

ABSTRACT
The aim of the study was to determine the acute contribution of fuel oxidation in mediating the increase in insulin secretion rate (ISR) in response to fatty acids. Measures of mitochondrial metabolism, as reflected by oxygen consumption rate (OCR) and cytochrome c reduction, calcium signaling, and ISR by rat islets were used to evaluate processes stimulated by acute exposure to palmitic acid (PA). The contribution of mitochondrial oxidation of PA was determined in the presence and absence of a blocker of mitochondrial transport of fatty acids (etomoxir) at different glucose concentrations. Subsequent to increasing glucose from 3 to 20 mM, PA caused small increases in OCR and cytosolic calcium (about 20% of the effect of glucose). In contrast, the effect of PA on ISR was almost 3 times that by glucose, suggesting that the metabolism of PA is not the dominant mechanism mediating PA’s effect on ISR. This was further supported by lack of inhibition of PA-stimulated OCR and ISR when blocking entry of PA into mitochondria (with etomoxir), and PA’s lack of stimulation of reduced cytochrome c in the presence of high glucose. Consistent with the lack of metabolic stimulation by PA, an inhibitor of calcium release from the endoplasmic reticulum, but not a blocker of L-type calcium channels, abolished the PA-induced elevation of cytosolic calcium. Notably, ISR was unaffected by thapsigargin showing the dissociation of endoplasmic reticulum calcium release and second phase insulin secretion. In conclusion, stimulation of ISR by PA was mediated by mechanisms largely independent of the oxidation of the fuel.
KEYWORDS: Palmitate, oxygen consumption, islets, cytochrome c, calcium, endoplasmic reticulum, L-type calcium channels
Introduction
Fatty acids (FA) have both acute and chronic effects on beta cell function and viability.1,2 Accordingly, much attention has been directed at understanding the mechanisms mediating their affect. A fundamental question that has not been definitively addressed is whether FAs can be a source of energy through oxidation, since this has implications both in regard to the mechanism of FA-stimulated insulin secretion, and the understanding of how insulin secretion is regulated. Previous studies overall support an oxidative role for endogenous fatty acids in energy generation.3-5 In addition, it is established that FAs can reduce glucose oxidation;4,6 however, a consensus from past studies has not emerged on the extent that exogenous FAs can acutely stimulate mitochondrial bioenergetics.6-10 Part of the controversy arises due to the difficulty in interpreting data obtained using radiolabeled substrate and measuring the release of CO2, as the measurements are greatly influenced by unknowable dilution of radiolabel by intracellular pools.5,11 In addition, many groups have observed that FAs can activate L-type calcium (Ca2+)channels,10,12-14 suggesting that FAs can close ATP-sensitive potassium channels by increased ATP/ADP ratio. To resolve this issue, comparison of oxygen consumption rate (OCR), a signal that at steady state closely approximates ATP synthesis in islets, in response to glucose and FAs is needed.
In this study, we focused on palmitic acid (PA), a FA that is particularly potent in stimulating insulin secretion rate (ISR).15 In vitro studies have nicely mirrored those seen in vivo: PA amplifies glucose-stimulated ISR by isolated islets,10,12,13,16 and most studies have reported that PA is ineffective in stimulating ISR in the presence of low glucose.10,16-19 Virtually all studies have found that PA can stimulate cytosolic Ca2+, and most have found that activation of L-type Ca2+ channels is involved.10,12-14 A contribution to the Ca2+ response has also been attributed to the release of Ca2+ from the endoplasmic reticulum (ER).6,10,12,20 A number of candidate processes have been suggested as drivers of the Ca2+ redistribution including increased ATP production due to beta-oxidation,3,5,8 KATP-independent stimulation of L-type Ca2+ channels,12 and direct activation of a G protein-coupled receptor GPR40.20,21
How PA increases ISR is also still not completely understood. There is evidence for the involvement of endogenous FAs via lipid signals generated from long-chain CoA such as diacylglycerol, which stimulates protein kinase C and ultimately glucose-activated ISR.16,22 This mechanism operates in concert with glucose-induced inhibition of transport of FA into the mitochondria by carnitine palmitoyltransferase 1 (CPT1) via increased malonyl CoA.23 There is also evidence for an important role for GPR40,24,25 and there is an ongoing challenge of determining the relative contribution of these pathways. Although it is commonly stated that an increase in cytosolic Ca2+ leads to an increase in ISR,26,27 we have recently reported on the lack of effect of Ca2+ release from the ER in mediating acetylcholine’s effect on ISR.28 This data supported previous studies showing that only Ca2+ entering the cell via L-type Ca2+ channels impinge on mechanisms mediating insulin secretion, which appear to be localized to a microdomain surrounding the channels.29,30
The data generated in the paper utilized a custom-made islet perifusion system that concomitantly measured OCR, and a measure of mitochondrial redox state (reduced cytochrome c) while collecting outflow fractions for subsequent measurement of ISR.31 OCR was accurately quantified in real time as the difference between the amount of oxygen flowing in and out of the islet perifusion chamber times the flow rate. Real-time CA2+ imaging was done using the same perfusion system but with an optical imaging dish rather than a perifusion column. The multi-parametric, real-time approach enables optimal comparison between parameters reflecting different stages of stimulus-secretion coupling and has been used for many mechanistically based studies of islet physiology.28,32-34
To take advantage of real-time methods, we utilized acute and pharmacologic approaches to assess rapid mechanisms mediating metabolic signaling and insulin secretion. Overall, based on the lack of effect of PA on ISR at low glucose, we predicted that PA would not be utilized significantly as a fuel to increase ATP production, nor increase Ca2+ influx via voltage-dependent Ca2+ channels. We further predicted, based on our previous studies showing that the release of Ca2+ from the ER does not mediate ISR,28 stimulation of ISR by PA would be mediated by mechanisms independent of its effects on Ca2+. These predictions were borne out by the present study and provide a biochemical explanation for there being a safeguard against over-production of insulin during normo- or hypoglycemia.
