

ABSTRACT
Understanding and overcoming resistance to cyclin-dependent kinase 4/6 (CDK4/6) inhibitors will be challenging. Recent work reveals that dysregulation of F-Box Protein 4 (FBXO4)-Cyclin D1 axis leads to mitochondrial dysfunction and drives glutamine-addiction in esophageal squamous cell carcinoma. This metabolism feature makes these tumors susceptible to combined treatment with glutaminase (GLS) inhibitor and metformin even when resisting to CDK4/6 inhibitors.
KEYWORDS: Glutamine-addiction, cyclin D1, glutaminase, CDK4/6 inhibitor, acquired resistance
Cyclin D1 (CCND1), a member of the D cyclin family (D1, D2, and D3), binds and activates cyclin-dependent kinase 4/6 (CDK4/6).1 Cyclin D1-CDK4/6 kinases initiate phosphorylation of Retinoblastoma Protein (RB) in early G1 phase, with the coordinated activity of the cyclin E-cyclin-dependent kinase 2 (CDK2), triggering the release of the E2F transcription factors (E2F), thereby promoting cell cycle progression. Cyclin D1-CDK4/6 kinases also regulate cell motility, DNA damage and stress responses, and cellular metabolism.1 As the result of gene amplification, increased transcription, or decreased protein degradation, cyclin D1 is overexpressed in human cancers, including pancreatic cancer, non-small cell lung carcinoma, breast cancer, head and neck squamous cell carcinoma, melanoma, endometrial cancer, esophageal squamous cell carcinoma (ESCC), colorectal carcinoma, mantle cell lymphoma, multiple myeloma.2 With regard to cyclin D1 degradation, loss of function mutations in F-Box Protein 4 (FBXO4), or mutations at threonine-286 in cyclin D1 inhibit its phosphorylation, also leading to the overexpression of cyclin D1; such mutations are directly linked with the development of upper gastrointestinal tumors in mice and ESCC in human.3,4 Because cyclin D1 and CDK4/6 are dysregulated in a variety of human cancers, they have been the subject of intense efforts in order to develop small molecule therapeutic inhibitors. Several potent CDK4/6 inhibitors have been developed and tested in pre-clinical and clinical studies. Specifically, palbociclib, abemaciclib, and ribociclib, with half maximal inhibitory concentrations (IC50s) at nanomoles/L range, have been approved by the Food and Drug Administration (FDA) to treat hormone-positive breast cancers.5 Currently, these compounds are still undergoing clinical evaluation in a variety of additional human tumors with cyclin D1 overexpression and/or accumulation.
During initiation and progression, tumor cells reprogram metabolism to provide energy and necessary metabolites for cell duplication.6 To achieve these goals, tumor cells enhance the utilization of glucose in a process known as the Warburg effect, and/or the metabolism of glutamine known as active glutaminolysis. Elucidation of metabolic characteristics associated with specific types of cancers and dissection of the genetic drivers that are associated with these metabolic characteristics will facilitate the development of new therapies.
Glutamine is metabolized in a process termed glutaminolysis. Following uptake, glutamine is initially converted to glutamate via glutaminase (GLS) that catalyzes the rate-limiting step. Previously, glutamine was considered as a non-essential amino acid (NEAA); however, recent research suggests that glutamine is a conditionally essential amino acid, particularly for highly proliferative cells. The importance of glutamine is highlighted by its contribution as (1) an energy source, as the metabolite of glutamine, α-ketoglutarate enters into the citric acid cycle for ATP production; (2) a nitrogen source, for the biosynthesis of NEAAs and nucleotides; and (3) a substrate-donor for the synthesis of glutathione, a factor to resist reactive oxygen species.5 As such, some tumor cells demonstrate a specific metabolic characteristic, glutamine-addiction, indicating tumor cells require glutamine for survival and proliferation. In accordance, tumor cells will increase glutamine uptake and elevate GLS expression. Glutamine-addiction is regulated through the activation of oncogenes (MYC proto-oncogene, bHLH transcription factor (MYC), MYCN proto-oncogene, bHLH transcription factor (MYCN), Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2)) or the loss of tumor suppressors (Tumor Protein P53 (TP53) and RB).7–9 However, it remains elusive whether other pathways also control tumor cells’ addiction to glutamine that can be targeted for therapy.
