

ABSTRACT
Bacterial pathogens can be very efficient at causing disease and are the cause of some of the worst epidemics that have affected humanity. However, most infections are prevented by the actions of our immune system. Immune activation depends on the rapid detection of bacteria by a diverse family of sensory proteins known as pattern recognition receptors. These receptors detect conserved features of bacteria that are not found in humans but are often necessary for survival within the host or environment. In this review, we discuss the strategies used by pattern recognition receptors to detect bacteria and their products. We also discuss emerging evidence that some pattern recognition receptors can be activated by bacterial pathogens specifically, through the surveillance of host activities that are commonly targeted by virulence factors. This collection of surveillance mechanisms provides an interconnected network of defense, which is important to maintain the germ-free environment of the inner organs of humans and other multicellular organisms.
INTRODUCTION
Outside a mammalian host, bacteria face numerous challenges that can result in life-threatening risks. These challenges include variations in temperature and osmolarity, predation, desiccation, and nutrient shortage. For bacteria with the ability to survive within a mammalian host, several of these threats are less severe, as host cells exist at a fixed temperature, osmolarity, and water and nutrient content. However, there is a cost associated with the benefits of an intracellular lifestyle. That cost is the threat of host-encoded immune defenses. Here, we describe the molecular mechanisms used by mammalian hosts to detect bacterial infection. We discuss the receptors encoded by the host immune system that recognize infection and the bacterial molecules that these receptors detect. Finally, we illustrate how these detection strategies, which have diverse mechanisms of action, share a thematically similar goal. This goal is to induce inflammatory responses that are typified by the recruitment of the biggest threat to bacterial viability to the sites of infection—polymorphonuclear leukocytes, also known as neutrophils.
BACTERIAL MOLECULES THAT INDUCE HOST DEFENSE
All multicellular organisms face the threat of microbial colonization, which poses a threat to the viability of the host. Studies over the last two decades, first by the late Charles Janeway, Jr., and subsequently by many others, established a common strategy used by multicellular organisms to detect bacterial encounters. This common strategy of bacterial detection comes from the use of germ line-encoded host proteins that recognize molecules present within large classes of bacteria. The microbial molecules sensed by the host include structural components of the bacterial cell wall, such as lipopolysaccharides (LPS), lipoproteins, peptidoglycan fragments, and flagellin subunits (Table 1). Additional microbial molecules include nucleic acids—DNA and RNA. As mammalian cells lack a cell wall, the presence of LPS and other cell wall components in the host provides a high-fidelity indicator of a bacterial encounter. Thus, the host-encoded sensors of cell wall components, referred to as pattern recognition receptors (PRRs), most certainly evolved under the selective pressure to rapidly detect bacterial encounters. Nucleic acids, in contrast, are not exclusively bacterial and can be found in all living cells and viruses. Thus, while PRRs that detect nucleic acids can detect bacteria, the true selective pressure that drove the evolution of these receptors may have been based on the need to detect viral infection. There is one exception to this statement, as recent studies revealed that a specific sequence found in bacterial rRNA is a potent inducer of host defenses (1, 2). This rRNA sequence is highly conserved and is not found in eukaryotic rRNAs or viral genomes. Therefore, the receptors that detect bacterial rRNA likely evolved to detect these microorganisms.
TABLE 1.
Mammalian PRRs and their targetsa
| Family | Sensor | PAMP | Localization |
|---|---|---|---|
| TLR | TLR2/1, TLR2/6 | Lipoproteins | Plasma membrane |
| TLR4 | LPS | Plasma membrane | |
| TLR3 | dsRNA | Endosomes | |
| TLR5 | Flagellin | Plasma membrane | |
| TLR7 | ssRNA | Endosomes | |
| TLR8 | ssRNA | Endosomes | |
| TLR9 | CpG DNA | Endosomes | |
| TLR13 | rRNA with specific sequence | Endosomes | |
| NLR | Nod1 | iE-DAP | Cytosol |
| Nod2 | Muramyl dipeptide | Cytosol | |
| NLRP3 | Peptidoglycan | Cytosol | |
| NAIP2 | T3SS rod | Cytosol | |
| NAIP5 | Flagellin | Cytosol | |
| NAIP6 | Flagellin | Cytosol | |
| CGAS | DNA | Cytosol | |
| ALR | AIM2 | DNA | Cytosol |
| RNA | RIG-I | Short dsRNA with 5′ terminus | Cytosol |
| MDA5 | Long branched chains of dsRNA | Cytosol |
ALR, AIM2-like receptor; ss, single stranded; ds, double stranded; iE-DAP, γ-d-glutamyl-meso-diaminopimelic acid.
