

ABSTRACT
Oncogene-induced replication stress (RS) plays an active role in tumorigenesis by promoting genomic instability but is also a challenge for cell proliferation. Recent evidence indicates that different types of cancer cells adapt to RS by overexpressing components of the ATR-CHK1 pathway that promote fork progression in a checkpoint-independent manner.
KEYWORDS: Replication fork stability, replication checkpoint, Claspin, timeless, replication stress
Proliferating cells are continuously exposed to a variety of events impeding the progression of replication forks, commonly referred to as replication stress (RS).1 The stability of replication forks is crucial for the maintenance of genome integrity because fork collapse can lead to the formation of DNA double-strand breaks (DSBs) that are particularly toxic for cells. Consequently, stalled forks must be sensed and kept under tight control by the ATR-CHK1 checkpoint pathway to prevent fork collapse.1 Full checkpoint activation requires the hyperphosphorylation of the effector kinase CHK1.2 This amplification of the checkpoint response depends on the CLAPSIN-TIMELESS-TIPIN complex, a conserved checkpoint mediator complex also known as the Fork Protection Complex (FPC). Studies in budding yeast have shown that the FPC also plays a structural role at forks by coordinating DNA polymerase and helicase activities and by maintaining fork stability in a checkpoint-independent manner.3 Whether this checkpoint independent function is conserved in human cells was less clear. Indeed, the depletion of FPC components leads to fork slowdown,4 but as normal fork progression also depends on CHK1,2 the checkpoint-dependent and -independent functions of FPC have remained difficult to separate.
Oncogene-induced RS is a double-edged sword for cancer cells. Although it contributes to cancer development by promoting genomic instability, it represents also a burden as it slows down replication and triggers senescence.5 Although checkpoint pathways act as barriers against tumor progression, they also protect cancer cells from oncogene-induced RS in a dosage-dependent manner.6,7 Understanding how tumor cells control this balance and grow in the presence of chronic RS is therefore of major importance for cancer therapy. CLASPIN and TIMELESS have a dual role in checkpoint signaling and in the maintenance of fork integrity. Since they are overexpressed in different cancers, they represent good candidates to control this balance. The functional implications of the overexpression of FPC components in the tolerance to oncogene-induced RS have therefore been recently addressed.8
Firstly, an integrated analysis of mRNA expression of different components the ATR-CHK1 pathway was performed in three different cancers. This analysis confirmed that downstream components of the pathway, CLASPIN, TIMELESS, and CHK1, are overexpressed in a highly correlated manner. However, this was not the case for upstream components of the pathway, such as ATR, RAD9, and RAD17. This correlation between downstream components of the pathway was also observed at the protein level in the proteomic landscape of 50 colon cancer cell lines. Together, these data indicate that CLASPIN, TIMELESS, and CHK1 define a functional module that is distinct from the rest of the ATR-CHK1 pathway and specifically enhanced in cancer cells.
To determine the impact of this overexpression on RS tolerance, the excess of CLASPIN and TIMELESS was reduced in the colon cancer cell line HCT116. This partial depletion impeded cell growth without interfering with ATR signaling. It also induced a slowdown of replication forks and an increase in sister fork asymmetry, indicative of fork stalling. These data indicate that the FPC promotes RS tolerance independently of checkpoint activation. Moreover, clones of immortalized fibroblasts expressing an oncogenic form of Ras (BJ-RasV12) and escaping senescence were isolated. Remarkably, several of these clones adapted to oncogene-induced RS by spontaneously overexpressing CLASPIN and TIEMELESS, independently of ATR signaling. This overexpression restored a normal fork progression in a FPC-dependent manner in cells overexpressing RasV12. Moreover, the overexpression of FPC components in BJ-RasV12 and in U2OS cells overexpressing CYCLIN E also increased tolerance to oncogene-induced RS.8
Altogether, these data support a model in which the overexpression of CLASPIN and TIMELESS represent a common strategy used by cancer cells to tolerate RS without hyper-activating the ATR pathway, which would be detrimental to tumor growth (Figure 1). This work raises the question of how cancer cells drive the overexpression of these factors. Transcriptome analyses of resistant clones revealed complex and heterogeneous gene expression profiles. These profiles are not consistent with a simple upregulation of the E2F pathway but rather reflect the overexpression of a large group of DNA Damage response genes involved in fork protection. As CLASPIN levels are mainly regulated at the post-translational level by proteolysis,4 understanding their regulation in cancer cells could open the way for the development of novel therapeutic strategies targeting CLASPIN and fork stability.
Figure 1.