Results
Effect of PA on OCR is small and not dependent on glucose concentration
In order to assess the acute contribution of metabolism to PA’s effect on ISR, the changes in three parameters, OCR, cytosolic Ca2+ and ISR, were measured in response to glucose followed by a 45-min exposure to 100 µM PA. Both glucose and PA increased OCR and ISR, but stimulation of ISR relative to OCR for the two agents was very different (Figure 1, Table 1). Whereas the increase in OCR caused by PA was only 17% of that by glucose, the increase in ISR was nearly threefold. The dramatically different ratio of ISR to change in OCR induced by PA indicates that the mechanism mediating PA’s stimulation of Ca2+ and ISR is different from that by glucose, which is known to involve increased metabolic rate.
Figure 1.
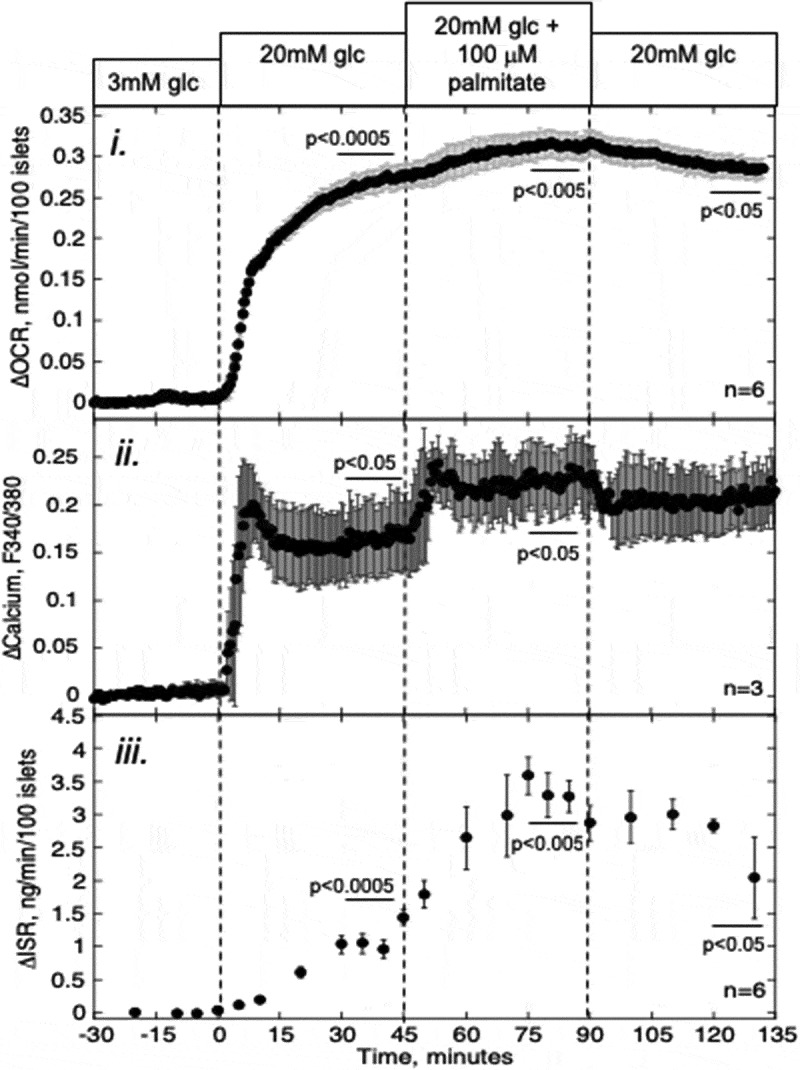
Effect of PA on glucose-stimulated OCR, Ca2+, and ISR. Islets were perifused in the presence of 3 mM glucose for 90 min. Subsequently, at time = 0 on the graph, glucose was increased to 20 mM for 45 min, followed by exposure to PA for 45 min and a 45-min washout period as indicated. i and iii: OCR, and ISR were measured concomitantly using the flow culture system. ii: Detection of cytosolic Ca2+ by fluorescence imaging (measured in separate experiments). Data are displayed as the change in signal relative to the steady-state value obtained at 3 mM glucose (determined by averaging data obtained in the final 15 min prior to the increase in glucose). Steady-state values of OCR and ISR at 3 mM glucose were 0.35 ± 0.065 nmol/min/100 islets (n = 6) and 0.23 ± 0.052 ng/min/100 islets (n = 6), respectively. Statistical analysis was carried out by comparing steady-state values (determined by averaging data obtained in the final 15 min of each experimental condition) before and after each change in media composition using a paired t-test.
Table 1.
The effect of substrates and agents on changes in steady-state values of OCR and ISR in rat pancreatic islets relative to the baseline of the previous condition. Data were taken from Figure 1–3 and 5,6, and each steady-state value shown in the table was the average of the final 15 min of the indicated condition. Statistical analysis was carried out by comparing steady-state values before and after each change in media composition using a paired t-test. Abbreviations: CPT1 = carnitine palmitoyl transferase1; PA = palmitic acid.