We recently elucidated the underlying mechanism how dysregulation of the FBXO4-cyclin D1-RB axis promotes glutamine-addiction, and through this study we identified a potential therapeutic weakness for ESCC and for overcoming CDK4/6 inhibitor resistance.5 ESCC is a particularly important tumor type for evaluation given the following reasons: (1) it an aggressive and lethal malignancy, with no effective therapy for late-stage ESCC; (2) cyclin D1 is considered as a driver of ESCC; (3) importantly, it remains unclear whether dysregulated FBXO4-cyclin D1-RB axis can be therapeutically targeted in ESCC. Our work revealed that cyclin D1-CDK4/6-RB dysregulation is necessary and sufficient to induce glutamine-addiction in ESCC-derived cells.
Mechanistically, cyclin D1-CDK4/6 kinases can promote cell proliferation and growth through inactivation of RB and activation of mammalian Target of Rapamycin Complex 1 (mTORC1); accordingly, these cells need to increase glutamine uptake in order to provide sufficient nutrients (Figure 1). Other oncogenes like MYC or MYCN not only enhance glutamine uptake, but also upregulate GLS expression, thereby contributing to glutamine-addiction. Hyperactivation of cyclin D1-CDK4/6 also increases glutamine uptake and GLS expression. While the precise mechanism of GLS upregulation remains unclear, current data demonstrate this increase is independent of MYC. Given the dependency on RB, it is tempting to speculate that GLS might be an E2F target. However, this remains to be determined.
Figure 1.
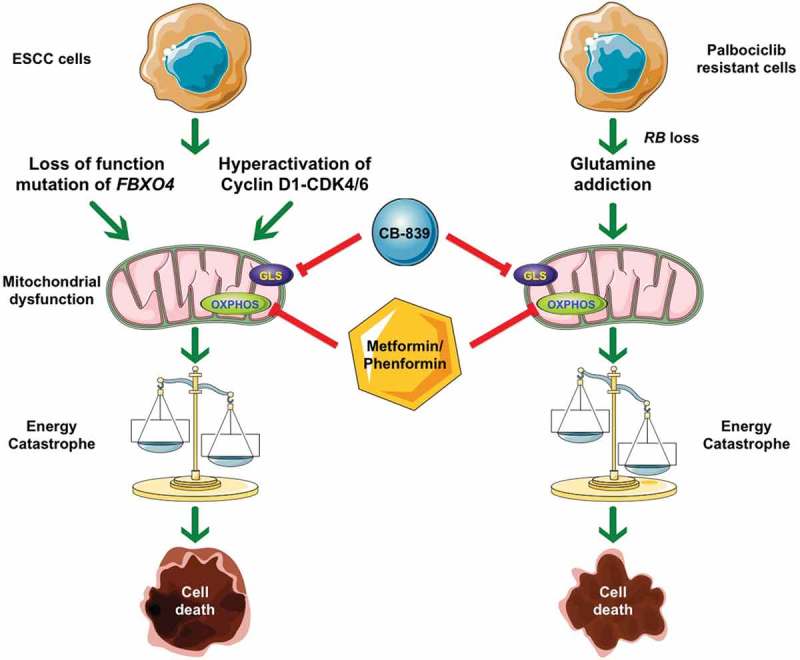
Molecular mechanisms of combined treatment with CB-839 and metformin/phenformin. 1) Loss of function mutations in F-Box Protein 4 (FBXO4) or hyperactivation of the Cyclin D1 (CCND1)-cyclin-dependent kinase 4/6 (CDK4/6) leads to mitochondrial dysfunction and glutamine-addiction. Taking advantage of these metabolism characteristics, targeting both glutaminase (GLS) and oxidative phosphorylation (OXPHOS) causes the imbalance between energy production and consumption, thereafter, energetic catastrophe, and finally cell death. 2) Palbociclib-resistant cells demonstrate RB transcriptional corepressor 1 (RB) loss that drives glutamine-addiction; moreover, these cells are more sensitive to combined treatment when compared with their parental counterparts. ESCC: esophageal squamous cell carcinoma.