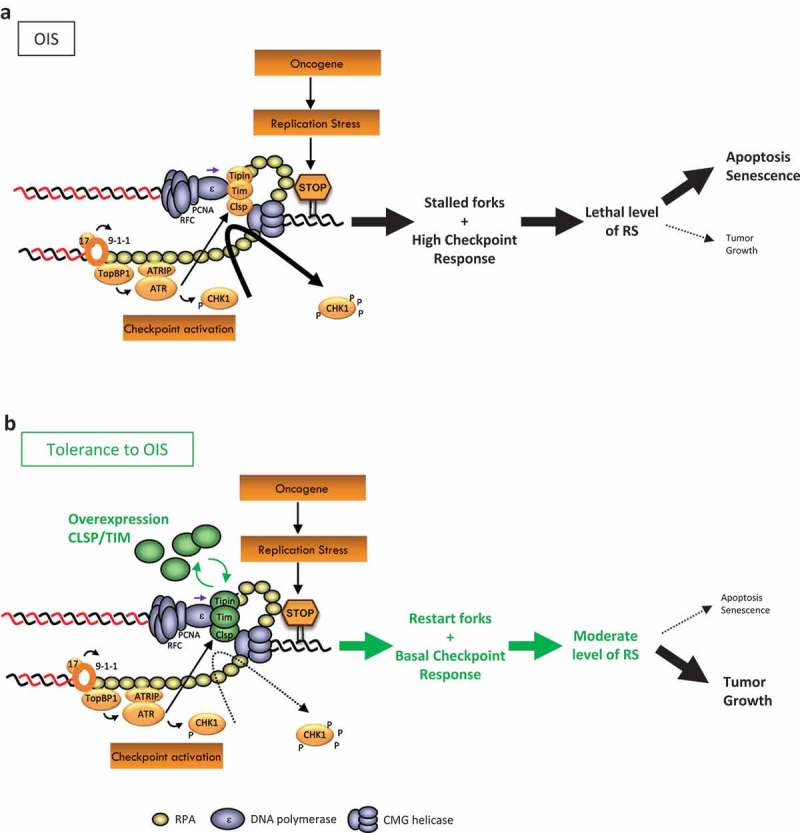
Overexpression of CLASPIN and TIMELESS confers a proliferative advantage by protecting replication forks from chronic replication stress in cancer cells.
(a) Oncogene-induced replication stress (RS) increases fork stalling and checkpoint activation in cancer cells. Sensing of stalled forks involves RPA-coated ssDNA and ATR activation by TOPBP1, RAD17 and the 9-1-1 complex. The ATR-dependent phosporylation of CLASPIN recruits and activates CHK1 at the fork. Cancer cells growing with a high level of RS need to modulate the intensity of checkpoint activation to escape senescence or apoptosis. (b) A strategy used by cancer cells to tolerate high RS is to overexpress fork protection factors such as CLASPIN (CLSP) and TIMELESS (TIM) to help them restart forks efficiently and complete the replication of their genome within the length of the S phase. Thick and thin black arrows indicate high and basal activation, respectively. P: phosphorylation event.
An important question that remains to address is the mechanism by which an excess of CLASPIN and TIMELESS promotes fork stability. Recent evidence indicates that, in cancer cells, TIMELESS is displaced from the replisome under RS conditions to slowdown forks and prevent genome instability.9 Under these conditions, an excess of free CLASPIN and TIMELESS could promote the reassembly of a functional FPC at forks. In RS conditions, this complex could sense the uncoupling between DNA polymerases and helicases activities to transduce a signal to remodel and restart forks. Indeed, the CLASPIN-TIMELESS-CHK1 module promotes PCNA ubiquitination in an ATR-independent manner, which is required for translesion DNA synthesis.10 Enhanced levels of FPC in cancer cells may, therefore, constitute a reservoir to promote the fast reassembly of a functional FPC and favor fork restart.
Importantly, overexpression of CLASPIN, TIMELESS and CHK1 (but not ATR, RAD9, and RAD17) in untreated low-grade Non-Small Cell Lung Cancer correlated with the aggressiveness of the tumor, showing that their overexpression impact significantly on prognosis. CLASPIN and TIMELESS represent therefore promising targets for anticancer therapies targeting replication forks. Further analysis of cancer replisomes could also lead to the identification of novel biomarkers besides CLASPIN and TIMELESS and the design of new anticancer drugs for individualized therapy.
Funding Statement
This work is funded by CNRS, INSERM, ANR, INCa, MSDAvenir Fund, SIRIC Montpellier Cancer and by Ligue contre le Cancer (équipe labellisée).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Zeman MK, Cimprich KA.. Causes and consequences of replication stress. Nat Cell Biol. 2014. January;16(1):2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.González Besteiro MA, Gottifredi V.. The fork and the kinase: a DNA replication tale from a CHK1 perspective. Mutat Res Rev Mutat Res. 2015. January-Mar;763:168–180. doi: 10.1016/j.mrrev.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tourrière H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst). 2007. July 1;6(7):900–913. doi: 10.1016/j.dnarep.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Smits VAJ, Cabrera E, Freire R, Gillespie DA. Claspin - checkpoint adaptor and DNA replication factor. FEBS J. 2019. February;286(3):441–455. doi: 10.1111/febs.14594. [DOI] [PubMed] [Google Scholar]
- 5.Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- 6.Bartek J, Mistrik M, Bartkova J. Thresholds of replication stress signaling in cancer development and treatment. Nat Struct Mol Biol. 2012. January 5;19(1):5–7. doi: 10.1038/nsmb.2220. [DOI] [PubMed] [Google Scholar]
- 7.Lecona E, Fernández-Capetillo O. Replication stress and cancer: it takes two to tango. Exp Cell Res. 2014. November 15;329(1):26–34. doi: 10.1016/j.yexcr.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bianco JN, Bergoglio V, Lin YL, Pillaire MJ, Schmitz AL, Gilhodes J, Lusque A, Mazières J, Lacroix-Triki M, Roumeliotis TI et al. Overexpression of Claspin and timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat Commun. 2019. February 22;10(1):910. doi: 10.1038/s41467-019-08886-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Somyajit K, Gupta R, Sedlackova H, Neelsen KJ, Ochs F, Rask MB, Choudhary C, Lukas J. Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science. 2017. November 10;358(6364):797–802. doi: 10.1126/science.aao3172. [DOI] [PubMed] [Google Scholar]
- 10.Yang XH, Zou L. Dual functions of DNA replication forks in checkpoint signaling and PCNA ubiquitination. Cell Cycle. 2009. January 15;8(2):191–194. doi: 10.4161/cc.8.2.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]