| Process affected | Conditions | ΔOCR (nmol/min/100 islets) | ΔISR (ng/min/100 islets) |
|---|---|---|---|
| Effect of PA in high glucose (Figure 1) | Cond 1: 20 mM glucose | +0.27 ± 0.035 (n = 6, p < .0005) | +1.1 ± 0.13 (n = 6, p < .0005) |
| Cond 2: 20 mM glucose +100 μM PA | +0.039 ± 0.008 (n = 6, p < .005) | +2.1 ± 0.33 (n = 6, p < .005) | |
| Effect of PA and CPT1 in low glucose (Figure 2) | Cond 1: 3 mM glucose + 100 μM PA | +0.062 ± 0.008 (n = 6, p < .001) | +0.13 ± 0.02 (n = 6, p < .005) |
| Cond 2: 3 mM glucose + 100 μM PA + 200 µM etomoxir | −0.066 ± 0.006 (n = 6, p < .0001) | +0.36 ± 0.078 (n = 6, p < .01) | |
| Cond 3: 20 mM glucose | +0.35 ± 0.022 (n = 6, p < .0001) | +2.0 ± 0.42 (n = 6, p < .01) | |
| Effect of PA while blocking CPT1 in high glucose (Figure 3A) | Cond 1: 20 mM glucose | +0.40 ± 0.031 (n = 7, p < .0001) | +2.3 ± 0.28 (n = 7, p < .0005) |
| Cond 2: 20 mM glucose + 200 µM etomoxir | −0.022 ± 0.009 (n = 7, p = .05) | +1.3 ± 0.36 (n = 7, p < .01) | |
| Cond 3: 20 mM glucose + 200 µM etomoxir + 100 μM PA | +0.036 ± 0.012 (n = 7, p < .05) | +1.6 ± 0.41 (n = 7, p < .01) | |
| Effect of PA while blocking CPT1 in low glucose (Figure 3B) | Cond 1: 3 mM glucose + 200 µM etomoxir | −0.083 ± 0.009 (n = 8, p < .0001) | +0.024 ± 0.007 (n = 8, N.S.) |
| Cond 2: 3 mM glucose + 200 µM etomoxir + 100 μM PA | +0.048 ± 0.008 (n = 8, p = .001) | +0.16 ± 0.021 (n = 8, p < .005) | |
| Cond 3: 20 mM glucose | +0.35 ± 0.052 (n = 8, p < .0005) | +1.47 ± 0.24 (n = 8, p < .0005) | |
| Effect of PA after depleting the endoplasmic reticulum calcium stores (Figure 5) | Cond 1: 20 mM glucose | +0.35 ± 0.062 (n = 4, p < .05) | +2.0 ± 0.29 (n = 4, p < .01) |
| Cond 2: 20 mM glucose + 5 µM thapsigargin | −0.013 ± 0.006 (n = 4, N.S.) | +1.0 ± 0.13 (n = 4, p < .005) | |
| Cond 3: 20 mM glucose + 5 µM thapsigargin + 100 µM PA | −0.038 ± 0.002 (n = 4, N.S.) | +1.6 ± 0.38 (n = 4, p < .05) | |
| Effect of PA during blockade of calcium influx (Figure 6) | Cond 1: 20 mM glucose | +0.28 ± 0.023 (n = 6, p < .001) | +1.2 ± 0.22 (n = 4, p = .01) |
| Cond 2: 20 mM glucose + 5 µM nimodipine | −0.062 ± 0.013 (n = 6, p < .01) | − 1.2 ± 0.21 (n = 4, p = .01) | |
| Cond 3: 20 mM glucose + 5 µM nimodipine + 100 µM PA | +0.038 ± 0.011 (n = 6, p < .05) | −0.05 ± 0.05 (n = 4, N.S.) |
Since at high glucose, malonyl CoA (an inhibitor of CPT1) is likely to be elevated,35 PA might have had little effect on OCR because its entry into the mitochondria was inhibited. We surmised that PA might be metabolized to a greater extent by islets at low glucose, and if so, would lead to an increase in both OCR and ISR. Indeed, the increase in OCR in response to 100 µM PA at 3 mM glucose was about 60% greater than that seen at 20 mM glucose (Figure 2, Table 1). Despite this increase in OCR, ISR was only slightly stimulated (by less than 10% of the response to glucose).
Figure 2.
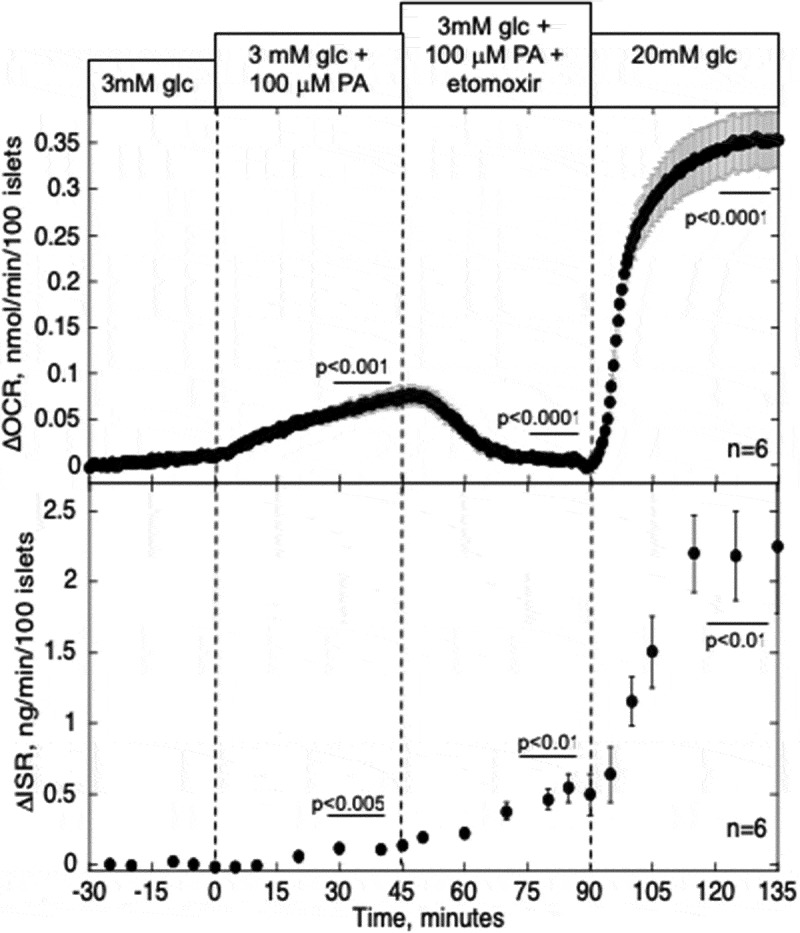
Effect of PA and blockade of CPT1 on OCR and ISR in the presence of low glucose. Islets were perifused in the presence of 3 mM glucose for 90 min. Subsequently, 100 µM PA was added to the inflow, and after 45 min a blocker of CPT1, etomoxir (200 µM), was also added. OCR, and ISR were measured concomitantly using the flow culture system. Steady-state values of OCR and ISR at 3 mM glucose were 0.43 ± 0.009 nmol/min/100 islets (n = 6) and 0.16 ± 0.052 ng/min/100 islets (n = 6), respectively. Data are displayed and processed as described in the legend of Figure 1.