The evolutionary focus of this discussion prompts consideration of the selective pressures placed on hosts and bacteria that directed the design of PRRs and their aforementioned microbial ligands. These microbial ligands are collectively referred to as pathogen-associated molecular patterns (PAMPs) (Table 1). Within the human genome, there are genes encoding proteins that regulate numerous activities, including tissue development, body patterning, and neurological, cardiac, and hormonal activities. The molecular cues (ligands) that control these processes are encoded by the same genome that encodes the receptors and pathways that these cues stimulate. Thus, in developmental and metabolic systems, receptors and ligands are under positive selection to reinforce the fidelity of the network. In contrast, PRRs and their microbial ligands are synthesized by different organisms. Moreover, robust detection of a PAMP by a PRR drives defensive responses that eliminate the bacterium that produced the very ligand that the receptor evolved to detect. Based on the separation of genomes that encode PAMPs and PRRs, and the antagonistic outcome of the receptor-ligand interaction, it is logical that host proteins involved in immune defense are encoded by some of the most rapidly evolving genes present in the human genome. Probably due to the rapid evolving nature of the receptors that inform our understanding of host-bacterium encounters, different multicellular organisms encode different repertoires of PRRs. These receptors, consequently, can induce defensive responses to infection that are tailored to suit the needs of the host. For example, PRRs that operate in humans link microbial detection to the initiation of T and B cell-mediated adaptive immunity (3). As the fruit fly Drosophila melanogaster does not contain T and B cells, PRRs within this organism link bacterial detection to the production of antimicrobial peptides. Despite this diversity of receptor design and defensive strategy, multicellular organisms from every kingdom of life appear to have evolved to use a similar set of PAMPs as indicators of infection. Below, we focus on the best-characterized experimental systems to study PRR activities in the context of bacterial encounters—the human and the mouse. We refer the reader to several excellent reviews that describe host defense strategies that operate in insects and plants (4–6).
HOST-ENCODED SENSORS OF BACTERIAL MOLECULES
Several families of PRRs exist in humans and mice. Within each family are proteins that exhibit significant structural similarity, but there is limited similarity across PRR families. Thus, the unifying feature that links these diverse families of proteins is function—the ability to detect and respond to bacterial encounters. The first-described and best-characterized family of PRRs consists of the Toll-like receptors (TLRs) (7–11). TLRs are type I transmembrane proteins that are structurally characterized as containing a large extracellular leucine-rich repeat domain that recognizes PAMPs and an intracellular signaling domain that shares homology with the intracellular domain of the interleukin 1 (IL-1) receptor and various plant resistance proteins. Consequently, the cytosolic tail of TLRs is classified as a Toll–IL-1 receptor resistance domain (7). Several TLRs are considered specific sensors of bacteria, in that they recognize specific components of the cell wall. These cell wall components include LPSs, which are detected by TLR4 (8–11), lipoproteins (detected by TLR2/1 or TLR2/6 heterodimers) (12, 13), and flagellin (TLR5) (14). TLR9, the receptor for unmethylated CpG-containing DNA, is also implicated in the detection of bacteria (15). However, as described above, TLR9, along with the other nucleic acid-sensing TLRs (TLR3, -7, and -8), probably evolved to detect viruses. TLR13 is the most recently defined member of this family and perhaps the most intriguing, in the context of bacterial detection. TLR13 recognizes an RNA sequence that corresponds to that found in bacterial rRNA (1, 2). In contrast, all other RNA-sensing TLRs do not detect their cognate PAMPs in a sequence specific manner. Interestingly, the sequence detected by TLR13 corresponds to the sequence targeted by several naturally occurring fungal metabolites that display potent antibacterial activity. This sequence of RNA is highly conserved across bacteria, which may explain why it is targeted by mammalian TLRs and fungal metabolites. Further evidence in support of the unusual nature of the target of TLR13 is based on the surprising finding that TLR13 is found only in mice. Despite the lack of TLR13, humans mount potent inflammatory responses to the RNA ligands that activate this receptor. Based on these findings, one must conclude that there is substantial selective pressure to detect bacterial encounters, and multiple PRRs likely evolved independently in diverse species to serve this purpose.
