

ABSTRACT
Mutations caused by DNA damage are a main driver of cancer. We discovered that recognition of newly synthesised histone H4 directs breast cancer type 1 susceptibility protein (BRCA1) to post-replicative chromatin. The switch from mutagenic to error-free DNA double strand break repair by homologous recombination is therefore controlled by chromatin.
KEYWORDS: BRCA1, BARD1, 53BP1, DNA double strand break repair, homologous recombination
DNA double strand breaks (DSBs) can be repaired by two alternative pathways, non-homologous end joining (NHEJ) and homologous recombination (HR). The NHEJ mechanism fuses two DNA ends and is potentially mutagenic since small deletions or insertions, and in the worst case, chromosomal translocations can occur. HR on the other hand uses the genetic information present on another homologous DNA molecule to repair the break and is virtually error-free, as missing information is copied from an intact template. This mechanism, however, only works if a second copy of the genetic information is available.1 Therefore, controlling the switch from NHEJ to HR, and linking this decision to the cell cycle, is a key challenge for the cell as HR must only be initiated during S and G2 phases when a second copy of the genetic material is present in the form of a newly replicated sister chromatid.1
In humans two key players in this process are tumor protein 53-binding protein 1 (TP53BP1, or short 53BP1), a factor that promotes NHEJ by antagonising DNA end-resection, and the breast cancer type 1 susceptibility protein (BRCA1) DNA-repair complex that promotes end-resection and thus HR.1 Cell cycle signalling is known to regulate the transition from NHEJ in G1 to HR in S/G21. However, in S phase the nucleus contains both, already replicated regions that are compatible with HR, and regions that still need to be replicated and thus should not be repaired by HR. This enigma argues against a mechanism that solely relies on a cell cycle switch. Until now it was elusive how the transition between 53BP1 and BRCA1 could work at the molecular level. Our recent work provides a simple solution to this problem by demonstrating that the BRCA1 complex is a sensor for the availability of a sister chromatid that signals this information to the DNA repair machinery.2
We identified the BRCA1 complex in a screen for proteins that distinguish between nucleosomes containing unmodified histone H4 and H4 di-methylated at lysine 20 (H4K20me2). In this screen BRCA1 showed a preference for H4 not methylated at lysine 20 (H4K20me0).2 H4K20-methylation oscillates during the cell cycle.3,4 In G1 virtually all H4K20 is di-methylated (H4K20me2), while H4 incorporated into newly replicated chromatin during S-phase is un-methylated (H4K20me0). H4K20me0 thereby marks ‘new’ chromatin, and thus the presence of a sister chromatid,4 and this is sensed by BRCA1. As a consequence, BRCA1 is recruited to post-replicative chromatin in S/G2 and resides there until H4K20me0 is converted to the mono-, di-, and tri-methylated states as the cells progress into mitosis and divide.2 These findings lead to a strikingly simple explanation of the cross-talk between BRCA1 and 53BP1 based on the competition between both factors on pre- and post-replicative chromatin.
In G1 BRCA1 cannot access ‘old’ chromatin due to pervasive H4K20me2 modification, while 53BP1 can directly bind this mark throughout the cell cycle.5 Therefore, 53BP1’s anti-resection activity ‘wins’ in G1 chromatin and NHEJ is initiated as the default repair pathway. In S/G2 dilution of H4K20me2 with H4K20me0 leads to reduced 53BP1 recruitment to DSBs in post-replicative chromatin.6,7 However, dilution alone cannot explain the shift to HR,8 and an additional 53BP1-repressive mechanism must exist. This activity depends on BRCA1,8 and its recruitment to ‘new’ chromatin via H4K20me0.2
53BP1 binds H4K20me2 with rather low affinity5 and probably ‘scans’ chromatin in a dynamic fashion. At DSBs ubiquitylation of histone H2A by RNF8 (RING finger protein 8) and RNF168 (RING finger protein 168) acts as a second recruitment-signal that strengthens binding of 53BP1 at the break.9 RNF8/RNF168-dependent ubiquitylation also stimulates BRCA1 recruitment to DSBs.10 However, BRCA1 is present on post-replicative chromatin already before the damage occurs.2 We envisage that the combination of reduced 53BP1 recruitment due to H4K20me2 dilution and the presence of the competing (H4K20me0-binding) activity of BRCA1 allows BRCA1 to accumulate at DSBs more quickly and ‘seize’ the break before 53BP1 (Figure 1). This competition only happens on replicated chromatin in S/G2, as the BRCA1 binding platform (H4K20me0) is absent in G1. Therefore BRCA1 excludes 53BP1 and promotes end-resection specifically at DSBs in replicated sister chromatids. The switch between NHEJ and HR is thus determined by the rivalry between both factors for the same binding site on histone H4 that is differentially methylated on ‘new’ and ‘old’ histone H4.
Figure 1.
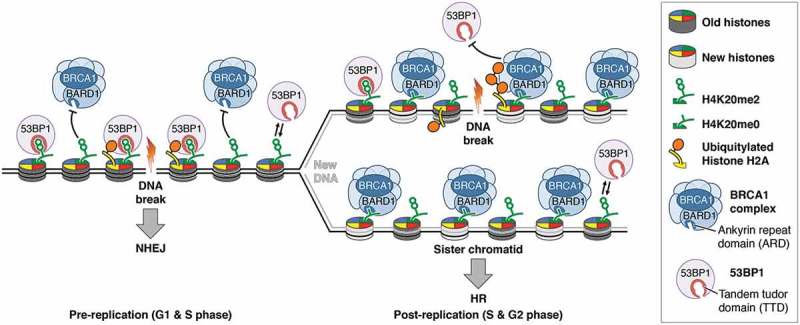
Sister chromatid sensing by BRCA1/BARD1 directs homologous recombination to post-replicative chromatin.