Effect of blocking CPT1 on PA-stimulated OCR and ISR
In order to more directly assess whether the small increase in OCR in response to acute exposure of PA was due to its metabolism by beta-oxidation, transport of PA into the mitochondria was blocked by an inhibitor of CPT1, the major mitochondrial transporter of fatty acids. In the presence of 3 mM glucose, etomoxir decreased OCR by a similar amount that was increased by PA (Figure 2, Table 1), and remarkably, ISR was increased to almost 50% of a glucose response.
At high glucose, 200 µM etomoxir had a greatly reduced effect on OCR relative to its effect at 3 mM glucose (Figure 3A) and increased ISR (as seen in the previous studies36). In the presence of etomoxir, PA had similar increases in OCR and ISR as it did in its absence. Thus, in the presence of high levels of glucose, it does not appear that entry into, and metabolism by, mitochondria are necessary for PA to stimulate OCR or ISR.
Figure 3.
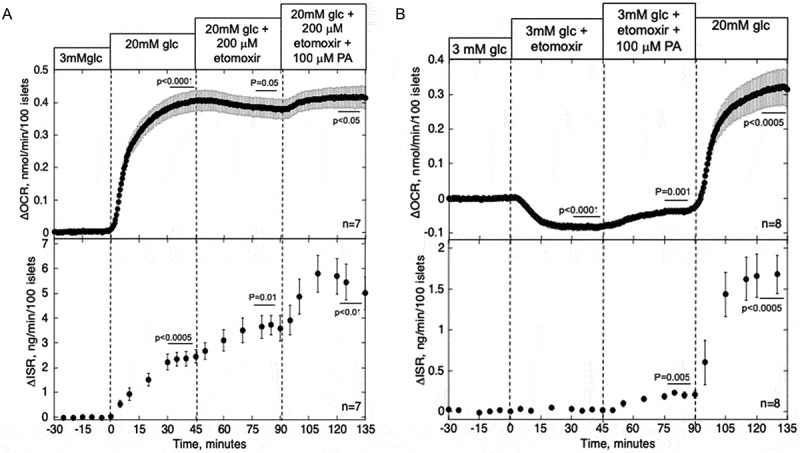
Effect of blockade of CPT1 and PA on OCR and ISR in the presence of high (A) and low (B) glucose. Islets were perifused in the presence of 3 mM glucose for 90 min. Islets were then exposed to etomoxir (200 µM) for 45 min and both etomoxir and PA (100 µM) for 45 min either after (A) or before (B) glucose was increased to 20 mM for 45 min. OCR and ISR were measured concomitantly using the flow culture system. Steady-state values of OCR and ISR at 3 mM glucose were 0.41 ± 0.056 nmol/min/100 islets (n = 7) and 0.14 ± 0.059 ng/min/100 islets (n = 7), respectively for the sets shown in A, and 0.50 ± 0.040 nmol/min/100 islets (n = 8) and 0.24 ± 0.064 ng/min/100 islets (n = 8), respectively for the sets shown in B. Data are displayed and processed as described in the legend of Figure 1.
To confirm that etomoxir blocks CPT1, its effect was tested at low glucose, a condition where CPT1 activity will not be suppressed by malonyl CoA. Etomoxir in the presence of 3 mM glucose inhibited OCR 3.8-fold over that observed in the presence of 20 mM glucose (Figure 3B, Table 1). PA-stimulated OCR was less than in the absence of etomoxir, but only by 23% (0.048 ± 0.008 vs. 0.062 ± 0.008).
PA-stimulated OCR is mediated by increased ATP usage rather than a push from mitochondrial oxidation
The previous results indicated that PA-stimulated OCR was little affected by mitochondrial metabolism of PA at high glucose, and only slightly at low glucose. Accordingly, we next considered what was driving the PA-induced changes in OCR. OCR can be stimulated by increased metabolic supply of NADH (referred to as a push system), or alternatively by increased ATP usage thereby elevating ADP levels and pulling electrons through the electron transport chain.37 To distinguish the two cases, we measured real-time changes in the reductive state of cytochrome c, which increases in response to NADH and remains unaltered in the face of changes in OCR mediated by increased ATP usage.28 Glucose increased both OCR and the reductive state of cytochrome c, reflecting the increased generation of NADH (increments were 0.29 nmol/min/100 islets (p < .001) and 25% (p < .01) respectively) (Figure 4). In contrast, the increase in OCR induced by PA (0.055 nmol/min/100 islets (p < .001)) was concomitant with an increase in cytochrome c reduction of only 2% (p < .01). The ratio of changes in cytochrome c reduction/OCR induced by PA is very small compared to that for glucose, suggesting that PA-induced increase in OCR is not due to increased generation of NADH production by the oxidation of PA.
Figure 4.
