

ABSTRACT
We have recently discovered that nicotinamide adenine dinucleotide metabolism controls the pro-inflammatory senescence-associated secretory phenotype during cellular senescence. This newly discovered epigenetic-metabolic signaling axis, mediated by high mobility group A and nicotinamide phosphoribosyltransferase, drives key metabolic changes and pro-inflammatory responses of senescent cells that fuel cancer progression.
KEYWORDS: Cellular senescence, senescence-associated secretory phenotype (SASP), NAD+, NAMPT, HMGA
Cellular senescence is a tumor suppressive response that prevents the division of damaged and potentially cancerous cells. A variety of stimuli induce cellular senescence including genomic and oncogenic stresses.1 Cellular senescence induced by shortened telomeres following replicative exhaustion, known as replicative senescence, is thought to contribute towards tissue aging, while oncogene-induced senescence is a tumor suppressive mechanism. Key hallmarks of senescent cells include a stable growth arrest and acquisition of the senescence-associated secretory phenotype (SASP). The SASP encompasses a collection of secreted factors, such as cytokines, chemokines, growth factors, and proteases, that stimulate tissue remodeling and inflammation. Acute responses to the SASP are beneficial, such as in the cases of wound healing and immune surveillance of damaged cells. However, chronic exposure to the SASP promotes low-grade inflammation and has deleterious effects that contribute towards tissue aging and pathology.2
Paradoxically, the SASP creates a pro-tumorigenic microenvironment and aids cancer phenotypes, and several studies have highlighted that selective elimination of senescent cells suppresses cancer development. Thus, a potential anti-cancer strategy is to specifically target the SASP while maintaining the tumor suppressive growth arrest of senescent cells. Also, specifically targeting the SASP avoids potential unintended side effects that are associated with eliminating senescent cells. The SASP is dynamically regulated based on the activation of key transcription factors, CCAAT/enhancer binding protein beta (C/EBPβ) and nuclear factor kappa B (NFkB).3 NFkB is responsible for the upregulation of pro-inflammatory SASP factors, such as interleukin-1β (IL1β), interleukin-6 (IL6), and interleukin-8 (IL8). Several signaling pathways have been linked to the activation of NFkB, including DNA damage, p38 mitogen-activated protein kinase (p38 MAPK), and mammalian target of rapamycin (mTOR) signaling pathways. We have uncovered a novel epigenetic-metabolic link that selectively governs activation of NFkB and the pro-inflammatory SASP during cellular senescence (Figure 1).4
Figure 1.
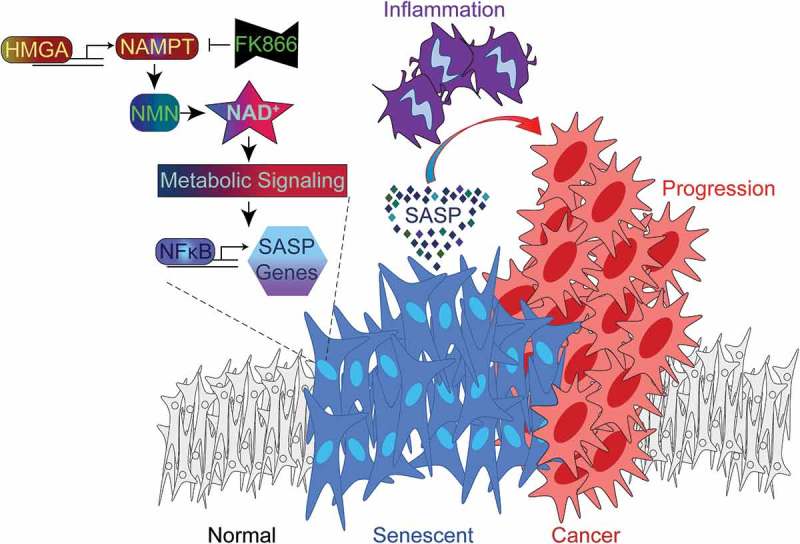
HMGA-NAMPT-NAD+ signaling promotes a pro-inflammatory SASP and drives cancer progression. During cellular senescence, high mobility group A (HMGA) proteins upregulate nicotinamide phosphoribosyltransferase (NAMPT) expression and stimulate nicotinamide adenine dinucleotide (NAD+) metabolism. This metabolic change suppresses AMP-activated protein kinase-p53 signaling that would serve to restrain nuclear factor-kappa B (NFkB) activity and the pro-inflammatory senescence-associated secretory phenotype (SASP). As a result, NAD+ metabolism stimulates the SASP of senescent cells, which can be inhibited by FK866 treatment or exacerbated by nicotinamide mononucleotide (NMN) treatment. In the pancreatic ductal adenocarcinoma (PDAC) mouse model, pancreas-specific oncogenic K-Ras expression induces pancreatic intraepithelial neoplasia (PanIN) lesions that are enriched in senescent cells. The PanIN lesions create a pro-inflammatory microenvironment that drives PDAC progression. PDAC progression is enhanced by exacerbating the SASP through stimulation of NAD+ metabolism using NMN treatment.