Whereas most TLRs detect bacterial PAMPs directly, one notable exception derives from studies of LPS detection by TLR4. TLR4 interacts weakly with LPS, yet picomolar concentrations of this PAMP can induce TLR4-dependent inflammatory responses (16). This high degree of sensitivity to LPS has been explained by the finding that TLR4 is not the sole sensor of extracellular LPS. Indeed, three LPS-binding proteins act upstream of TLR4 to promote high-efficiency detection. The extracellular LPS-binding protein is the first of these receptors to act, as it binds directly to the outer membrane of Gram-negative bacteria, bacterial outer membrane vesicles, or micelles. These interactions somehow disrupt the tight packaging of LPS and facilitate the extraction of a monomer of LPS by the glycosylphosphatidylinositol-anchored protein CD14. CD14 then transfers LPS to the small molecule MD-2, which is constitutively associated with the extracellular domain of TLR4. Only upon binding MD-2 can LPS form contacts with TLR4, which results in its dimerization. The dimerized ectodomain is the first step in the signaling process, whereby the intracellular Toll–IL-1 receptor resistance domains also dimerize and promote NF-κB-dependent inflammatory responses. The directionality of LPS transfer to TLR4 is ensured by the increasing affinity of LPS-binding protein, CD14, and MD-2 for LPS (17–22).
Interestingly, despite the symmetrical operation of many stages of microbial detection and inflammation induction across different PRR families, this complex process of PAMP binding is unique to TLR4. All other well-characterized PRRs detect their ligands directly, with high affinity. One possible explanation why PAMP detection by TLR4 is so complex is based on the nature of the ligand itself. Like other PAMPs derived from the bacterial cell wall, LPS is surface exposed. However, it is not the exposed substructure of LPS that is bound by MD-2 and TLR4, but rather the hydrophobic acyl chains that are buried in the membrane. Thus, the nonexposed structures within LPS must be extracted by CD14 and presented to downstream PRRs. But why extract LPS in the first place? Why not simply couple initial detection of the exposed LPS substructures with inflammation-inducing activities? While the answer to this question is unknown, it is worth noting that the very nature of LPS detection by TLR4 involves a single molecule of LPS being extracted by a bacterium that contains thousands of such molecules. Importantly, it has been estimated that 1,000 molecules of LPS can be extracted from an individual Escherichia coli cell, each of which could (in principle) be used to activate TLR4-induced inflammatory responses on 1,000 different macrophages (16). The use of the term “overwhelming force,” which is often associated with military dominance, is appropriate for this discussion, as we can consider the unusual strategy of LPS detection a means to amplify the number of cells that respond to a single bacterial encounter.
In recent years, new LPS receptors have been identified, as well as new functions for known LPS receptors. For example, caspase-11 in mice and caspase-4 and -5 in humans are now known to bind directly to LPS (23, 24). Recent biophysical analysis illustrated that, unlike the aforementioned receptors that bind monomers of LPS, caspase-4 (and likely the functional orthologues) do not bind monomers. Rather, these caspases appear to bind large aggregates of LPS (25). Additionally, the G-protein-coupled receptor BAI-1 was identified as a protein that binds the surface-exposed core oligosaccharides of LPS. Finally, CD14, in addition to transferring LPS to TLR4, acts to promote endocytosis, thereby promoting the transfer of TLR4 dimers into endosomes, which is an important site of inflammatory signal transduction (26).
