

Abstract
Controlled generation of cytotoxic agents with near-IR light is a current focus of photoactivated cancer therapy, including that involving cytotoxic platinum species. Herein, we present a novel heptamethine cyanine scaffolded Pt(II) complex, IR797-Platin (1), which exhibits unprecedented Pt-O bond scission and enhancement in DNA platination in near-IR light. Complex 1 also displayed significant singlet oxygen quantum yield thereby qualifying as a near-IR photodynamic therapeutic agent. Complex 1 showed 30–60 fold enhancements of cytotoxicity in near-IR light in various cancer cell lines. The cellular imaging properties of 1 were also leveraged to observe its significant co-localization in cytoplasmic organelles. This is the first demonstration of a near-IR light-initiated therapy involving the cytotoxic effects of both active cisplatin and singlet oxygen.
Keywords: Platinum, photodynamic therapy, cancer, cyanines, imaging agents
Graphical Abstract
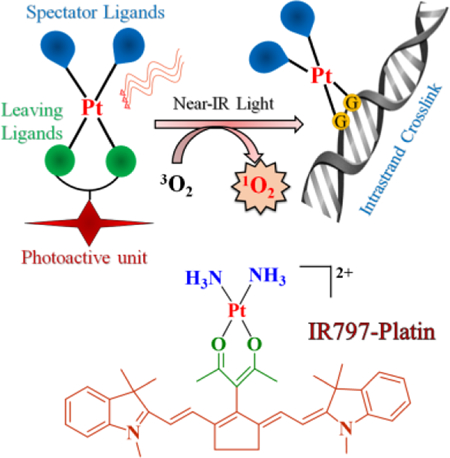
A platinum(II) complex of heptamethine cyanine, IR797-Platin, showed near-IR light induced Pt-O bond dissociation leading to enhanced DNA platination. IR797-Platin also showed significant singlet oxygen generation which resulted in mitochondria-targeted PDT effects in cancer cells.
The FDA validated platinum based anticancer drugs cisplatin, oxaliplatin and carboplatin form the centerpiece of metal-based anticancer drug therapy.[1] The putative mechanism of cytotoxicity relies on an initial Pt-X (X = Cl, O) bond scission to form aquated Pt(II) species. These activated Pt(II) species can then react with nuclear DNA and form cyto-lethal Pt-DNA crosslinks ultimately leading to apoptotic cell death.[2] However, the anarchic hydrolytic behavior of these drugs leads to undesired systemic toxicity and limits the administration of higher drug-dosage levels. Controlled generation of the bioactive (aquated) form of platinum(II) drugs specifically within the targeted tumor is, therefore, an ideal way to prevent the off-target toxicity.[3] To gain control over the kinetics of metal-ligand bond rupture, Pt(IV) complexes were introduced as inert prodrugs which produce DNA damaging platinum(II) species only upon reduction by cell-abundant glutathione.[4,5] These Pt(IV) complexes showed promising tumor-targeted anticancer activities and oftentimes resulted in delivery of multiple drugs and adjuvants. Though efficacious, this strategy relies on endogenous entities for drug activation and thus the regulation of cytotoxicity is lost once the drug is administered. In another approach, platinum(II) cytotoxins were generated from photosensitive Pt(IV) diazido molecules with light.[6] The exploitation of exogenous light for uncaging “active” chemotherapeutics offers a spatio-temporal control over drug activation.[7–9] Such photo-initiated chemotherapy has advantages over clinically established photodynamic therapy (PDT), which relies on generation of cytotoxic singlet oxygen and therefore fails in the treatment of deep-seated hypoxic tumors.[10]
Recently, there have been a few successful efforts to photo-release bioactive Pt(II) species.[11a] Carboplatin and its 7-azaindole based analogues were shown to form bifunctional Pt-DNA adducts on UVA light exposure.[11b,c] Another similar example constitutes the novel Pt(II) complexes of curcumin which demonstrated visible light enhanced formation of Pt-DNA crosslinks.[11d,e] However, these approaches are limited by i) collateral tissue damage caused by UVA light and ii) the poor tissue penetration of light at these wavelengths.[8e] Thus, an ideal Pt(II) based photo-initiated chemotherapeutic agent should be activated in the “biological window” of 650–850 nm which has improved tissue compatibility and penetrating properties.[7, 8e]
From these observations, we reasoned that it might be possible to release a bioactive Pt(II) species from its complex with a ligand that absorbs strongly in the near-IR region. Among potential organic chromophores, the heptamethine cyanine dyes have strong near-IR absorption, are easy to access synthetically, and are widely used as imaging and PDT agents.[12, 13] Moreover, these cyanine dyes accumulate preferentially in the mitochondria and lysosomes. Targeting these cytoplasmic organelles is known to overcome drug-resistance and afford improved cytotoxicity of traditionally used anticancer agents.[14, 15]
Based on the precedent of existing platinum(II) photoactivatable complexes,[11a] we posited that a diammine bound Pt(II) system having a photodetachable O^O bidentate donor in direct conjugation with the heptamethine cyanine framework would release an active cisplatin on near-IR light exposure. To realize our hypothesis, we successfully designed a Pt(II) conjugate of heptamethine cyanine, which we call IR797-Platin, 1 (Figure 1a). Herein, we describe the synthesis and evaluation of photo-enhanced DNA binding properties and cellular cytotoxicity of 1.
