassaneetrithep B; Burgess TH; Granelli-Piperno A; Trumpfheller C; Finke J; Sun W; Eller MA; Pattanapanyasat K; Sarasombath S; Birx DL; Steinman RM; Schlesinger S; Marovich MA, DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med 2003, 197 (7), 823–9. DOI 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meertens L; Carnec X; Lecoin MP; Ramdasi R; Guivel-Benhassine F; Lew E; Lemke G; Schwartz O; Amara A, The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12 (4), 544–57. DOI 10.1016/j.chom.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz-Oliveira C; Freire JM; Conceicao TM; Higa LM; Castanho MA; Da Poian AT, Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol Rev 2015, 39 (2), 155–70. DOI 10.1093/femsre/fuu004. [DOI] [PubMed] [Google Scholar]
- 13.Krishnan MN; Sukumaran B; Pal U; Agaisse H; Murray JL; Hodge TW; Fikrig E, Rab 5 is required for the cellular entry of dengue and West Nile viruses. J Virol 2007, 81 (9), 4881–5. DOI 10.1128/JVI.02210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acosta EG; Castilla V; Damonte EB, Functional entry of dengue virus into Aedes albopictus mosquito cells is dependent on clathrin-mediated endocytosis. J Gen Virol 2008, 89 (Pt 2), 474–84. DOI 10.1099/vir.0.83357-0. [DOI] [PubMed] [Google Scholar]
- 15.van der Schaar HM; Rust MJ; Chen C; van der Ende-Metselaar H; Wilschut J; Zhuang X; Smit JM, Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog 2008, 4 (12), e1000244 DOI 10.1371/journal.ppat.1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allison SL; Schalich J; Stiasny K; Mandl CW; Kunz C; Heinz FX, Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J Virol 1995, 69 (2), 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bressanelli S; Stiasny K; Allison SL; Stura EA; Duquerroy S; Lescar J; Heinz FX; Rey FA, Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J 2004, 23 (4), 728–38. DOI 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modis Y; Ogata S; Clements D; Harrison SC, Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427 (6972), 313–9. DOI 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt AG; Lee K; Yang PL; Harrison SC, Small-Molecule Inhibitors of Dengue-Virus Entry. PLOS Pathogens 2012, 8, e1002627 DOI 10.1371/journal.ppat.1002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark Margaret J; Miduturu C; Schmidt Aaron G; Zhu X; Pitts Jared D; Wang J; Potisopon S; Zhang J; Wojciechowski A; Hann Chu Justin J; Gray Nathanael S; Yang Priscilla L, GNF-2 Inhibits Dengue Virus by Targeting Abl Kinases and the Viral E Protein. Cell Chemical Biology 2016, 23, 443–452. DOI 10.1016/j.chembiol.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lian W; Jang J; Potisopon S; Li PC; Rahmeh A; Wang J; Kwiatkowski NP; Gray NS; Yang PL, Discovery of Immunologically Inspired Small Molecules That Target the Viral Envelope Protein. ACS Infect Dis 2018, 4 (9), 1395–1406. DOI 10.1021/acsinfecdis.8b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Wispelaere M; Lian W; Potisopon S; Li PC; Jang J; Ficarro SB; Clark MJ; Zhu X; Kaplan JB; Pitts JD; Wales TE; Wang J; Engen JR; Marto JA; Gray NS; Yang PL, Inhibition of Flaviviruses by Targeting a Conserved Pocket on the Viral Envelope Protein. Cell Chem Biol 2018, 25 (8), 1006–1016 e8 DOI 10.1016/j.chembiol.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGovern SL; Caselli E; Grigorieff N; Shoichet BK, A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem 2002, 45 (8), 1712–22. [DOI] [PubMed] [Google Scholar]
- 24.Seidler J; McGovern SL; Doman TN; Shoichet BK, Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem 2003, 46 (21), 4477–86. DOI 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 25.Feng BY; Shelat A; Doman TN; Guy RK; Shoichet BK, High-throughput assays for promiscuous inhibitors. Nat Chem Biol 2005, 1 (3), 146–8. DOI 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 26.Gaudin Y, Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell Biochem 2000, 34, 379–408. [DOI] [PubMed] [Google Scholar]