Whereas the TLRs are the primary sensors of extracellular bacteria, several PRRs detect cytosolic bacteria. In terms of number of receptors that participate in cytosolic bacteria detection, the nucleotide-binding leucine-rich repeat proteins (NLRs) are most notable. Among the NLRs, NOD1 detects γ-d-glutamyl-meso-diaminopimelic acid (27, 28), NOD2 detects muramyl dipeptide, and NLRP3 detects the activities of the N-acetylglucosamine fragment of peptidoglycan (29, 30). The NLRs NAIP2, -5, and -6 also detect bacterial products directly, with NAIP2 detecting a bacterial secretion system structure and NAIP5 and NAIP6 detecting a substructure within flagellin distinct from that detected by TLR5 (31). Members of the RIG-I-like receptor (RLR) family also detect bacteria, as do the DNA sensors cGAS and AIM2. Each of these receptors surveys the cytosol for nucleic acid ligands. The first-described RLR, RIG-I, detects short double-stranded RNA sequences with a di- or triphosphorylated 5′ terminus, and the RLR MDA5 detects long branched chains of double-stranded RNA (32–36).
Genetic analysis has implicated RLRs in the detection of bacterial infections, most notably those caused by the cytosolic pathogen Listeria monocytogenes (37, 38). Rather than sensing RNA sequences in the cytosol, cGAS and AIM2 detect DNA in this subcellular compartment (39, 40). The presence of DNA in the cytosol is unusual, as all cellular sources of DNA should be confined to the nucleus of mitochondria. cGAS and AIM2 each contain high-affinity DNA-binding domains, which can detect any DNA that leaks into the cytosol, such as during genotoxic stress or mitochondrial dysfunction of bacterial lysis. While bacteriolysis in the cytosol would be considered a rare event, the number of infections known to activate cGAS is increasing. For example, cGAS appears to detect Mycobacterium tuberculosis, Chlamydia species, and Legionella pneumophila (41, 42). Several of the aforementioned studies proposed that the DNA detected during these infections is derived from living (not lysed) intracellular bacteria. At present, it is unclear how DNA would be released from bacteria without lysis, although bacterial type III, IV, and VII secretion systems (T3SS, T4SS, and T7SS) may provide conduits to the cytosol.
HOST-ENCODED SENSORS OF PATHOGEN ACTIVITIES
While most PRRs detect microbes by directly sensing the presence of PAMPs, others detect infection through sensing of activities carried out by bacteria as part of their virulence strategies. The consequences of these activities can be cellular stress, damage, and biochemical modification of host factors. Inflammasomes are sensors that detect pathogen activities, and they are discussed in greater detail in other articles. Of note, the NLRP3 inflammasome detects disturbances in cellular homeostasis, including damage to membranes caused by bacterial toxins and secretion systems, including T3SSs and T4SSs. Secretion systems are a common virulence mechanism of many bacterial pathogens of plants and animals. Bacteria use these organelles to inject factors (known as effector proteins) into target cells. T3SSs and T4SSs are molecular syringes that span bacterial membranes and insert themselves into the host cell membrane (and cell wall in the case of plant cells). The pore formed by the insertion of translocon components into host membranes causes activation of the NLRP3 inflammasome, leading to pyroptosis via caspase-1 activation (43).
In addition to the physical disruption caused by the presence of bacteria and their secretion systems, the biochemical activities of secreted effectors themselves are signals detected by host PRRs. Translocated effectors carry out a number of biochemical functions to manipulate host cell functions, including affecting membrane trafficking, affecting cytoskeletal dynamics, and inhibiting the host immune response. The injection of this arsenal of toxins does not go unnoticed, and we now know that host cells are able to detect certain modification of host targets. This phenomenon of “indirect interaction” was long thought to be unique to plant innate immunity and is often discussed in that context under the name “guard hypothesis.” This theory stipulates that important host proteins that are targeted by bacterial effectors are “guarded” by sensors. Sensor activation leads to the activation of an innate response (4, 44). For example, Arabidopsis thaliana RIN4 is an important negative regulator of plant immunity. It is phosphorylated and inhibited by a number of bacterial factors, including the Pseudomonas syringae effectors AvrB and AvrRpm1 (45). This phosphorylation event is detected by two plant proteins, the guards RPM1 and RPS2, activating an immune response (46–48).