Figure 1.
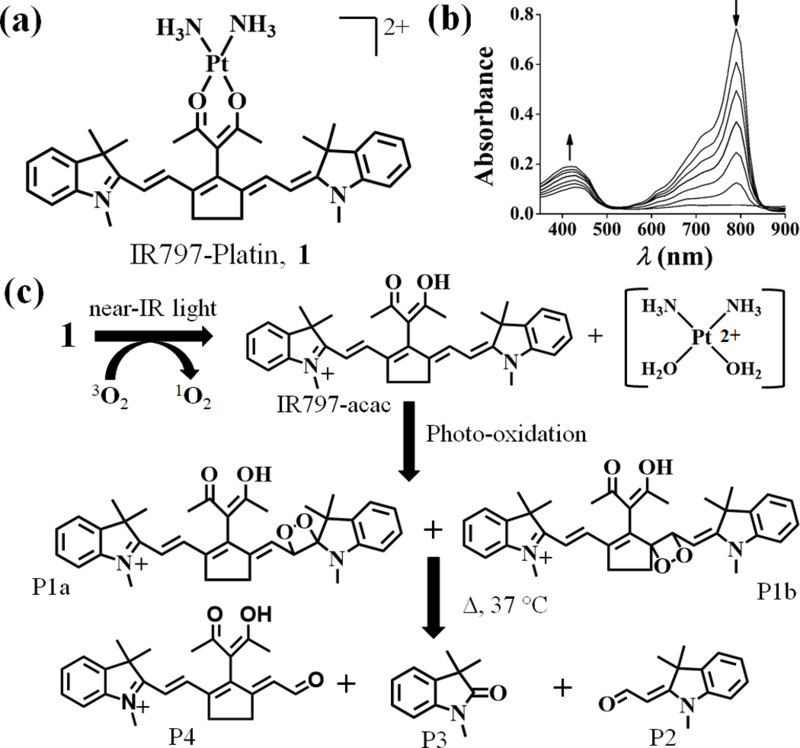
(a) Chemical structure of cationic IR797-Platin, 1. (b) Absorption spectral traces of complex 1 (3 μM in 0.1% DMSO-PBS) exposed to light (readings at 30 sec and 1 min intervals) showing decrease in intensity at 790 nm and increase in intensity at 430 nm. (c) Proposed mechanism of photo-degradation of 1 when exposed to near-IR light (panel of LEDs, 720–740 nm, 3.5 ±1.5 mW/cm2).
The new acetylacetone (Hacac) derivative of heptamethine cyanine, IR797-acac was successfully synthesized in one step from a chloro cyanine precursor (Figure S1a, Supporting information). IR797-Platin, 1 was prepared by reacting this ligand with cisplatin after pre-activation with AgNO3. To understand the role of the organic chromophore and light, we also prepared a cyanine-lacking analog, [Pt(NH3)2(acac)](NO3)], 2 and used it as a control in our experiments (Figure S1b, Supporting information). The newly synthesized compounds were characterized by analytical and spectroscopic techniques (Figures S2-S8, Supporting information). 1H NMR spectra of 1 displayed signals assignable to the cyanine scaffold (Figure S5, Supporting information). A notable upfield shift of 0.3 ppm observed for the methyl protons of acac in 1 supported coordination of the oxygen atoms to Pt(II) center. A single mass spectra signal at m/z value of 380.6 with a Pt isotopic distribution pattern and a bi-positive charge of the fragment implicated the successful formation of 1 (Figure S7, Supporting information). The purity of 1 was also verified using analytical HPLC (Figure S8, Supporting information).