- 27.Roche S; Bressanelli S; Rey FA; Gaudin Y, Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313 (5784), 187–91. DOI 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- 28.Roche S; Rey FA; Gaudin Y; Bressanelli S, Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315 (5813), 843–8. DOI 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- 29.Shresta S; Sharar KL; Prigozhin DM; Beatty PR; Harris E, Murine model for dengue virus-induced lethal disease with increased vascular permeability. J Virol 2006, 80 (20), 10208–17. DOI 10.1128/JVI.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perry ST; Buck MD; Plummer EM; Penmasta RA; Batra H; Stavale EJ; Warfield KL; Dwek RA; Butters TD; Alonzi DS; Lada SM; King K; Klose B; Ramstedt U; Shresta S, An iminosugar with potent inhibition of dengue virus infection in vivo. Antiviral Res 2013, 98 (1), 35–43. DOI 10.1016/j.antiviral.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Zellweger RM; Prestwood TR; Shresta S, Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe 2010, 7 (2), 128–39. DOI 10.1016/j.chom.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prestwood TR; Morar MM; Zellweger RM; Miller R; May MM; Yauch LE; Lada SM; Shresta S, Gamma interferon (IFN-gamma) receptor restricts systemic dengue virus replication and prevents paralysis in IFN-alpha/beta receptor-deficient mice. J Virol 2012, 86 (23), 12561–70. DOI 10.1128/JVI.06743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stein DA; Perry ST; Buck MD; Oehmen CS; Fischer MA; Poore E; Smith JL; Lancaster AM; Hirsch AJ; Slifka MK; Nelson JA; Shresta S; Fruh K, Inhibition of dengue virus infections in cell cultures and in AG129 mice by a small interfering RNA targeting a highly conserved sequence. J Virol 2011, 85 (19), 10154–66. DOI 10.1128/JVI.05298-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dejnirattisai W; Supasa P; Wongwiwat W; Rouvinski A; Barba-Spaeth G; Duangchinda T; Sakuntabhai A; Cao-Lormeau V-M; Malasit P; Rey FA; Mongkolsapaya J; Screaton GR, Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nature Immunology 2016, 17, 1102–1108. DOI 10.1038/ni.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fowler AM; Tang WW; Young MP; Mamidi A; Viramontes KM; McCauley MD; Carlin AF; Schooley RT; Swanstrom J; Baric RS; Govero J; Diamond MS; Shresta S, Maternally Acquired Zika Antibodies Enhance Dengue Disease Severity in Mice. Cell Host Microbe 2018, 24 (5), 743–750 e5 DOI 10.1016/j.chom.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Priyamvada L; Quicke KM; Hudson WH; Onlamoon N; Sewatanon J; Edupuganti S; Pattanapanyasat K; Chokephaibulkit K; Mulligan MJ; Wilson PC; Ahmed R; Suthar MS; Wrammert J, Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proceedings of the National Academy of Sciences 2016, 113, 7852–7857. DOI 10.1073/pnas.1607931113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savidis G; McDougall WM; Meraner P; Perreira JM; Portmann JM; Trincucci G; John SP; Aker AM; Renzette N; Robbins DR; Guo Z; Green S; Kowalik TF; Brass AL, Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep 2016, 16 (1), 232–46. DOI 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 38.Stettler K; Beltramello M; Espinosa DA; Graham V; Cassotta A; Bianchi S; Vanzetta F; Minola A; Jaconi S; Mele F; Foglierini M; Pedotti M; Simonelli L; Dowall S; Atkinson B; Percivalle E; Simmons CP; Varani L; Blum J; Baldanti F; Cameroni E; Hewson R; Harris E; Lanzavecchia A; Sallusto F; Corti D, Specificity, cross-reactivity and function of antibodies elicited by Zika virus infection. Science 2016, aaf8505 DOI 10.1126/science.aaf8505. [DOI] [PubMed] [Google Scholar]