Such a relationship has now been described in metazoans, including insects and vertebrates. In both cases, the guarded targets identified are small GTPases of the Rho family (RhoA, Rac1/2, and Cdc42). Rho GTPases sit at the apex of a number of signaling pathways that regulate critical cellular functions, including cytoskeletal dynamics and activation of inflammation. As such, they are targeted by a range of bacterial factors. For example, E. coli cytotoxic necrotizing factor 1 activates the Rho GTPase Rac2. This modification is detected by the immune adaptor IMD in flies and Rip1-Rip2 in mammalian cells, leading to the expression of proinflammatory cytokines (49). Similarly, the intracellular pathogen Salmonella activates Rac1 and Cdc42 in order to induce its internalization into nonphagocytic cells. This process of Rac1 manipulation is detected by Nod1, in a complex with Hsp90 and Rip2 (50). Finally, RhoA inhibition by a number of pathogens is sensed by the cytosolic protein pyrin, leading to subsequent activation of pyroptosis mediated by the inflammasome (51).
NEUTROPHIL RECRUITMENT TO THE SITES OF INFECTION—A COMMON OUTCOME OF BACTERIAL DETECTION
The varying sensors of bacterial molecules or virulence factor activities lead to the activation of defense responses that can be generally classified as inflammatory (Fig. 1). These responses involve the upregulation of genes that promote numerous aspects of host defense. Many downstream host defense responses are context dependent, but a common response associated with all innate immune responses is the induction of inflammation at the site of bacteria detection. Upon bacterial detection, all of the PRR pathways described herein induce the production of chemokines that promote the recruitment of large numbers of neutrophils to infected tissues. Many neutrophil chemokines are expressed by the action of the transcription factors NF-κB and AP-1, which are commonly activated by transcription-inducing PRR pathways and inflammasome pathways via the release of IL-1 (3, 52). In most cases of bacterial encounters, neutrophils are the first cells recruited from the bloodstream to the infected tissue. Neutrophils are the most potent antibacterial phagocytes known to operate in the human body. Unlike dendritic cells, monocytes, and macrophages, which are also professional phagocytes, neutrophils rarely live longer than 24 hours and are not thought to play a role in antigen presentation to T lymphocytes (53, 54). Rather, the primary function of neutrophils is to kill bacteria and other microorganisms. These cells display several activities supporting such an antibacterial function, including potent phagocytic activity and the ability to produce copious amounts of reactive oxygen species. Neutrophils also contain secretory granules and lysosome-like organelles that are rich in antimicrobial peptides and enzymes that operate to degrade virtually every component of the bacterial cell wall. Finally, neutrophils are adept at undergoing an unusual form of cell death known as NETosis, where the cells release genomic DNA into the extracellular space to ensnare bacteria. Not only do these DNA-rich nets capture bacteria and prevent bacterial spread, but the associated histones display potent antibacterial activity (55, 56).
FIGURE 1.
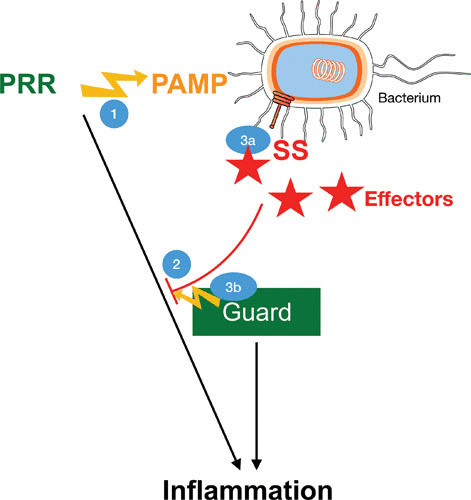
Evolutionary arms race between the host immune system and bacterial virulence mechanisms. (1) PRRs recognize PAMPs and activate conserved signaling pathways that induce transcriptional activation of inflammation with expression of cytokines and interferons. (2) Pathogenic bacteria use secretion systems (SS) to inject effector proteins into host cells. The diverse functions of these effectors include the ability to block different components of PRR-induced pathways. (3) The activities of pathogenic bacteria are detected by guard proteins. In vertebrates, these are mostly NLRs and other inflammasome stimulators. They detect the presence of T3SSs and T4SSs, usually through the formation of pores in host membranes (3a) and posttranslational modification of host proteins (3b). Consequently, NLRs and inflammasomes induce inflammation at the transcriptional and posttranslational levels through caspase-dependent activation of pyroptosis.