Complex 1 and ligand IR797-acac showed comparable absorption profiles with a maxima at 790 nm and a high molar extinction coefficient (ε) of 4–5 ×104 M−1.cm−1 (Figure S9a, Table S1, Supporting information). Such strong near-IR absorptions are rare for metal complexes.[8,9] It was previously noted that the N or O substitution in heptamethine cyanine dyes resulted in a large absorption blue-shift of ~100 nm.[16a,b] Interestingly, the C-substituted variants, IR797-acac and 1 retained the maximum absorbance in the near-IR region. The free ligand showed intense emission at 825 nm (φf = 0.53) reflecting a characteristic small Stokes shift of 25 nm (Figure S9b, Table S1, Supporting information). Complex 1 imitated the emissive features of IR797-acac (λemi = 820 nm, φf = 0.32), but showed partial quenching due to the diamagnetic Pt(II) center.
Near-IR light-triggered uncaging of C4’-amine-substitued cyanine analogs was recently demonstrated by Schnermann and co-workers.[16] This finding, coupled with the observations suggesting that the Pt(II)-O bonds are photolabile,[11] prompted us to examine the photo-degradation pathways of 1. We performed the stability studies in solvents relevant to cell-culture settings i.e. 1% DMSO in PBS or DMEM at biological pH of 7.4. The photo reactivity of 1 and IR797-acac was demonstrated by the rapid decrease in absorbance at 790 nm and a concomitant gradual increase at 410 nm when exposed to near-IR light (Figure 1b, Figure S10, Supporting information). The half-lives (t1/2L) of photodecomposition were found to be ~ 40 secs and 2 min for IR797-acac and 1 respectively. They exhibited considerable stability in dark conditions with t1/2D of ~ 10 h and 20 h respectively (Figure S11, Table S1, Supporting information).
The light affected reaction course was discerned from LC-MS chromatographic elution of the photo-exposed solutions of 1 at different time intervals. To our gratification, we observed the appearance of a new signal (m/z of 533.3) which corresponds to the free ligand IR797-acac within a brief irradiation time (Figure S12, Supporting information). A portion of this photo-exposed sample when subjected to HPLC depicted the appearance of a peak which was collected and characterized as free IR797-acac (inset of Figure S12, Supporting information). This clearly indicates the Pt-O bond in 1 rapidly dissociates and liberates the free ligand on near-IR light exposure (Figure 1c). On further irradiation, an increase in relative counts at m/z of 565.3 was observed (Figures S13, Supporting information). This signal is attributed to the isomeric 1,2-dioxetanes, P1a and P1b and is consistent with oxidative reaction of released IR797-acac and singlet oxygen.[16b] On warming the solutions to 37 °C for 1 h, new m/z signals at 202.2, 176.1 and 390.2 were seen which are assignable to the smaller degraded products P2-P4 (Figures S14-S16, Supporting information). Irradiation of 1 also lead to a peak of m/z 396.5 which is consistent with 1,2-dioxetane (P5a/ P5b) formation (Figure S17, Supporting Information). In the absence of light, 1 showed no decomposition (Figure S18a, Supporting information). The accumulation and disappearance of these products over time, as discerned from a light-dose-dependent LC-MS analysis, helped us to propose the sequential photolytic scheme (Figure 1c, Figure S18b, Supporting information). 1H NMR studies of 1 and IR797-acac further demonstrated the formation of similar photoproducts on near-IR light exposure (Figures S19, S20, Supporting information).[16a] The importance of singlet oxygen for degradation was confirmed from retention of absorption and emission intensity when irradiated under inert atmosphere or in presence of NaN3, a singlet oxygen quencher (Figure S21, Supporting information). When the same set of experiments was carried out with IR797-acac, we also observed formation of P1-P4 (Figure S22, Supporting information) in accordance with the known photooxidative degradation of cyanine dyes.[16b]
The widely perceived mechanism holds that “bioactive Pt(II) DNA-philes” are responsible for cisplatin cytotoxicity.[1b, 2] In order to track the fate of the released Pt(II) species upon irradiation of 1, we chose to perform the release experiments in the presence of pyridine (py), a simple monodentate N donor which should trap any reactive Pt(II) species. A solution containing 1 (20 μM) and pyridine (50 μM) was exposed to near-IR light and subjected to mass spectral analysis. Predominant peaks at m/z of 193.6 and 256.4 assignable to [Pt(NH3)2(py)2]2+, P6, and [Pt(py)4]2+, P7, fragments were duly noted only in light exposed solutions (Figure 2a, Figure S23, Supporting information). Formation of P6 and P7 were not observed in light-unexposed controls. The formation of these Pt-pyridine adducts therefore likely proceeded via an initial diaqua diammine Pt(II) species. These reactive Pt(II) species also formed covalent adducts with ctDNA which increased over time. ICP-MS measurements revealed enhanced platination of isolated ctDNA (2–7 ng of Pt/ μM of ctDNA) when complex 1 (50 μM) was exposed to near-IR light compared to controls in the dark (Table S2, Supporting information). As expected, we did observe slow incorporation of Pt(II) into DNA upon extended incubation of ctDNA with 1 or 2 in the dark (Table S2, Supporting Information). The DNA melting experiments and Hoechst displacement assays revealed 1 and IR797-acac interact with the minor groove of ctDNA in the dark (Figure S24, Supporting information).[17a]
Figure 2.
