

ABSTRACT
Self-DNA has previously been thought to be protected from immune detection by compartmentalisation in the nucleus or mitochondria. Here, we describe the discovery of a signalling cascade that links the detection of DNA damage in the nucleus to the activation of the innate immune adaptor STING (STimulator of INterfern Genes).
KEYWORDS: DNA damage, innate immunity, etoposide, STING, IFI16
Maintaining DNA integrity is crucial for our cells to function. However, DNA damage is a common occurrence, as our cells are exposed to ultraviolet light, environmental toxins and reactive oxygen species generated by metabolic processes. Cells respond to DNA damage by arresting the cell cycle, promoting repair of the DNA lesion and, if that is not possible, by undergoing cell death. Tumour cells are particularly sensitive to the effects of DNA damage, and this feature is exploited by many conventional cancer therapies, such as radiotherapy or chemotherapy with genotoxic agents. In addition to causing cell cycle arrest and cell death, DNA damage can also activate the immune system. Indeed, the immune response to radio- and chemotherapy-induced damage is important for the effectiveness of cancer treatment, and this is at least in part mediated by a cell-intrinsic response of the tumour cells themselves.1,2 While it has been known for some time that damaged cells secrete type I interferons and pro-inflammatory cytokines,3 the molecular mechanisms that link the detection of DNA damage to innate immune signalling are only starting to be discovered.
When damaged DNA leaks into the cytosol, it can be detected by the DNA sensor cGAS (cyclic guanosine monophosphate adenosine monophosphate synthase). This occurs for instance several days after a cell’s recovery from irradiation-induced double strand DNA breaks, and involves the formation of micronuclei which then release DNA for recognition by cGAS.4,5 cGAS usually detects cytosolic DNA during infection with intracellular pathogens. Upon DNA binding, cGAS produces the second messenger cGAMP (cyclic guanosine monophosphate adenosine monophosphate), which activates the adaptor protein STING (STimulator of INterferon Genes, also known as transmembrane protein 173, TMEM173).6 STING then translocates from the endoplasmic reticulum to peri-nuclear foci where it facilitates signalling by TBK1 (TANK binding kinase 1) and IRF3 (interferon regulatory factor 3).6 This ultimately results in the production of type I interferons.
While it had initially been hypothesised that nuclear DNA might be sheltered from detection by innate immune sensors, we have recently discovered an additional DNA sensing mechanism that can detect damaged DNA in the nucleus.7 This response occurs within hours of treatment with the genotoxin Etoposide, a topoisomerase II poison, which causes the formation of protein-DNA adducts and the generation of double strand breaks. Using gene targeting, RNA interference and inhibitors, we found that the innate immune response to Etoposide required STING but, unexpectedly, was independent of cGAS.7 We also did not detect any production of the second messenger cGAMP in Etoposide-treated cells, or the classical signs of STING activation such as its translocation to peri-nuclear foci or its phosphorylation at Serine 366. Instead, we found that STING was activated in a non-canonical manner by the DNA repair proteins proteins ATM (ataxia telangiectasia mutated) and PARP1 (poly-ADP-ribose polymerase 1), together with the DNA sensor IFI16 (interferon-inducible protein 16). IFI16 shuttles between the nucleus and the cytosol and also helps cGAS in the activation of STING during cytosolic DNA recognition in human cells.8,9 However, even though IFI16 is also involved in conventional cytosolic DNA sensing, it promotes a different mode of STING activation after Etoposide-induced damage. The detection of DNA damage in the nucleus induces the formation of an alternative STING signalling complex that includes IFI16, the tumour suppressor protein TP53 (also known as p53) and the E3 ubiquitin ligase TRAF6 (tumor necrosis factor receptor associated factor 6). Using STING- and IFI16-deficient cells, we observed that IFI16 and p53 associate first with each other, and IFI16 then promotes the recruitment of p53 and TRAF6 to STING. TRAF6 catalyses the formation of K63-linked ubiquitin chains on STING, which promotes the activation of nuclear factor κB (NF-κB), see Figure 1. As TRAF6 is dispensable for the response to cytosolic DNA,7 the activation of STING via TRAF6-dependent ubiquitylation is a specific signal for nuclear DNA damage detection. This alternative mode of STING activation, which results in the predominant activation of NF-κB, with only a minor contribution from IRF3, consequently also induces a different set of cytokines and chemokines than conventional DNA sensing, even though both modes of STING signalling induce the production of interferon-β.7
Figure 1.