It is worth noting that several of the activities described here are also observed in other phagocytic cells, such as the ability to produce reactive oxygen species and ingest bacteria. However, these antibacterial activities appear to be most active in neutrophils and, in the case of NETosis, appear to be unique to this cell type. When this is considered in the context of the rapid accumulation of large numbers of these cells in infected tissues, it is reasonable to suggest that the biggest threat to bacterial survival in the host is the antimicrobial activities of neutrophils. Consistent with this suggestion are numerous findings that bacteria that have the ability to replicate within other phagocytes are readily killed by neutrophils. Thus, the mechanisms that link bacterial detection by PRRs to antibacterial activity of neutrophils are of prime importance for host defense. Other aspects of immunity, such as the activation of antigen-specific T lymphocytes and production of antigen-specific antibodies, are also important, but these activities are induced after neutrophil-mediated activities are mobilized to contain the infection.
PERSPECTIVES
In this essay, we discuss several means by which mammalian cells detect and respond to bacterial encounters. These means include the use of structurally diverse receptors and mechanistically distinct signaling pathways. Despite this diversity, the evolutionary pressures to develop these detection systems are likely constant, as they must meet the need to ensure host survival by preventing bacterial replication. It is worth noting that much of our knowledge in this area derives from studies that have focused on the interactions between bacteria and phagocytes or fibroblasts, neither of which are the first cells to encounter bacteria in the natural course of events. Epithelial cells in the skin, airway, and digestive tract represent the barrier to the environment. As such, these cells are poised for rapid detection of bacterial infection, and the innate immune pathways that operate in epithelial cells likely influence many downstream innate and adaptive responses to infection. Indeed, there is ample evidence that epithelia are the first cells that experience the activities of bacterial T3SSs, in particular, those involved in invasion. Studies over the next several years will likely expand our knowledge of the innate immune system in phagocytes and fibroblasts to include similar inquiries into the biology of barrier tissues. This broadening of the scope of host-microbe analysis will allow the classification of innate immune responses as being either common to all cell types or tissue specific. The emergence of human and mouse organoid models that represent an increasingly diverse set of tissues should facilitate such inquiries and may allow a more detailed view of the earliest stages of any host-bacterium interaction to emerge.
ACKNOWLEDGMENTS
We thank the members of the Kagan and Odendall labs for helpful discussions. This work was supported by NIH grants AI133524, AI093589, AI116550, and P30DK34854 to J.C.K. J.C.K. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. C.O. is supported by a King’s College London Prize Fellowship and a Sir Henry Dale Fellowship from the Royal Society and the Wellcome Trust (grant number 206200/Z/17/Z).