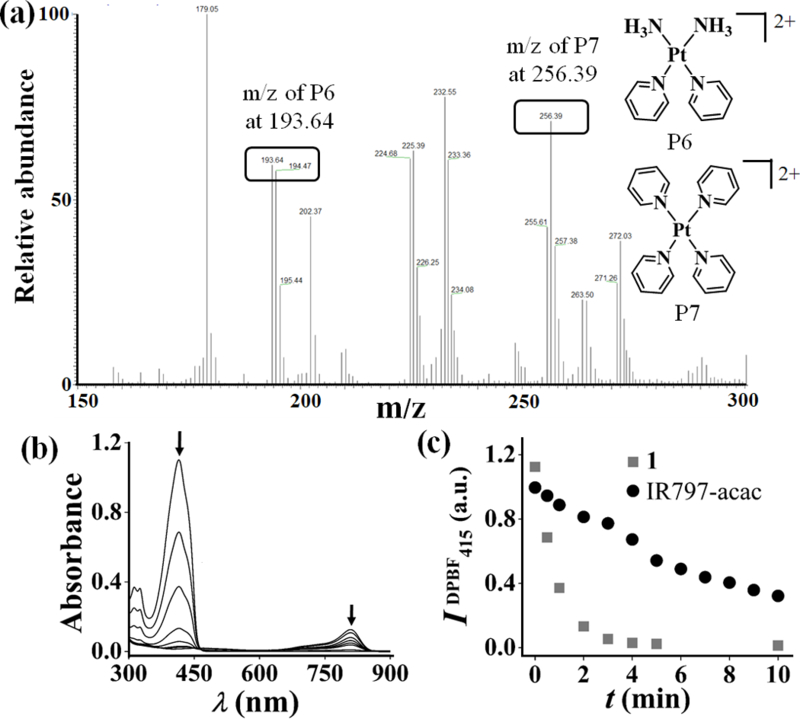
(a) ESI-MS of photolysed samples of 1 and pyridine showing m/z peaks assignable to P6 and P7 (for full spectra, vide SI). (b) Decrease in absorbance of DPBF treated with complex 1 (3 μM in 0.1% DMSO-PBS) and irradiated with light of 720–740 nm (first reading at 30s and then at intervals of 1 min). (c) Scatter plot comparing decrease in absorbance of DPBF for 1 (gray squares) and IR797-acac (black circles) at 415 nm with time of irradiation in near-IR light.
Unmodified heptamethine cyanines display poor singlet oxygen generation quantum yields.[17b] However, heavy atoms when aptly incorporated in a fluorogenic scaffold tend to increase the singlet oxygen generation via populated triplet excited states.[13b,18] The near-IR light induced rapid decrease in the absorbance of 1,3-diphenylisobenzofuran (DPBF) at 415 nm in presence of complex 1 (t1/2DPBF = 30 sec, φΔ = 0.15) demonstrated the generation of singlet oxygen (Figure 2b and c, Figure S25, Supporting information). In comparison, free IR797acac showed much weaker singlet oxygen production (t1/2DPBF = 6 min, φΔ = 0.017). Therefore, complex 1 is expected to have dual-modes of cytotoxicity through reactive Pt(II) and PDT.
The cleavage of Pt(II)-O bonds with near-IR light is unprecedented. To better understand this process, we performed theoretical calculations. DFT (density functional theory) optimized geometry of 1 revealed a square planar Pt(II) and one of the indole rings in a parallel orientation with the acac moiety (Figure 3a, Tables S3). The computed transitions showed that near-IR excitation of 1 involves charge transfer from orbitals having significant localization on both cyanine and Pt(II) (Figure 3b-d, Table S4, Supporting information). Characterization of the photo-dissociating excited triplet state depicted involvement of dσ* orbitals of Pt and elongation of the Pt(II)-O and Pt(II)-N bonds by ~0.15 Å in the optimized geometry (Figure S26, Table S5, Supporting information). This rationalizes the observed photo-induced bond-dissociation in 1.[19] Also predicted transitions of the 1,2-dioxetanes (P1a/ P1b) resulted in weak near-IR absorption which indicates that they can only thermally degrade to form the products P2-P4 (Figure S27, Table S6, Supporting information).
