

ABSTRACT
Apoptosis can promote inflammation by triggering activation of the NLRP3 inflammasome (NLR family, pyrin domain containing 3). However, the molecular mechanisms regulating these processes are ill-defined. We recently reported that pannexin-1 is required to promote NLRP3 inflammasome assembly. We further demonstrate that differential cleavage of gasdermin D (GSDMD) by apoptotic caspases regulates inflammatory cell lysis. Here, we discuss our findings and perspectives for future studies.
KEYWORDS: Apoptosis, inflammation, Gasdermins, NLRP3 inflammasome, pannexin
Introduction
Apoptosis is a form of caspase-dependent cell death that is traditionally classified as immunologically silent. In support of this, emerging studies indicate that apoptotic caspases cleave a number of cellular proteins to inactivate innate immune signaling pathways and to promote the removal of dying cells. For example, apoptotic caspase-3 and -7 cleave the plasma membrane glycoprotein, pannexin-1, to release ‘find-me’ and ‘eat-me’ signals to enhance clearance of apoptotic cells.1 Despite these studies, exposure of innate immune cells to ‘classical apoptosis activators’ such as staurosporine, cycloheximide, UV treatment, FasL, SMAC mimetics and BH3 mimetics was all reported to promote inflammation by activating the NLRP3 inflammasome (NLR family, pyrin domain containing 3).2,3 NLRP3 assembly activates the cysteine protease caspase-1, which processes pro-interleukin (IL)-1β and pro-IL-18 into their mature active forms, and also cleaves the pore-forming protein gasdermin D (GSDMD) to drive a form of lytic cell death known as pyroptosis.4 However, the molecular mechanisms by which apoptosis triggers NLRP3 assembly are ill-defined, and whether the apoptotic caspase cascade suppresses GSDMD-dependent cell lysis has not been explored in detail.
Study
Recent studies from the Alnemri and Bachovchin laboratories demonstrated that caspase-3, and to a lesser extent caspase-7, cleave human GSDMD at position D87 (D88 in mouse) in the pore-forming domain, thereby inactivating its lytic function.5,6 However, under which conditions GSDMD needs to get inactivated by apoptotic effector caspases had not been examined. We speculated that it might be a mechanism to restrict the NLRP3-dependent cell lysis and cytokine release during apoptosis. Indeed, we found that induction of extrinsic apoptosis in macrophages resulted in cell lysis that was in part GSDMD-dependent (pyroptosis) and in part driven by caspase-3/-7 (secondary necrosis). To study the effects of caspase-3/7-driven inactivation of GSDMD, we next engineered a GsdmdD88A knock-in mouse, in which we replaced aspartate at position 88 of GSDMD to alanine. Confirming previous findings, we observed that murine GsdmdD88A cells resist caspase-3/7 cleavage. Consistent with our hypothesis, GsdmdD88A knock-in macrophages, where the pore-forming domain of GSDMD could not be inactivated, were indeed more susceptible to cell lysis upon extrinsic apoptosis.7
While investigating GSDMD processing during extrinsic apoptosis, we made the surprising observation that GSDMD processing is largely intact in Casp-1/11- and Nlrp3-deficient cells. Unexpectedly, we found that caspase-8 directly cleaves GSDMD, albeit less efficiently than caspase-1, to induce cell lysis during apoptosis, consistent with an earlier report.8 In the non-canonical inflammasome pathway, caspase-11-driven GSDMD pores promote potassium efflux and NLRP3 assembly.4 By contrast and in disagreement with Orning et al.,8 we found no evidence that caspase-8-dependent GSDMD cleavage promotes NLRP3 activation. Since caspase-1 and caspase-3 activation were blunted in cells lacking receptor-interacting serine-threonine kinase 3 during apoptosis, and a recent study reported that caspase-3 and -7 drive NLRP3 inflammasome assembly following mitochondrial apoptosis,9 we investigated whether any known caspase-3/7 substrates promote NLRP3 activation during apoptosis. In doing so, we identified pannexin-1, a channel-forming glycoprotein, as a universal driver of NLRP3 assembly during both extrinsic and intrinsic apoptosis.7
In conclusion, our study demonstrates the complex regulation of GSDMD by apoptotic caspases during apoptosis and reveals pannexin-1 as the bridge between apoptosis and NLRP3 inflammasome activation.
Perspectives
Apoptosis was long regarded as an immunologically silent form of cell death, and activation of the apoptotic caspase cascade was thought to universally shut down proinflammatory signaling pathways. However, administration of tumor cells expressing Fas ligand was observed to drive IL-1β maturation and neutrophil recruitment in mice more than 20 years ago;10 and more recently, various studies reported that exposure of innate immune cells to pro-apoptotic factors triggers cell lysis and activate the NLRP3 inflammasome.3 These studies indicate that the apoptotic caspase cascade perform has a dual function, either promoting or suppressing inflammation. Our study agrees with this emerging concept and demonstrates that caspase-8 and -3 have both pro- and anti-inflammatory functions. While caspase-8 was initially classified as an apoptotic caspase, an increasing number of studies document that caspase-8 promotes inflammation, for example, by driving TLR-induced cytokine production or directly maturing IL-1β.2,3 Our study adds to this emerging literature and demonstrates that caspase-8 cleaves GSDMD to trigger cell lysis during extrinsic apoptosis. In agreement with a dual function of caspase-3/7 during apoptosis, our study revealed that caspase-3/7-dependent cleavage and inactivation of GSDMD at position aspartate 88 suppress lytic cell death during extrinsic apoptosis; by contrast, caspase-3/7 also promote pannexin-1 activation and potassium efflux, presumably by cleaving-off the C-terminal region,1 and thus drive NLRP3 inflammasome activation during apoptosis. Since GSDMD is rapidly inactivated by caspase-3/7, and pro-IL-1β and NLRP3 are not constitutively expressed, we assume that caspase-8-dependent GSDMD activation and pannexin-1-driven NLRP3 assembly are unlikely to drive inflammation during tissue homeostasis (Figure 1). Instead, these pathways are likely activated during specific pathogen challenge, where caspase-8 is strongly activated, and when NLRP3 and pro-IL-1β expression are induced, as reported during Yersinia infection.8 Future studies should establish the impact of caspase-3/7 and caspase-8-dependent GSDMD cleavage during host defence in vivo. Nevertheless, modulating pannexin-1 activity could impact the outcome of SMAC mimetic or BH3 mimetic-based cancer chemotherapy.