Senescent cells undergo drastic epigenetic remodeling whereby high mobility group A (HMGA) proteins regulate chromatin accessibility to transcription factors. Interestingly, HMGA proteins are not only expressed in senescent cells but also in cancer cells and are associated with a poor prognosis in multiple types of cancer.5 We explored the functionality of HMGA1 during cellular senescence by identifying its association with the loci of target genes. A top candidate gene was nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme for the nicotinamide adenine dinucleotide (NAD+) salvage pathway. We found that HMGA proteins bind the enhancer region of NAMPT, leading to NAMPT upregulation and enhanced NAD+ production. NAD+ is essential for many cellular functions and energy-producing metabolic pathways, such as glycolysis and mitochondrial oxidative phosphorylation. Interestingly, senescent cells have long been known to exhibit increased glycolytic and mitochondrial oxidative phosphorylation activities despite also displaying a stable proliferative arrest.6 Consistent with previous reports, we found elevated activity in glycolysis and oxidative phosphorylation. However, these metabolic changes rely on increased NAD+ metabolism through enhanced NAMPT expression. Notably, the HMGA/NAMPT regulated pro-inflammatory SASP is independent of senescence-associated growth arrest. Thus, the increase in glycolysis and oxidative phosphorylation was primarily used for production of SASP factors.
Importantly, we found that NAMPT linked cellular metabolism to activation of the SASP in senescent cells. The metabolic state driven by NAMPT suppressed AMP-activated protein kinase (AMPK)-mediated activation of tumor protein p53 (TP53, best known as p53). A key tumor suppressive role of p53 is suppression of NFkB activity and the pro-inflammatory SASP.7 Indeed, inhibition of NAMPT suppressed the SASP in an AMPK-p53 signaling-dependent manner and prevented the growth stimulatory effects of senescent cells on cancer cells. Meanwhile, treatment with the NAD+ precursor nicotinamide mononucleotide (NMN) in multiple types of senescence, including oncogene-induced, replicative, and chemotherapy-induced senescence, amplified the pro-inflammatory SASP and growth stimulatory effect on cancer cells. Therefore, we tested the role of NAD+ metabolism in mediating the SASP on cancer progression using a pancreatic ductal adenocarcinoma (PDAC) mouse model. This mouse model develops pancreatic intraepithelial neoplasia (PanIN) lesions that are enriched in senescent cells following pancreas-specific expression of activated K-Ras. It has been demonstrated that the SASP of the PanIN lesions creates a pro-inflammatory microenvironment that is a major driver of PDAC progression.8 Indeed, NAD+ metabolism facilitated the SASP of PanIN lesions, resulting in an inflammatory response and PDAC progression.
The pro-inflammatory role of NAD+ metabolism was demonstrated using pharmacological inhibition of NAMPT using FK866 or supplementation with NMN, both of which have therapeutic implications. Pharmacological NAMPT inhibition, such as by FK866 treatment, has shown efficacy and reached clinical trial testing in multiple types of cancer. Based on these new findings, the effects of pharmacological NAMPT inhibition on pro-inflammatory signaling and the immune microenvironment should be considered during anti-cancer therapy. For instance, pharmacological NAMPT inhibition may improve the efficacy of chemotherapy and suppress mechanisms of chemoresistance.9 This may be especially important for types of cancer that exhibit elevated expression of HMGA or NAMPT. Clinical trials have also employed NAD+ precursors, such as NMN or nicotinamide riboside, to suppress pathologic changes that accompany aging.10 However, inflammation is a key driver of the aging process and there is a possibility that the administration of NAD+ precursors may have pro-inflammatory effects based on the disease state, especially in the case of pre-cancerous lesions. Therefore, NAD+ precursors should be administered with precision to avoid potential unintended side effects associated with inflammation and tumorigenesis.
Funding Statement
This work was supported by the National Cancer Institute [R01CA160331]; National Institute on Aging [P01AG031862]; National Cancer Institute/National Institutes of Health [T32CA009191].
Acknowledgments
The review was supported by US National Institutes of Health grants R01CA160331 and P01AG031862 to R. Z.; and T32CA009191 to T. N. Support of Core Facilities was provided by Cancer Centre Support Grant (CCSG) CA010815 to The Wistar Institute.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
AMPK AMP-activated protein kinase
C/EBP-β CCAAT/enhancer binding protein beta
HMGA high mobility group A
PanIN pancreatic intraepithelial neoplasia
PDAC pancreatic ductal adenocarcinoma
NAD nicotinamide adenine dinucleotide
NAMPT nicotinamide phosphoribosyltransferase
NFkB nuclear factor-kappa B
NMN nicotinamide mononucleotide
SASP Senescence-associated secretory phenotype
References
- 1.Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM.. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16:718–735. doi: 10.1038/nrd.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sieben CJ, Sturmlechner I, van de Sluis B, van Deursen JM.. Two-step senescence-focused cancer therapies. Trends Cell Biol. 2018;28:723–737. doi: 10.1016/j.tcb.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito Y, Hoare M, Narita M. Spatial and Temporal Control of Senescence. Trends Cell Biol. 2017;27:820–832. doi: 10.1016/j.tcb.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, Aird KM, Iwasaki O, Kossenkov AV, Schultz D, et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. 2019;21:397–407. doi: 10.1038/s41556-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumter TF, Xian L, Huso T, Koo M, Chang YT, Almasri TN, Chia L, Inglis C, Reid D, Resar LM. The High Mobility Group A1 (HMGA1) transcriptome in cancer and development. Curr Mol Med. 2016;16:353–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiley CD, Campisi J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 2016;23:1013–1021. doi: 10.1016/j.cmet.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, Rodriguez-Justo M, Serrano M, Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 10.Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27:529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]