Figure 3.
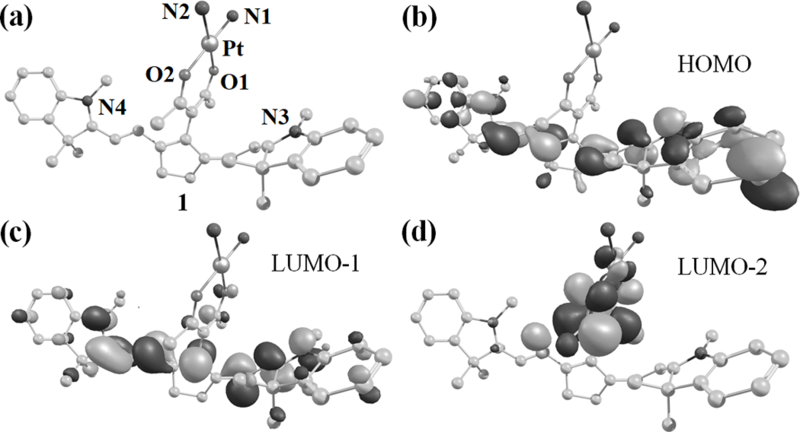
Computational studies using B3LYP/LANL2DZ (for Pt) and 6–31+G (for O,N,C and H) functionals performed on 1 to obtain (a) energy-minimized structure and (b-d) the electronic distributions of molecular orbitals involved in transitions in the near-IR region. The heteroatoms are labelled and hydrogen atoms are omitted for clarity.
Encouraged by the ability of 1 to both produce singlet oxygen and release DNA-binding Pt(II) species in near-IR light, we next assessed the cytotoxic effects of 1 in a panel of human cancer cell lines (Figure 4a and b, Table 1, Figures S28-S32, Table S7, Supporting information). Complex 1 exhibited excellent near-IR light-mediated cytotoxicity (IC50 = 0.12–2 μM) with a 30–60-fold enhancement in potency with light (Figure 4a and b, Table 1). The toxicity caused by 1 in unexposed cells was comparable to 2 and cisplatin under similar conditions. Consistent with other heptamethine cyanine dyes, IR797-acac was toxic on its own. [16b] However, neither IR797-acac alone or in combination with cisplatin showed much enhancement in potency with light. (Table 1 and S7, Supporting information). The cytotoxic activity of 1 relied on light-induced reactive oxygen species, which initiated cellular apoptosis as demonstrated by DCFDA (dichlorofluorescein diacetate) and annexinV-FITC (fluorescein isothiocyanate) assays (Figures S33, S34, Supporting information). Confocal microscopic experiments showed notable co-localization of 1 in the mitochondria and lysosomes (Figure 4c, Figure S35 Supporting information). This emphasizes the important role of heptamethine cyanine backbone which successfully directs 1 towards the cytoplasmic organelles. Furthermore, 1-treated and light-exposed cells had higher Pt content (quantified by ICP-MS) in the nuclear fractions (Table S8, Supporting information) as compared to dark controls. This supports light-promoted release and migration of active Pt(II) species to the nuclear DNA following a similar mechanism known for cisplatin, carboplatin and oxaliplatin.[2]
Figure 4.