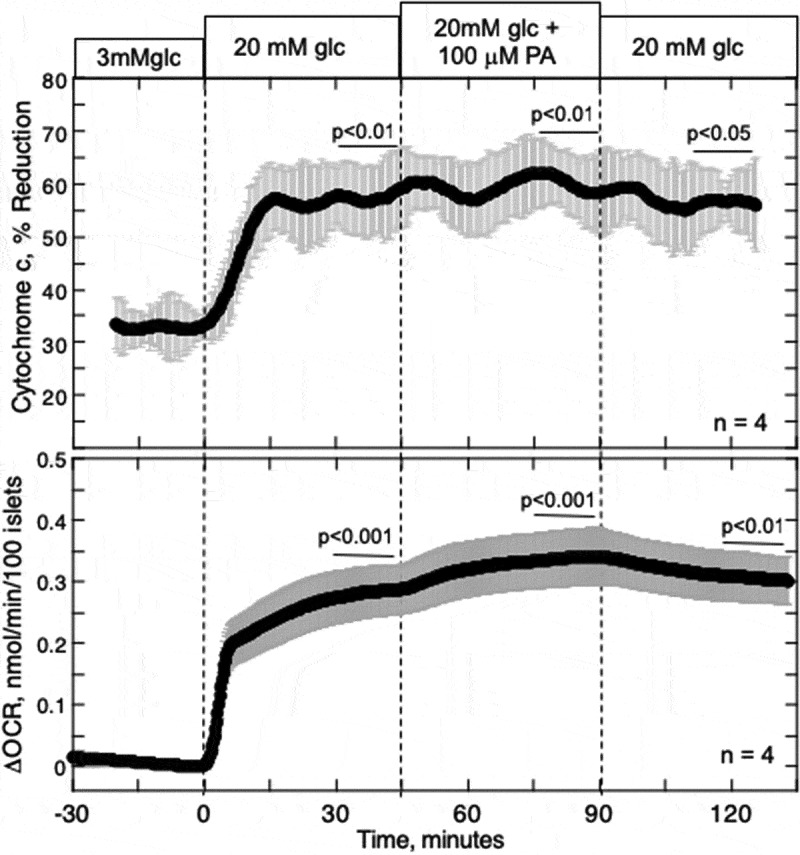
Effect of PA on glucose-stimulated reduced cytochrome c and OCR. Islets were perifused in the presence of 3 mM glucose for 90 min. Subsequently, glucose was increased to 20 mM for 45 min, followed by exposure to PA for 45 min and a 45-min washout period as indicated. Cytochrome c reduction and OCR were measured simultaneously using our flow culture system.
Increased intracellular Ca2+ induced by PA is not mediated by increased L-type Ca2+ channel activity
As another test of PA’s ability to stimulate metabolism in islets, the source of Ca2+ that mediated PA-induced cytosolic Ca2+ was determined. Secretagogues that are metabolized by islets, including glucose, glyceraldehyde, and KIC,32 all increase cytosolic Ca2+ via L-type Ca2+ channels. As PA only had a small effect on metabolism, we expected that the increase in Ca2+ would be primarily due to its release from the ER. To test this, thapsigargin, a blocker of sarcoendoplasmic reticulum Ca2+ ATPase (SERCA), was used to empty the ER, and subsequently, islets were stimulated with PA (Figure 5). As we have found previously,28 thapsigargin caused a rapid increase in cytosolic Ca2+, but only a minor and delayed increase in ISR. In the presence of thapsigargin, 80% of Ca2+ response to PA was inhibited, consistent with our expectation that ER Ca2+ mediates the PA-induced rise in cytosolic Ca2+. PA-stimulated ISR was similar to that seen in the absence of thapsigargin, supporting previous observations that ER Ca2+ has a little acute effect on insulin secretion.28
Figure 5.
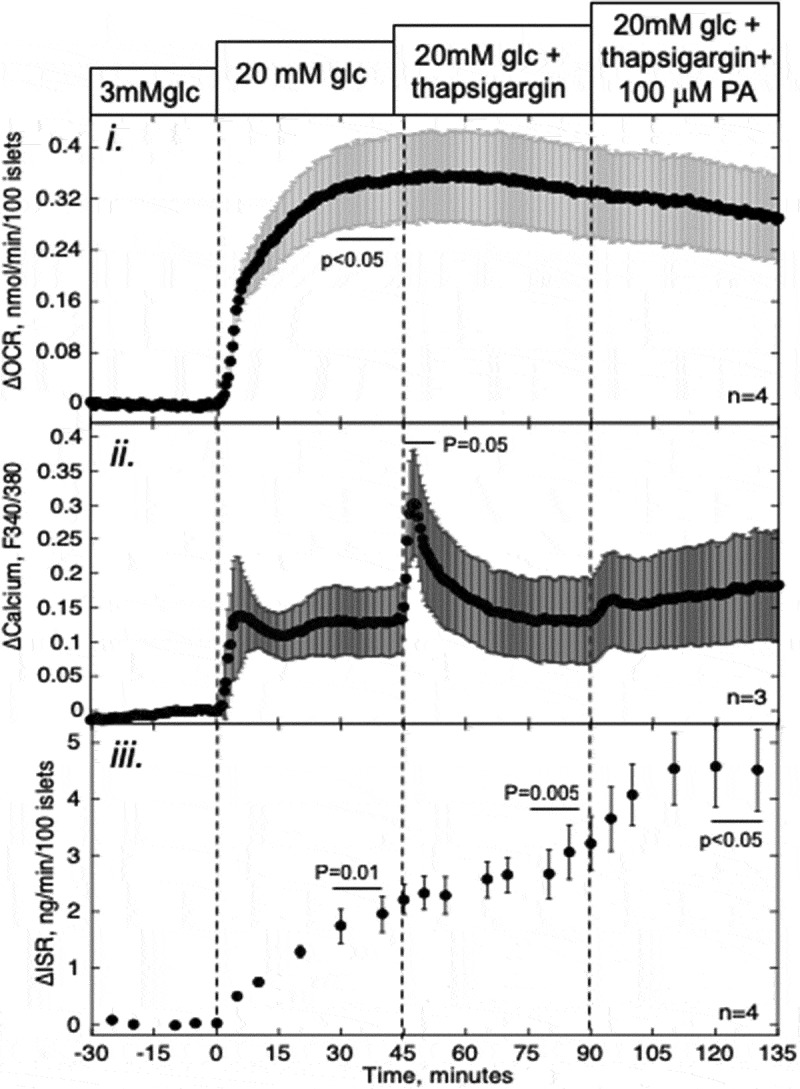
Effect of PA on glucose-stimulated OCR, Ca2+, and ISR after depleting ER Ca2+ pools. Islets were perifused in the presence of 3 mM glucose for 90 min. Subsequently, glucose was increased to 20 mM for 45 min, followed by a 45-min exposure to a blocker of the SERCA (thapsigargin (5 μM)) and stimulation by PA for 45 min as indicated. i and iii: OCR, and ISR were measured concomitantly using the flow culture system. ii: Detection of cytosolic Ca2+ by fluorescence imaging (measured in separate experiments). Steady-state values of OCR and ISR at 3 mM glucose were 0.49 ± 0.046 nmol/min/100 islets (n = 4) and 0.18 ± 0.028 ng/min/100 islets (n = 4), respectively. Data are displayed and processed as described in the legend of Figure 1.