In addition to drive glutamine-addiction, overexpression of cyclin D1 also compromised mitochondrial activity and oxidative phosphorylation (OXPHOS), leading to defective energy production. The above mentioned metabolic characteristics make tumor cells with hyperactivated cyclin D1-CDK4/6 kinases sensitive to combined treatment with CB-839 (GLS inhibitor) and metformin, which triggers an energy catastrophe, resulting in cell death in cell culture and in mouse xenografts.
Although CDK4/6 inhibitors are efficient and tumor cells respond well initially, tumors will invariably acquire resistance. As such, there is an urgent need to develop an alternative treatment to bypass this resistance.10 With regard to CDK4/6 inhibitors, there are concerted efforts to elucidate the molecular mechanisms through which tumor cells can develop resistance. One documented mechanism that triggers resistance to CDK4/6 inhibitors is the loss of RB. RB loss obviates the need for cyclin D1-dependent kinases, and as such endears cells with resistance to CDK4/6 inhibitors. Indeed, we found that acquired resistance to CDK4/6 inhibitors is typically associated with RB loss in ESCC. Consistent with this new work focusing on hyperactivation of cyclin D1 and glutamine-addiction, we observed dysregulation of glutamine metabolism enzymes in palbociclib-treated cells. An upregulation of GLS and increased glutamine uptake was consistently noted in palbociclib-resistant cells relative to their parental controls. As expected with this signature, CDK4/6 inhibitor-resistant cells are sensitive to CB-839+metformin in vitro and in vivo.
Collectively, this new work revealed that dysregulated FBXO4-cyclin D1 axis reprograms cellular metabolism through RB inactivation and mTORC1 activation. Moreover, cyclin D1 overexpression leads to mitochondrial deficiency, thereafter, insufficient energy production. These metabolism characteristics make ESCC cells susceptible to combined treatment with GLS inhibitor plus metformin/phenformin in vitro and in xenograft tumors. Importantly, CB-839+metformin has therapeutic efficacy for ESCC cells with acquired resistance to CDK4/6 inhibitors.
Funding Statement
This study is supported by the National Institutes of Health (P01 CA098101 J.A.D.; T32 DE017551 S.Q.); Foundation for the National Institute of Dental and Craniofacial Research [T32 DE017551]; Foundation for the National Cancer Institute [P01 CA098101].
Acknowledgments
The authors appreciate the assistance from Core Facilities and Shared Resources at the Medical University of South Carolina and Hollings Cancer Center. The work of the authors is supported by the National Institutes of Health (P01 CA098101, J.A.D.; T32 DE017551 S.Q.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Qie S, Diehl JA.. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl). 2016;94:1313–1326. doi: 10.1007/s00109-016-1475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL.. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 3.Barbash O, Zamfirova P, Lin DI, Chen X, Yang K, Nakagawa H, Lu F, Rustgi AK, Diehl JA. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell. 2008;14:68–78. doi: 10.1016/j.ccr.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qie S, Majumder M, Mackiewicz K, Howley BV, Peterson YK, Howe PH, Palanisamy V, Diehl JA. Fbxo4-mediated degradation of Fxr1 suppresses tumorigenesis in head and neck squamous cell carcinoma. Nature Communications. 2017;8:1534. doi: 10.1038/s41467-017-01199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qie S, Yoshida A, Parnham S, Oleinik N, Beeson GC, Beeson CC, Ogretmen B, Bass AJ, Wong KK, Rustgi AK, et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat Commun. 2019;10:1296. doi: 10.1038/s41467-019-09179-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin C, Qie S, Sang N. Carbon source metabolism and its regulation in cancer cells. Crit Rev Eukaryot Gene Expr. 2012;22:17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qie S, Chu C, Li W, Wang C, Sang N. ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J Cell Biochem. 2014;115:498–509. doi: 10.1002/jcb.24684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang Y, Stine ZE, Xia J, Lu Y, O‘Connor RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest. 2015;125:2293–2306. doi: 10.1172/JCI75836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolay BN, Gameiro PA, Tschop K, Korenjak M, Heilmann AM, Asara JM, Stephanopoulos G, Iliopoulos O, Dyson NJ. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013;27:182–196. doi: 10.1101/gad.206227.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34:9–20. doi: 10.1016/j.ccell.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]