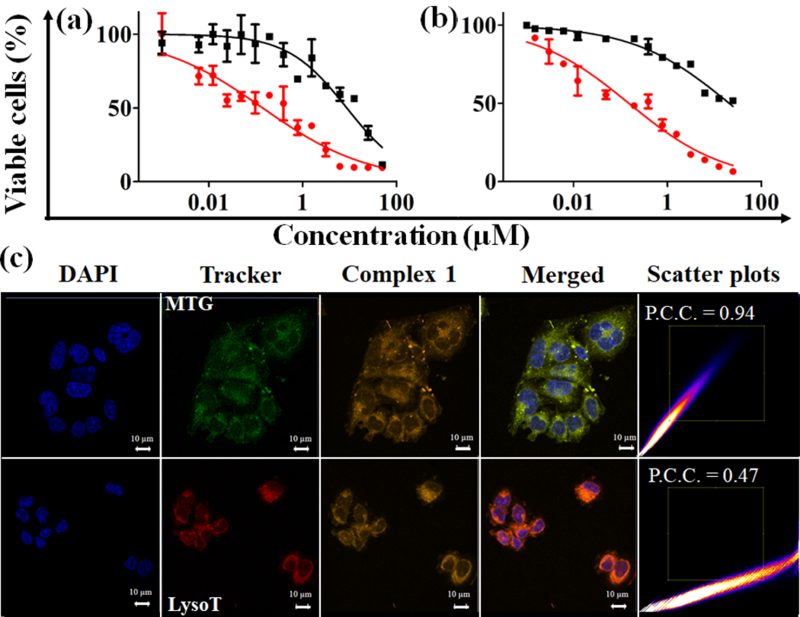
Cell viability plots showing % viability as obtained in 1 treated (a) C-33 A and (b) MCF-7 cells for 4 h in dark and either photoexposed (near-IR light, 45 mins, red circles) or unexposed (dark, black squares) conditions. c) Confocal microscopic images in C-33 A cells showing DAPI(4’,6-diamidino-2-phenylindole, nuclear stain) in 1st column, trackers (Mito-Tracker® Green, 1st row; Lyso Tracker® Red DND-99, 2nd row) in 2nd column, complex 1 in 3rd column and merged of all three channels in 4th column. The near-IR emission of complex 1 is reproduced in orange colour. Scale bar = 10 μm. The scatter plots show degree of overlap between images in 2nd and 3rd column. Higher Pearson correlation coefficient (P.C.C.) indicates higher co-localization.
Table 1.
IC50 values (μM) of compounds in light and dark[a]
| Compound | C-33 A | MCF-7 | ||
|---|---|---|---|---|
| Light[b] | Dark | Light[b] | Dark | |
| Complex 1 | 0.14 ± 0.05 | 8.4 ± 1.4 | 0.65 ± 0.23 | 18.2 ± 3.2 |
| IR797-acac | 0.30 ± 0.07 | 1.2 ± 0.1 | 4.7 ± 0.8 | 5.9 ± 0.3 |
| CP[c] | 16.1 ± 1.5 | 17.7 ± 1.5 | 22.5 ± 3.2 | 25.0 ± 2.2 |
| CP + IR797-acac | 0.25 ± 0.15 | 1.4 ± 0.8 | 0.88 ± 0.17 | 2.6 ± 1.4 |
Cells treated with compounds for 4 h in the dark.
Cells treated with compounds for 4 h in dark and exposed to light (45 mins, 720–740 nm, 3.5 ± 1.5 mW.cm−2).
Cells incubated with cisplatin (CP) for 48 h in the dark.
In conclusion, here we demonstrate both active Pt(II)-and PDT-mediated cytotoxicity in near-IR light of a new Pt(II)-cyanine conjugate. The dual roles of platinum in triggering singlet oxygen production and bond-dissociation offer an innovative strategy for simultaneous realization of PDT and uncaging of anticancer agents. Moreover, the retention of strong absorption and emission features in the near-IR region in these C-substituted heptamethine cyanines makes them attractive theranostic agents for clinical applications. Also, the subcellular distribution of 1 is important considering the recent paradigm shift for targeting anticancer agents to cytoplasmic organelles. We are hopeful that our findings will open up heretofore unexplored avenues of metal-based photo-chemotherapeutics offering non-surgical and externally regulated modalities for cancer treatment.
Supplementary Material
Acknowledgements
This research was supported by grant funding from Virginia’s Commonwealth Health Review Board (236-03-16). The ESI instrument is part of the MCC Proteomics Resources supported by CCSG grant NCI 5P30CA16059-35. The authors thank Mr. N. A. Abrigo, Dr. F. White, Dr. J. Turner and Dr. R. Cao for their help with HPLC, confocal, ICP-MS and lifetime measurements.