To further verify that ER Ca2+, and not L-type Ca2+ channels, mediate PA-induced stimulation of cytosolic Ca2+, the effect of PA was tested in the presence of nimodipine, a blocker of L-type Ca2+ channels. Nimodipine blocked about 70%, 30%, and 95% of glucose-stimulated cytosolic Ca2+, OCR and ISR (Figure 6). Subsequently, PA increased Ca2+ and OCR by almost identical amounts as that seen in the absence of nimodipine. Thus, blocking ER Ca2+ release, but not L-type Ca2+ channels, inhibited PA-stimulation of cytosolic Ca2+, providing further support that PA’s effect is not mediated by a significant effect on islet metabolism.
Figure 6.
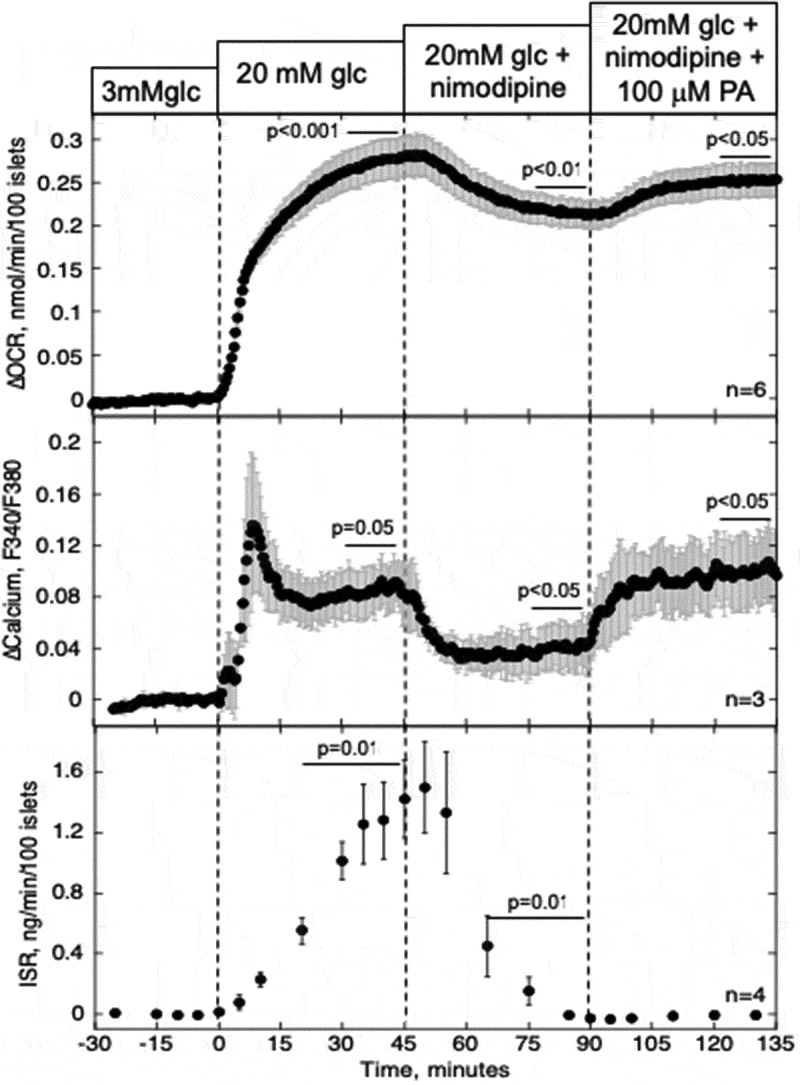
Effect of PA on glucose-stimulated OCR, Ca2+, and ISR after blocking Ca2+ influx through L-type Ca2+ channels. Data collection and analysis was done identically to that described in Figure 5 except that nimodipine (5 μM) was used instead of thapsigargin. Steady-state values of OCR and ISR at 3 mM glucose were 0.29 ± 0.042 nmol/min/100 islets (n = 6) and 0.11 ± 0.044 ng/min/100 islets (n = 4), respectively.
Discussion
As with glucose, FAs are elevated in diabetes38,39 and have major effects on islet function and viability. As an important fuel for many tissues,40 FAs may impact islets through alterations in mitochondrial metabolism.4,8,41 The present study was designed to assess the relative contribution of metabolism to the increased ISR induced by a representative FA, PA. In addition, we aimed to evaluate the role of Ca2+ signaling in mediating PA’s effects. The major findings of our study are that PA increased OCR by stimulating ATP usage rather than increased beta-oxidation; the increase in metabolic rate was not an important mechanism for stimulating ISR; increased Ca2+ in response to PA occurs via release from the ER, not activation of L-type Ca2+ channels; and the acute effect of PA on ISR (3–45 min) is mediated downstream of Ca2+ by non-oxidatively generated signals. These results have implications on the role of FA in governing insulin secretion and fundamental mechanisms mediating stimulus-secretion coupling.
PA can amplify but not trigger insulin secretion
In general, nutrients that are metabolized by pancreatic beta cells can independently stimulate ISR. Glucose, glyceraldehyde, KIC and some amino acids such as leucine, stimulate metabolism that leads to both the closure of KATP channels and generation of essential metabolic cofactors that together are sufficient to elicit ISR. A common factor amongst this class of secretagogues is the ability to increase the reductive state of cytochrome c, without which activation of insulin secretion does not occur.32,34 A second class of secretagogues such as acetylcholine and GLP-1, are unable to stimulate ISR independently, but can potentiate nutrient-stimulated ISR. The results of this study place PA in the latter category. The increase in OCR in response to PA in the presence of high glucose was small (less than 20% of the increase induced by glucose) and did not appear to be mediated by increased metabolic generation of NADH that would have come about from beta-oxidation. The latter was based on a lack of effect of a blocker of CPT1 on PA-stimulated OCR at high glucose, and only a very limited response in reduced cytochrome c (a marker of metabolic generation of NADH). OCR can be increased either by substrate oxidation, or increased ADP caused by cellular work (i.e. activation of steps that lead to greater ATP usage).31,42 The lack of increase in cytochrome c reduction indicates that PA-induced increases in OCR were not due to increased oxidation. The most likely factor mediating the increased OCR in response to PA was ATP usage, which can enhance OCR in the absence of changes in reduced cytochrome c by the action of ADP.43 In this study, we did not directly address what ATP-utilizing processes were stimulated by PA, although this will be addressed in future studies. It is possible that PA could cause a small amount of mitochondrial uncoupling resulting in increased OCR, which has been previously observed in response to chronic exposure to FA.44,45 However, uncoupling mitochondria in response to FCCP acutely increases cytochrome c reduction (unpublished results), and this was not observed in these studies. In addition, uncoupling by free FAs, in general, takes 2 days,45 and the acute effects of other free fatty acids such as the unsaturated FAoleate and stearate leads to increases in OCR and ISR but have less effect on mitochondrial uncoupling.46 In total, based on all these observations, we favor stimulated ATP usage as the mediator of PA-stimulated OCR.