Figure 1.
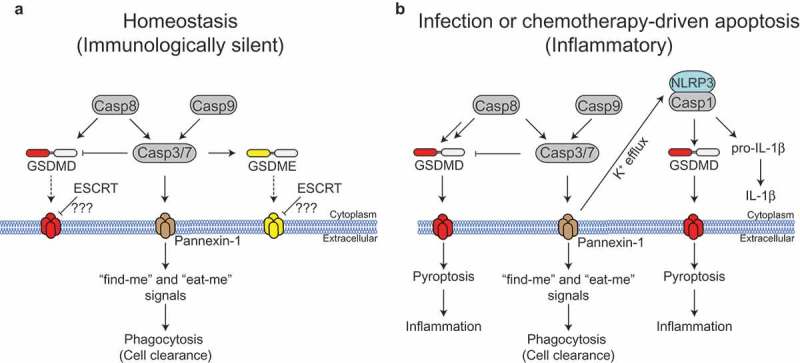
Immunologically silent versus inflammatory apoptosis. (a) During apoptosis, active caspase-3 and −7 cleave and activate the plasma membrane glycoprotein pannexin-1 to enhance clearance of apoptotic cells. Caspase-8 can directly cleave Gasdermin D (GSDMD) to induce cell lysis, but this is counteracted by caspase-3/7. Caspase-3 processes Gasdermin E (GSDME) and liberates its pore-forming domain, but this does not drive cell lysis. Whether endosomal sorting complexes required for transport (ESCRT)-mediated membrane repair suppresses GSDMD or GSDME-driven pore formation during apoptosis is unknown. (b) Infection with certain pathogens or exposure to chemotherapeutic drugs may trigger enhanced caspase-8 activation, and induce the expression of NLRP3 (NLR family, pyrin domain containing 3) and pro-interleukin (IL)-1β. This promotes caspase-8-driven GSDMD activation and resultant cell lysis. Furthermore, pannexin-1 activation promotes NLRP3 inflammasome assembly via potassium efflux, driving IL-1β and GSDMD processing by caspase-1.
Casp8, caspase-8; Casp9, caspase-9; Casp1, caspase-1, Casp3, caspase-3;Casp7, caspase-7.
An unexpected result from our study is that during extrinsic apoptosis neither GSDMD nor Gasdermin E (GSDME) is required for potassium efflux-induced NLRP3 activation, although both are activated, by caspase-8 or caspase-3, respectively, and form membrane pores under these conditions. This may in part, be driven by the endosomal sorting complexes required for transport (ESCRT)-dependent repair, which removes damaged membrane areas and could act as an additional mechanism to suppress cell lysis during apoptosis. Future studies should investigate if ESCRT-dependent membrane repair suppresses NLRP3 activation during apoptosis.
Funding Statement
This work was supported by the European Research Council [770988].
References
- 1.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, et al. Pannexin 1 channels mediate ‘find-me‘ signal release and membrane permeability during apoptosis. Nature. 2010. October 14;467(7317):863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen KW, Lawlor KE, von Pein JB, Boucher D, Gerlic M, Croker BA, Bezbradica JS, Vince JE, Schroder K.. Cutting edge: blockade of inhibitor of apoptosis proteins sensitizes neutrophils to TNF- but not lipopolysaccharide-mediated cell death and IL-1beta secretion. J Immunol. 2018. May 15;200(10):3341–3346. doi: 10.4049/jimmunol.1701620. [DOI] [PubMed] [Google Scholar]
- 3.Feltham R, Vince JE, Lawlor KE.. Caspase-8: not so silently deadly. Clin Transl Immunol. 2017. January;6(1):e124. doi: 10.1038/cti.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016. July;16(7):407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 5.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017. January 3;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol. 2017. April 20;24(4):507–514 e4. doi: 10.1016/j.chembiol.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P and Broz P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019. March 22. pii: e101638. doi: 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018. November 30;362(6418):1064–1069. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, Simpson D, Vijayaraj S, Lindqvist LM, Bouillet P, et al. The mitochondrial apoptotic effectors BAX/BAK activate caspase-3 and -7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1beta activation. Cell Rep. 2018. November 27;25(9):2339–2353 e4. doi: 10.1016/j.celrep.2018.10.103. [DOI] [PubMed] [Google Scholar]
- 10.Miwa K, Asano M, Horai R, Iwakura Y, Nagata S, Suda T. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat Med. 1998. November;4(11):1287–1292. doi: 10.1038/3276. [DOI] [PubMed] [Google Scholar]