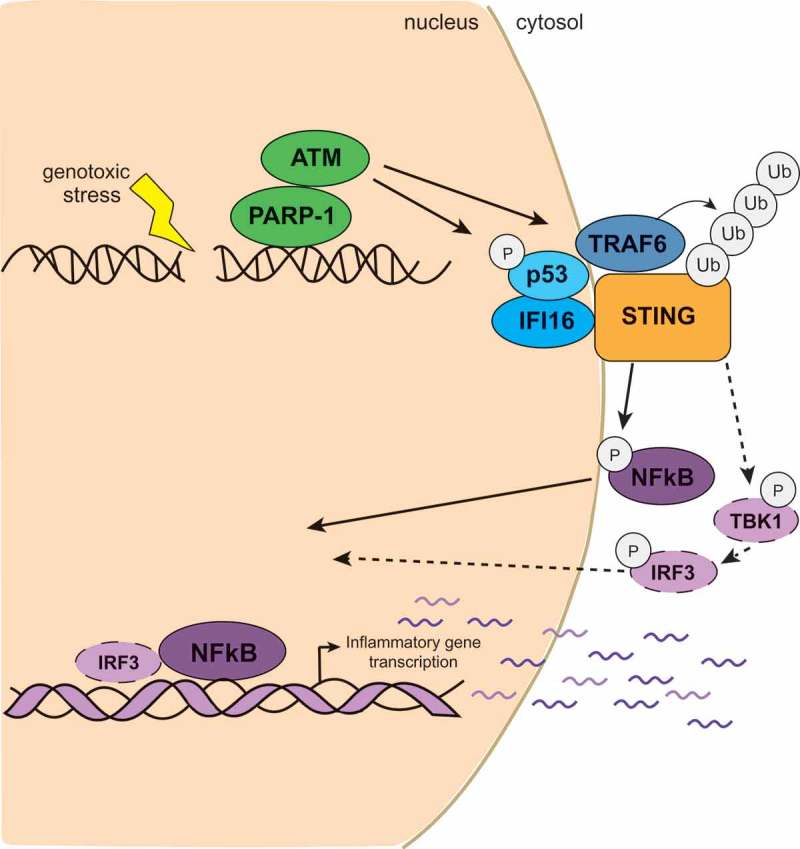
Non-canonical activation of STING (STimulator of INterferon Genes) after DNA damage. The detection of Etoposide-induced DNA damage in the nucleus by ATM (ataxia telangiectasia mutated), PARP1 (poly-ADP-ribose polymerase 1) and IFI16 (interferon-inducible protein 16) leads to the assembly of a non-canonical STING signalling complex at the endoplasmic reticulum. This complex contains IFI16, p53 (also known as tumour protein 53, TP53) and TRAF6 (tumor necrosis factor receptor associated factor 6). The assembly of K63-linked ubiquiting chains on STING mediated by TRAF6 leads to the predominant activation of NF-κB (nuclear factor κB) and the production of cytokines and chemokines. TBK1, TANK-binding kinase 1; IRF3, interferon regulatory factor 3.
While we were able to observe a response to nuclear DNA damage in multiple human cell types, this response is particularly potent in human epithelial cells such as keratinocytes. Thus, it is possible that this signalling pathway alerts the immune system to persistent replication stress and DNA damage that is an important feature of pre-cancerous and cancerous epithelial cells. Indeed, IFI16 and STING are often lost during tumour development, and have been proposed to function as tumour suppressors in some circumstances.10 As immune activation can promote tumour clearance, but can also drive tumourigenesis and metastasis,10 it will be important to examine the innate immune response induced by cancer treatments in detail. While recent years have seen unprecedented progress in the discovery of immune mechanisms that help clear tumour cells, and immunotherapy with T cell checkpoint inhibitors has already resulted in great clinical benefit for some patients, many open questions remain. We will need to understand which innate immune signalling cascades can promote the establishment of a T cell inflamed micro-environment required for immunotherapy success,2 and determine exactly which features of the response are most beneficial, while avoiding potentially counter-productive effects on inflammation-induced tumourigenesis or metastasis. In the future, our understanding of the molecular mechanisms that drive DNA damage-induced immune responses may provide opportunities for patient stratification or the development of personalised treatment regimes with therapy combinations that exploit the signalling pathways available in the tumour cells of the patient.
Funding Statement
Our work is funded by the Medical Research Council (MR/K00655X/1), the European Commission (MC-CIG 631718) and North West Cancer Research (CR1140).
Disclosures of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. 2014. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 20:1301–1309. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 2.Woo S-R, Corrales L, Gajewski TF.. 2015. Innate immune recognition of cancer. Annu Rev Immunol. 33:445–474. doi: 10.1146/annurev-immunol-032414-112043. [DOI] [PubMed] [Google Scholar]
- 3.Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. 2011. The DNA damage response induces IFN. J. Immunol. 187:5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. 2017. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 548:466–470. doi: 10.1038/nature23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. 2017. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 548:461–465. doi: 10.1038/nature23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Q, Sun L, Chen ZJ.. 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 17:1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 7.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, Jakobsen MR, Nevels MM, Bowie AG, Unterholzner L. 2018. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell. 71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almine JF, O’Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, et al. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun. 8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, Egedal JH, Kjolby M, Mohammadi P, Prabakaran T, et al. 2017. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun. 8:14391. doi: 10.1038/ncomms14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He L, Xiao X, Yang X, Zhang Z, Wu L, Liu Z. 2017. STING signaling in tumorigenesis and cancer therapy: a friend or foe? Cancer Lett. 402:203–212. doi: 10.1016/j.canlet.2017.05.026. [DOI] [PubMed] [Google Scholar]