This is not to say that PA is not oxidized, which has been shown by studies using radiolabeled PA.8,9,47 Indeed, studies have shown that increased FA metabolism is offset by a decrease in glucose metabolism.9,48 As the net change in metabolic rate induced by PA was very small, it follows that metabolism does not contribute significantly to its potentiation of glucose-stimulated ISR. Teleologically, this mechanism makes sense for survival, since it prevents an animal from becoming hypoglycemic when FAs are elevated in normal glucose, and lessens mitochondrial burden when glucose is elevated.
PA increased cytosolic Ca2+ by the release of Ca2+ from the ER
In contrast to most studies of islet response to PA where PA was found to increase Ca2+ influx,10,12-14 we found that PA-induced elevation of cytosolic Ca2+ was blocked by emptying the ER Ca2+ stores, but not by inhibition of L-type Ca2+ channels. We considered a number of factors that could explain the difference in results between studies. Our studies were done on rat islets, whereas most studies showing that PA activates Ca2+ influx were carried out on mouse islets, which are generally more electrically excitable than rat islets. The concentration of PA that was used in our study (0.1 mM PA) was on the low end of what is typically used in in vitro studies (ranging from 0.1 to 1 mM.10,12-14 However, it was well above what is needed to elicit a robust insulin secretory response and above the physiologic range.49,50 Whatever the differences may be, the results obtained in this study are consistent with the inability of PA to stimulate metabolism and suggests that stimulation of L-type Ca2+ channels is not a major process mediating PA-stimulated cytosolic Ca2+.
The role of Ca2+ release from the ER in stimulating ISR
It is commonly stated that an increase in cytosolic Ca2+ results in stimulation of ISR. However, we, and others, have published data supporting the exclusive role of L-type Ca2+ channels in the control of second phase ISR.28,30,51 The findings that neither thapsigargin- nor PA-induced elevation of cytosolic Ca2+ via changes in exchange between the ER and the cytosol affected ISR, mirrored those previously obtained using acetylcholine.28 It was considered whether ER stress, known to be induced by the use of thapsigargin, could have affected our results. However, thapsigargin typically takes 24 h before ER stress is activated,52 and the lack of effect of thapsigargin on OCR also provides support that ER stress was not activated during these acute experiments. In addition, nimodipine, a blocker of L-type Ca2+ channels, completely suppressed glucose- and PA-stimulated ISR. These findings illustrate an important safeguard of beta cell function that prevents the secretion of insulin unless glucose stimulates an energy-dependent process. Acetylcholine and FAs cannot cause hypoglycemia because of an inappropriate release of Ca2+ from the ER, and there is an absolute requirement for Ca2+ influx through L-type Ca2+ channels. Conclusions drawn from many studies have been based on the assumption that an increase in cytosolic Ca2+ will lead to increased ISR, irrespective of the source of the Ca2+,53,54 and this critical issue should receive more attention.
CPT1 mediated PA-stimulated ISR
Our data were consistent with the previously established role of CPT1 as a metabolic switch. CPT1 has been elegantly shown to shut off at high glucose via inhibition by elevated malonyl CoA. and 35,36,55,56 Accordingly, there was a larger decrease in OCR that occurred in response to etomoxir at low glucose vs. high glucose reflecting suppression of CPT1 by high glucose. Although there is evidence that etomoxir can inhibit complex 1,57 this effect has not been shown to be glucose-dependent. However, since we are using high levels of etomoxir, we cannot rule out that this is the cause of some of the inhibition of OCR. Nonetheless, etomoxir also acutely increased ISR which would also argue against a significant loss of ATP production due to inhibition of complex 1. Importantly, an increase in ISR caused by etomoxir is consistent with a redirection of FAs away from beta-oxidation and toward the generation of long-chain CoA that function as amplifying signals for ISR. Apparently, any small contribution that endogenous FAs may have made to increase ISR via increased ATP production was not significant.
Providing further support for the role of non-oxidative signals generated from FAs, PA in the presence of etomoxir stimulated ISR at low glucose. Although the increase was not as large as with glucose, we interpret this as a demonstration that signals generated from PA independently of mitochondrial entry impact ISR, although we cannot rule out the involvement of PPARα.58 There are very few agents that increase ISR at sub-stimulatory levels of glucose, and the data begs the question of what is the PA-generated signal that is mediating its effect? Although we did not address this in our study, the esterified lipid product that is thought to be an important contributor to FA-stimulated ISR is diacylglycerol, which directly activates protein kinase C.59,60 Other compounds that activate protein kinase C, such as acetylcholine and TPA, stimulate ISR at low glucose to a very similar degree as the combination of PA and etomoxir.28,61 We did not investigate the role of GPR40 in this study, but previous reports have established a role for this receptor,24,25 and GPR40 could contribute to non-oxidative signaling mediating effects of palmitate.