References
- [1].a) Johnstone TC, Suntharalingam K, Lippard SJ, Chem. Rev 2016, 116, 3436–3486; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bruno PM, Liu Y, Park GY, Murai J, Koch CE, Eisen TJ, Pritchard JR, Pommier Y, Lippard SJ, Hemann MT, Nat. Med 2017, 23, 461–471; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Harper BW, Krause-Heuer AM, Grant MP, Manohar M, Garbutcheon-Singh KB, Aldrich-Wright JR, Chem. Eur. J 2010, 16, 7064–7077; [DOI] [PubMed] [Google Scholar]; d) Mjos KD, Orvig C, Chem. Rev 2014, 114, 4540–4563; [DOI] [PubMed] [Google Scholar]; e) Wang X, Wang X, Guo Z, Acc. Chem. Res 2015, 48, 2622–2631. [DOI] [PubMed] [Google Scholar]
- [2].a) Wang D, Lippard SJ, Nat. Rev. Drug Discov 2005, 4, 307–320; [DOI] [PubMed] [Google Scholar]; b) Gibson D, Dalton Trans 2009, 10681–10689. [DOI] [PubMed]
- [3].a) Ankenbruck N, Courtney T, Naro Y, Deiters A, Angew. Chem. Int. Ed 2018, 57, 2–33; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) MacEwan SR, Chilkoti A, Angew. Chem. Int. Ed 2017, 56, 6712–6733; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mahato R, Tai W, Cheng K, Adv. Drug Deliv. Rev 2011, 63, 659–670; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kratz F, Müller IA, Ryppa C, Warnecke A, ChemMedChem 2008, 3, 20–53. [DOI] [PubMed] [Google Scholar]
- [4].a) Awuah SG, Zheng Y-R, Bruno PM, Hemann MT, Lippard SJ, J. Am. Chem. Soc 2015, 137, 14854–14857; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zheng Y-R, Suntharalingam K, Johnstone TC, Lippard SJ, Chem. Sci 2015, 6, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Basu U, Banik B, Wen R, Pathak RK, Dhar S, Dalton Trans 2016, 45, 12992–13004; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wexselblatt E, Yavin E, Gibson D, Angew. Chem. Int. Ed 2013, 52, 6059–6062; [DOI] [PubMed] [Google Scholar]; c) Gaviglio L, Gross A, Metzler-Nolte N, Ravera M, Metallomics 2012, 4, 260–266; [DOI] [PubMed] [Google Scholar]; d) Novohradsky V, Zanellato I, Marzano C, Pracharova J, Kasparkova J, Gibson D, Gandin V, Osella D, Brabec V, Sci. Rep 2017, 7, 3751–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Müller P, Schröder B, Parkinson JA, Kratochwil NA, Coxall RA, Parkin A, Parsons S, Sadler PJ, Angew. Chem. Int. Ed 2003, 42, 335–339; [DOI] [PubMed] [Google Scholar]; b) Butler JS, Woods JA, Farrer NJ, Newton ME, Sadler PJ, J. Am. Chem. Soc 2012, 134, 16508–16511; [DOI] [PubMed] [Google Scholar]; c) Farrer NJ, Woods JA, Salassa L, Zhao Y, Robinson KS, Clarkson G, Mackay FS, Sadler PJ, Angew. Chem. Int. Ed 2010, 49, 8905–8908. [DOI] [PubMed] [Google Scholar]
- [7].a) Dcona MM, Mitra D, Goehe RW, Gewirtz DA, Lebman DA, Hartman MCT, Chem. Commun 2012, 48, 4755–4757; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dcona MM, Yu Q, Capobianco JA, Hartman MCT, Chem. Commun 2015, 51, 8477–8479; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Goodman AM, Neumann O, Nørregaard K, Henderson L, Choi M-R, Clare SE, Halas NJ, Proc. Natl. Acad. Sci. U.S.A 2017, 114, 12419–12424; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wegener M, Hansen MJ, Driessen AJM, Szymanski W, Feringa BL, J. Am. Chem. Soc 2017, 139, 17979–17986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Li A, Turro C, Kodanko JJ, Chem. Commun 2018, 54, 1280–1290; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) White JK, Schmehl RH, Turro C, Inorg. Chim. Acta 2017, 454, 7–20; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Loftus LM, Li A, Fillman KL, Martin PD, Kodanko JJ, Turro C, J. Am. Chem. Soc 2017, 139, 18295–18306; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Howerton BS, Heidary DK, Glazer EC, J. Am. Chem. Soc 2012, 134, 8324–8327; [DOI] [PubMed] [Google Scholar]; e) Sun W, Thiramanas R, Slep LD, Zeng X, Mailänder V, Wu S, Chem. Eur. J 2017, 23, 10832–10837. [DOI] [PubMed] [Google Scholar]