REFERENCES
- 1.Oldenburg M, Krüger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, Bathke B, Lauterbach H, Suter M, Dreher S, Koedel U, Akira S, Kawai T, Buer J, Wagner H, Bauer S, Hochrein H, Kirschning CJ. 2012. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337:1111–1115 10.1126/science.1220363. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Li X-D, Chen ZJ. 2012. Sequence specific detection of bacterial 23S ribosomal RNA by TLR13. eLife 1:e00102 10.7554/eLife.00102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palm NW, Medzhitov R. 2009. Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227:221–233 10.1111/j.1600-065X.2008.00731.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Jones JDG, Dangl JL. 2006. The plant immune system. Nature 444:323–329 10.1038/nature05286. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Padmanabhan M, Cournoyer P, Dinesh-Kumar SP. 2009. The leucine-rich repeat domain in plant innate immunity: a wealth of possibilities. Cell Microbiol 11:191–198 10.1111/j.1462-5822.2008.01260.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchon N, Silverman N, Cherry S. 2014. Immunity in Drosophila melanogaster—from microbial recognition to whole-organism physiology. Nat Rev Immunol 14:796–810 10.1038/nri3763. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunne A, O’Neill LAJ. 2003. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE 2003:re3. [DOI] [PubMed] [Google Scholar]
- 8.Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397 10.1038/41131. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088 10.1126/science.282.5396.2085. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Poltorak A, Smirnova I, He X, Liu MY, Van Huffel C, Birdwell D, Alejos E, Silva M, Du X, Thompson P, Chan EK, Ledesma J, Roe B, Clifton S, Vogel SN, Beutler B. 1998. Genetic and physical mapping of the Lps locus: identification of the Toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis 24:340–355 10.1006/bcmd.1998.0201. [DOI] [PubMed] [Google Scholar]
- 11.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:3749–3752. [PubMed] [Google Scholar]
- 12.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451 10.1016/S1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 13.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. 1999. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285:736–739 10.1126/science.285.5428.736. [PubMed] [DOI] [PubMed] [Google Scholar]
- 14.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103 10.1038/35074106. [PubMed] [DOI] [PubMed] [Google Scholar]
- 15.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745 10.1038/35047123. [PubMed] [DOI] [PubMed] [Google Scholar]
- 16.Gioannini TL, Weiss JP. 2007. Regulation of interactions of Gram-negative bacterial endotoxins with mammalian cells. Immunol Res 39:249–260 10.1007/s12026-007-0069-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Tobias PS, Soldau K, Ulevitch RJ. 1986. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med 164:777–793 10.1084/jem.164.3.777. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. 1990. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249:1431–1433 10.1126/science.1698311. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Tobias PS, Soldau K, Kline L, Lee JD, Kato K, Martin TP, Ulevitch RJ. 1993. Cross-linking of lipopolysaccharide (LPS) to CD14 on THP-1 cells mediated by LPS-binding protein. J Immunol 150:3011–3021. [PubMed] [Google Scholar]
- 20.Haziot A, Ferrero E, Köntgen F, Hijiya N, Yamamoto S, Silver J, Stewart CL, Goyert SM. 1996. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 4:407–414 10.1016/S1074-7613(00)80254-X. [DOI] [PubMed] [Google Scholar]
- 21.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782 10.1084/jem.189.11.1777. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viriyakosol S, Tobias PS, Kitchens RL, Kirkland TN. 2001. MD-2 binds to bacterial lipopolysaccharide. J Biol Chem 276:38044–38051. [DOI] [PubMed] [Google Scholar]
- 23.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, Forsberg LS, Carlson RW, Dixit VM. 2013. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341:1246–1249 10.1126/science.1240248. [PubMed] [DOI] [PubMed] [Google Scholar]
- 24.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. 2013. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341:1250–1253 10.1126/science.1240988. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, Martin A, Di Lorenzo F, Py BF, Molinaro A, Henry T. 2018. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat Commun 9:242 10.1038/s41467-017-02682-y. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC. 2011. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147:868–880 10.1016/j.cell.2011.09.051. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, Ogura Y, Kawasaki A, Fukase K, Kusumoto S, Valvano MA, Foster SJ, Mak TW, Nuñez G, Inohara N. 2003. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 4:702–707 10.1038/ni945. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Girardin SE, Boneca IG, Carneiro LAM, Antignac A, Jéhanno M, Viala J, Tedin K, Taha M-K, Labigne A, Zähringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. 2003. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587 10.1126/science.1084677. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nuñez G. 2003. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 278:5509–5512 10.1074/jbc.C200673200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 30.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. 2003. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278:8869–8872 10.1074/jbc.C200651200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Kofoed EM, Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595 10.1038/nature10394. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang D-C, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. 2002. mda-5: an interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci USA 99:637–642 10.1073/pnas.022637199. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737 10.1038/ni1087. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C-S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105 10.1038/nature04734. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann K-K, Schlee M, Endres S, Hartmann G. 2006. 5′-triphosphate RNA is the ligand for RIG-I. Science 314:994–997 10.1126/science.1132505. [PubMed] [DOI] [PubMed] [Google Scholar]
- 36.Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001 10.1126/science.1132998. [PubMed] [DOI] [PubMed] [Google Scholar]
- 37.Hagmann CA, Herzner AM, Abdullah Z, Zillinger T, Jakobs C, Schuberth C, Coch C, Higgins PG, Wisplinghoff H, Barchet W, Hornung V, Hartmann G, Schlee M. 2013. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS One 8:e62872 10.1371/journal.pone.0062872. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, Boulant S, Gehrke L, Cossart P, Kagan JC. 2014. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol 15:717–726 10.1038/ni.2915. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518 10.1038/nature07725. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791 10.1126/science.1232458. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prantner D, Darville T, Nagarajan UM. 2010. Stimulator of IFN gene is critical for induction of IFN-beta during Chlamydia muridarum infection. J Immunol 184:2551–2560 10.4049/jimmunol.0903704. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lippmann J, Müller HC, Naujoks J, Tabeling C, Shin S, Witzenrath M, Hellwig K, Kirschning CJ, Taylor GA, Barchet W, Bauer S, Suttorp N, Roy CR, Opitz B. 2011. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol 13:1668–1682 10.1111/j.1462-5822.2011.01646.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. 2010. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7:376–387 10.1016/j.chom.2010.04.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marathe R, Dinesh-Kumar SP. 2003. Plant defense: one post, multiple guards?! Mol Cell 11:284–286 10.1016/S1097-2765(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 45.Lee D, Bourdais G, Yu G, Robatzek S, Coaker G. 2015. Phosphorylation of the plant immune regulator RPM1-INTERACTING PROTEIN4 enhances plant plasma membrane H+-ATPase activity and inhibits flagellin-triggered immune responses in Arabidopsis. Plant Cell 27:2042–2056 10.1105/tpc.114.132308. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung E-H, da Cunha L, Wu A-J, Gao Z, Cherkis K, Afzal AJ, Mackey D, Dangl JL. 2011. Specific threonine phosphorylation of a host target by two unrelated type III effectors activates a host innate immune receptor in plants. Cell Host Microbe 9:125–136 10.1016/j.chom.2011.01.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackey D, Belkhadir Y, Alonso JM, Ecker JR, Dangl JL. 2003. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell 112:379–389 10.1016/S0092-8674(03)00040-0. [DOI] [PubMed] [Google Scholar]
- 48.Mackey D, Holt BF III, Wiig A, Dangl JL. 2002. RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell 108:743–754 10.1016/S0092-8674(02)00661-X. [DOI] [PubMed] [Google Scholar]
- 49.Boyer L, Magoc L, Dejardin S, Cappillino M, Paquette N, Hinault C, Charriere GM, Ip WKE, Fracchia S, Hennessy E, Erturk-Hasdemir D, Reichhart J-M, Silverman N, Lacy-Hulbert A, Stuart LM. 2011. Pathogen-derived effectors trigger protective immunity via activation of the Rac2 enzyme and the IMD or Rip kinase signaling pathway. Immunity 35:536–549 10.1016/j.immuni.2011.08.015. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keestra AM, Winter MG, Auburger JJ, Frässle SP, Xavier MN, Winter SE, Kim A, Poon V, Ravesloot MM, Waldenmaier JFT, Tsolis RM, Eigenheer RA, Bäumler AJ. 2013. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 496:233–237 10.1038/nature12025. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong Y-N, Peng X, Xi JJ, Chen S, Wang F, Shao F. 2014. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature 513:237–241 10.1038/nature13449. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Vijay K. 2018. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol 59:391–412. CORRIGENDUM Int Immunopharmacol 62:338 10.1016/j.intimp.2018.03.002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175 10.1038/nri3399. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Galli SJ, Borregaard N, Wynn TA. 2011. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 12:1035–1044 10.1038/ni.2109. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 56.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. 2012. Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489. [PubMed] [DOI] [PubMed] [Google Scholar]