Recognition of di-methylated lysine 20 of histone H4 (H4K20me2) in ‘old’ chromatin by 53BP1 (tumour protein 53-binding protein 1) and sensing of un-modified lysine 20 of histone H4 (H4K20me0) in ‘new’ chromatin by the BRCA1 (breast cancer type 1 susceptibility protein) complex via the ankyrin repeat domain of its BARD1 (BRCA1-associated RING domain protein 1) subunit drives a chromatin-based switch between mutagenic non-homologous end joining (NHEJ) and error-free DNA double strand break repair by homologous recombination (HR) in pre- and post-replicative chromatin.
This mechanism solves several problems at once: it targets HR specifically to newly replicated chromatin while still allowing NHEJ to occur during S-phase in un-replicated regions, and it limits HR to S/G2 when sister chromatids are present. The BRCA1 complex recognises H4K20me0 via the ankyrin repeat domain (ARD) of its BARD1 (BRCA1-associated RING domain protein 1) subunit. Indeed, mutating the histone binding site in the ARD results in loss of chromatin localisation of BRCA1 and a shift of DSB repair during S-phase from HR to NHEJ, as predicted.2 Our findings resolve the conundrum of how BRCA1 ‘knows’ when to initiate HR. They also demonstrate that in addition to the cell cycle, chromatid sensing directs DSB repair pathway choice, and argue that the critical decision between NHEJ and HR is made at the chromatin level before cell cycle-regulated end resection and loading of BRCA2 (breast cancer type 2 susceptibility protein) and RAD51 (DNA repair protein RAD51 homolog 1) take place.1
Chromatid sensing appears to be a more general mechanism as it is employed by the TONSL/MMS22L (Tonsoku-like protein; MMS22-like protein) and SLF (SMC5-SMC6 complex localisation factor) DNA repair complexes, in addition to BRCA1/BARD1.2,4 All three complexes recognise H4K20me0 through subunits with ankyrin repeats.2,4 H4K20me0-binding ARDs are potentially ‘druggable’ domains. Our findings have therefore important clinical implications. Tumours with mutations in BRCA1 are vulnerable to PARP (Poly [ADP-ribose] polymerase) inhibitor treatment. Our study demonstrates that H4K20me0 recognition is a central function of BARD1 in HR and that mutating its ARD sensitises cancer cells to PARP inhibitors.2 The development of compounds that specifically target the BARD1 ARD and block H4K20me0 binding would open up the possibility to chemically inactivate BRCA1 in cancers that depend on HR due to high levels of replication stress. These are exciting prospects for a new class of ‘epidrugs’ with the potential to benefit many cancer patients.
Funding Statement
Research in the T.B. lab is funded by the Helmholtz Association and the European Research Council (ERC StG no. 309952). The Groth lab is supported by the Danish Cancer Society, the Novo Nordisk Foundation, the Lundbeck Foundation, the European Research Council (ERC CoG no. 724436), the Independent Research Fund Denmark, and the NEYE Foundation.
Abbreviations
53BP1 tumour protein 53-binding protein 1
ARD Ankyrin repeat domain
BARD1 BRCA1-associated RING domain protein 1
BRCA1 Breast cancer type 1 susceptibility protein
BRCA2 Breast cancer type 2 susceptibility protein
DSB DNA double strand break
H4K20me0 Histone H4 not methylated at lysine 20
H4K20me2 Histone H4 di-methylated at lysine 20
HR homologous recombination
MMS22L Methyl methanesulfonate-sensitivity protein 22-like
NHEJ non-homologous end joining
PARP Poly [ADP-ribose] polymerase
RAD51 DNA repair protein RAD51 homolog 1
RNF8 RING finger protein 8
RNF168 RING finger protein 168
SLF SMC5-SMC6 complex localisation factor
TONSL Tonsoku-like protein
Disclosure of Potential Conflicts of Interest
A.G. is an inventor on a filed patent application covering the therapeutic targeting of ARD interactions with H4K20me0 for cancer therapy. A.G. is a co-founder and CSO of Ankrin Therapeutics.
References
- 1.Hustedt N, Durocher D.. The control of DNA repair by the cell cycle. Nat Cell Biol. 2016;19(1):1–9. doi: 10.1038/ncb3452. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura K, Saredi G, Becker JR, Foster BM, Nguyen NV, Beyer TE, Cesa LC, Faull PA, Lukauskas S, Frimurer T, et al. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat Cell Biol. 2019;21(3):311–318. doi: 10.1038/s41556-019-0282-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pesavento JJ, Yang H, Kelleher NL, Mizzen CA.. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol. 2008;28(1):468–486. doi: 10.1128/MCB.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saredi G, Huang H, Hammond CM, Alabert C, Bekker-Jensen S, Forne I, Reverón-Gómez N, Foster BM, Mlejnkova L, Bartke T, et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature. 2016;534(7609):714–718. doi: 10.1038/nature18312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pellegrino S, Michelena J, Teloni F, Imhof R, Altmeyer M. Replication-coupled dilution of H4K20me2 guides 53BP1 to pre-replicative chromatin. Cell Rep. 2017;19(9):1819–1831. doi: 10.1016/j.celrep.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simonetta M, de Krijger I, Serrat J, Moatti N, Fortunato D, Hoekman L, Bleijerveld OB, Altelaar AFM, Jacobs JJL. H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell Cycle. 2018;17(1):124–136. doi: 10.1080/15384101.2017.1404210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci. 2012;125(Pt 15):3529–3534. doi: 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499(7456):50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316(5828):1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]