- 39.Zimmerman MG; Quicke KM; O’Neal JT; Arora N; Machiah D; Priyamvada L; Kauffman RC; Register E; Adekunle O; Swieboda D; Johnson EL; Cordes S; Haddad L; Chakraborty R; Coyne CB; Wrammert J; Suthar MS, Cross-Reactive Dengue Virus Antibodies Augment Zika Virus Infection of Human Placental Macrophages. Cell Host Microbe 2018, 24 (5), 731–742 e6 DOI 10.1016/j.chom.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paz-Bailey G; Rosenberg ES; Doyle K; Munoz-Jordan J; Santiago GA; Klein L; Perez-Padilla J; Medina FA; Waterman SH; Gubern CG; Alvarado LI; Sharp TM, Persistence of Zika Virus in Body Fluids - Preliminary Report. N Engl J Med 2017. DOI 10.1056/NEJMoa1613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sotelo JR; Sotelo AB; Sotelo FJB; Doi AM; Pinho JRR; Oliveira RC; Bezerra A; Deutsch AD; Villas-Boas LS; Felix AC; Romano CM; Machado CM; Mendes-Correa MCJ; Santana RAF; Menezes FG; Mangueira CLP, Persistence of Zika Virus in Breast Milk after Infection in Late Stage of Pregnancy. Emerg Infect Dis 2017, 23 (5), 856–857. DOI 10.3201/eid2305.161538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calvet GA; Kara EO; Giozza SP; Botto-Menezes CHA; Gaillard P; de Oliveira Franca RF; de Lacerda MVG; da Costa Castilho M; Brasil P; de Sequeira PC; de Mello MB; Bermudez XPD; Modjarrad K; Meurant R; Landoulsi S; Benzaken AS; de Filippis AMB; Broutet NJN; Team ZS, Study on the persistence of Zika virus (ZIKV) in body fluids of patients with ZIKV infection in Brazil. BMC Infect Dis 2018, 18 (1), 49 DOI 10.1186/s12879-018-2965-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim SP; Wang Q-Y; Noble CG; Chen Y-L; Dong H; Zou B; Yokokawa F; Nilar S; Smith P; Beer D; Lescar J; Shi P-Y, Ten years of dengue drug discovery: Progress and prospects. Antiviral Research 2013, 100, 500–519. DOI 10.1016/j.antiviral.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 44.Cecilia D; Gould EA, Nucleotide changes responsible for loss of neuroinvasiveness in Japanese encephalitis virus neutralization-resistant mutants. Virology 1991, 181 (1), 70–7. [DOI] [PubMed] [Google Scholar]
- 45.Hasegawa H; Yoshida M; Shiosaka T; Fujita S; Kobayashi Y, Mutations in the envelope protein of Japanese encephalitis virus affect entry into cultured cells and virulence in mice. Virology 1992, 191 (1), 158–65. [DOI] [PubMed] [Google Scholar]
- 46.Lee E; Weir RC; Dalgarno L, Changes in the dengue virus major envelope protein on passaging and their localization on the three-dimensional structure of the protein. Virology 1997, 232 (2), 281–90. DOI 10.1006/viro.1997.8570. [DOI] [PubMed] [Google Scholar]
- 47.Beasley DW; Aaskov JG, Epitopes on the dengue 1 virus envelope protein recognized by neutralizing IgM monoclonal antibodies. Virology 2001, 279 (2), 447–58. DOI 10.1006/viro.2000.0721. [DOI] [PubMed] [Google Scholar]
- 48.Hurrelbrink RJ; McMinn PC, Attenuation of Murray Valley encephalitis virus by site-directed mutagenesis of the hinge and putative receptor-binding regions of the envelope protein. J Virol 2001, 75 (16), 7692–702. DOI 10.1128/JVI.75.16.7692-7702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monath TP; Arroyo J; Levenbook I; Zhang ZX; Catalan J; Draper K; Guirakhoo F, Single mutation in the flavivirus envelope protein hinge region increases neurovirulence for mice and monkeys but decreases viscerotropism for monkeys: relevance to development and safety testing of live, attenuated vaccines. J Virol 2002, 76 (4), 1932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bulich R; Aaskov JG, Nuclear localization of dengue 2 virus core protein detected with monoclonal antibodies. J Gen Virol 1992, 73 (Pt 11), 2999–3003. DOI 10.1099/0022-1317-73-11-2999. [DOI] [PubMed] [Google Scholar]
- 51.van Meer G; Voelker DR; Feigenson GW, Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 2008, 9 (2), 112–24. DOI 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaitseva E; Yang ST; Melikov K; Pourmal S; Chernomordik LV, Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog 2010, 6 (10), e1001131 DOI 10.1371/journal.ppat.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.