- [9].a) Hess J, Huang H, Kaiser A, Pierroz V, Blacque O, Chao H, Gasser G, Chem. Eur. J 2017, 23, 9888–9896; [DOI] [PubMed] [Google Scholar]; b) Mari C, Pierroz V, Leonidova A, Ferrari S, Gasser G, Eur. J. Inorg. Chem 2015, 3879–3891; [DOI] [PubMed]; c) Mari C, Pierroz V, Ferrari S, Gasser G, Chem. Sci 2015, 6, 2660–2686; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pierri AE, Huang P-J, Garcia JV, Stanfill JG, Chui M, Wu G, Zheng N, Ford PC, Chem. Commun 2015, 51, 2072–2075. [DOI] [PubMed] [Google Scholar]
- [10].a) Wilson WR, Hay MP, Nat. Rev. Cancer 2011, 11, 393–410; [DOI] [PubMed] [Google Scholar]; b) Chen H, Tian J, He W, Guo Z, J. Am. Chem. Soc 2015, 137, 1539–1547. [DOI] [PubMed] [Google Scholar]
- [11].a) Mitra K, Dalton Trans, 2017, 45, 19157–19171; [DOI] [PubMed] [Google Scholar]; b) Mlcouskova J, Stepankova J, Brabec V, J. Biol. Inorg. Chem 2012, 17, 891–898; [DOI] [PubMed] [Google Scholar]; c) Štarha P, Trávníček Z, Dvořák Z, Radošová-Muchová T, Prachařová J, Vančo J, Kašpárková J, PLoS ONE 2015, 10, e0123595; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mitra K, Gautam S, Kondaiash P, Chakravarty AR, Angew. Chem. Int. Ed 2015, 54, 13989–13993; [DOI] [PubMed] [Google Scholar]; e) Mitra K, Gautam S, Kondaiah P, Chakravarty AR, Eur. J. Inorg. Chem 2017, 1753–1763. [DOI] [PubMed]
- [12].a) Chen H, Dong B, Tang Y, Lin W, Acc. Chem. Res 2017, 50, 1410–1422; [DOI] [PubMed] [Google Scholar]; b) Owens EA, Henary M, Fakhri GE, Choi HS, Acc. Chem. Res 2016, 49, 1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Altman RB, Terry DS, Zhou Z, Zheng Q, Geggier P, Kolster RA, Zhao Y, Javitch JA, Warren JD, Blanchard SC, Nat. Methods 2012, 9, 68–73; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Atchison J, Kamila S, Nesbitt H, Logan KA, Nicholas DM, Fowley C, Davis J, Callan B, McHale AP, Callan JF, Chem. Commun 2017, 53, 2009–2012. [DOI] [PubMed] [Google Scholar]
- [14].a) Fulda S, Galluzzi L, Kroemer G, Nat. Rev. Drug Discov 2010, 9, 447–464; [DOI] [PubMed] [Google Scholar]; b) Weinberg SE, Chandel NS, Nat. Chem. Biol 2015, 11, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Lv W, Zhang Z, Zhang KY, Yang H, Liu S, Xu A, Guo S, Zhao Q, Huang W, Angew. Chem. Int. Ed 2016, 55, 9947–9951; [DOI] [PubMed] [Google Scholar]; b) Daum S, Reshetnikov MSV, Sisa M, Dumych T, Lootsik MD, Bilyy R, Bila E, Janko C, Alexiou C, Herrmann M, Sellner L, Mokhir A, Angew. Chem. Int. Ed 2017, 56, 15545–15549. [DOI] [PubMed] [Google Scholar]
- [16].a) Gorka AP, Nani RR, Zhu J, Mackem S, Schnermann MJ, J. Am. Chem. Soc 2014, 136, 14153–14159; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gorka AP, Nani RR, Schnermann MJ, Org. Biomol. Chem 2015, 13, 7584–7598; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gorka AP, Schnermann MJ, Curr. Opin. Chem. Biol 2016, 33, 117–125; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nani RR, Gorka AP, Nagaya T, Kobayashi H, Schnermann MJ, Angew. Chem. Int. Ed 2015, 127, 13839–13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Armitage BA, Top Curr. Chem 2005, 253, 55–76. [Google Scholar]; b) Stennett EMS, Ciuba MA, Levitus M, Chem. Soc. Rev 2014, 43, 1057–1075. [DOI] [PubMed] [Google Scholar]
- [18].Cui X, Zhao J, Mohmood Z, Zhang C, Chem. Rec 2016, 16, 173–188. [DOI] [PubMed] [Google Scholar]
- [19].a) Nisbett K, Tu Y-J, Turro C, Kodanko JJ, Schlegel HB, Inorg. Chem 2018, 57, 231–240; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Salassa L, Phillips HIA, Sadler PJ, Phys. Chem. Chem. Phys 2009, 11, 10311–10316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.