The effect of non-beta cells on the interpretation of whole islet data
All the measurement done in this study were using whole isolated rat islets giving rise to the possibility that contribution of signals generated by non-beta cells (if substantially different from beta cells) would confound the interpretation of the islet data with respect to regulation of ISR. ISR is a uniquely beta cell generated response, whereas cytosolic CA2+ OCR and cytochrome c are all generated by all cells within the islets. Even though the data that was obtained in this study was quantitatively affected by contributions of non-beta cells, we do not believe that the conclusions drawn from the whole islet data are dependent on knowing the exact contribution for the different cell types. Specifically, the low level of changes in OCR and cytochrome c reduction in response to PA indicates that neither beta-cells nor non-beta cells effectively utilize PA as a fuel. One could consider that the ER-release of Ca2+ seen in response to PA was only occurring in non-beta cells, making it incorrect to conclude that ER-Ca2+ release in beta cells did not greatly impact ISR. However, it would still be correct to state that ER-Ca2+ does not mediate PA’s effect on ISR. Moreover, it is fairly well established that PA can stimulate ER-Ca release in clonal beta cell lines62,63 providing support for a lack of coupling between ER-Ca2+ release in the beta cell and ISR as we have concluded from studies of acetylcholine.28
Summary
Metabolic analysis of the effects of PA and etomoxir revealed that mitochondrial metabolism of neither endogenous nor exogenous FAs contributes to the stimulation of ISR. Therefore, it seems that the established mechanism whereby PA results in the generation of non-oxidative signals when CPT1 is inhibited is wholly responsible for its increase in ISR. The lack of metabolic stimulation by palmitate is consistent with its inability to stimulate insulin secretion at low glucose. This study refines our understanding of how a class of fuels affects islet function under normal conditions. Future experiments will focus on generating data on human islets with altered exposure to FAs, since the quantitative role of FAs depends on the accumulation of lipids within the islet.64,65
Materials and methods
Chemicals
Krebs Ringer Bicarbonate (KRB) buffer was used for the perifusion analyses, prepared as described previously.37 PA and bovine serum albumin (BSA), potassium cyanide (KCN), antimycin A, nimodipine, thapsigargin, etomoxir, glucose, and PA were purchased from Sigma–Aldrich.
Preparation of palmitate/albumin complex
A solution of PA (100 mM) was prepared by dissolving PA in 0.1 N NaOH at 70°C. Subsequently, this solution was diluted 1:10 in 10% (w/v) BSA and incubated at 55°C for 10 min to achieve a stock solution with 10 mM PA that was stored at −20°C until use.66 This stock solution was added 1:100 into the KRB perifusion buffer for a final working concentration of 0.1% BSA/100 μM PA (free PA was estimated to be 248 nM67).
Rat islet isolation and culture
Islets were harvested from male Sprague–Dawley rats (≈ 250 g, Charles River, Wilmington, MA) anesthetized by intraperitoneal injection of Beuthanasia-D (35 mg pentobarbital sodium and 5.5 mg phenytoin sodium/230 g rat) purchased from Schering-Plough Animal Health Corp. (Union, NJ). All procedures were approved by the University of Washington Institutional Animal Care and Use Committee. Islets were prepared and purified as described,31,68 and then cultured at 37°C in RPMI Media 1640 supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen, Grand Island, NY) for 18 h prior to the experiments.
Measurement of OCR, cytochrome c reduction and ISR
A flow culture system was used that concomitantly measures OCR while collecting outflow fractions for subsequent measurement of ISR (described previously31,69,70). OCR was calculated as the flow rate (approximately 80 µL/min) times the difference between inflow and outflow oxygen tension measured by detecting the phosphorescence lifetime (Tau Theta, Inc., Fort Collins, CO) of an oxygen-sensitive dye that was painted on the inside of the perifusion chamber.37 Reduction of cytochrome c was measured as absorbance at 550 nm by the layer of intact islets in the perifusion chamber as we have previously described.31 Data were normalized to reference spectra obtained when cytochrome c was fully oxidized by antimycin A. Percent reduction of cytochrome c was calculated following Kashiwagura et al.71 as a percent of the maximal signal obtained in the presence of KCN when cytochrome c is fully reduced. Insulin was measured using an RIA kit (Linco Research Inc., St. Charles, MO).
Imaging and quantification of cytosolic Ca2+
Cytosolic Ca2+ was measured by fluorescence imaging of islets after loading them with Fura-2 AM (Invitrogen) as previously described.33 Dyed islets were pipetted into a temperature-controlled, 250-µl perifusion dish (Bioptechs, Butler, PA) that was mounted on to the stage of a Nikon Eclipse TE-200 inverted microscope, and KRB (containing 5 mM NaHCO3) was pumped through the dish at a flow rate of 150 µL/min. Fluorescent emission was detected at 510 nm by a Photometrics Cool Snap EZ camera (Tucson, AZ) during alternating excitation at either 340 or 380 nm. Results are displayed as the ratio of the fluorescent intensities during excitation at these two wavelengths (F340/F380).
Data analysis
All data were displayed as the average ± SEM of at least three separate perifusions. Statistical significance was determined using paired t-tests calculated with Excel (Microsoft, Redmond, WA). Statistical significance for the kinetic data was determined on changes in steady-state values calculated as the average of the parameter for the final 15 min at that condition. Steady-state values are tabulated for all real-time responses in Table 1.
Funding Statement
This work was supported by National Institutes of Health grants DK17047 (the DRC Cell Function Analysis Core and an R24 seed grant supplement) and R41 TR001196. This work was also supported by National Science Foundation Grant IIP-0750508.
Abbreviations
- BSA
Bovine serum albumin
- Ca2+
Calcium
- CMCP
Ca2+/metabolic coupling process
- CPT1
Carnitine palmitoyl transferase 1
- ER
Endoplasmic reticulum
- ISR
Insulin secretion rate
- K+
Potassium
- KATP
ATP-sensitive potassium channel
- KIC
α-ketoisocaproate
- KCN
Potassium cyanide
- OCR
Oxygen consumption rate
- PA
Palmitic acid
- SERCA
Sarcoendoplasmic reticulum Ca2+ ATPase.
Disclosure of potential conflicts of interest
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions
Author contributions: I.T.K and A.M.R. performed experiments, processed data and prepared figures; I.R.S interpreted results of experiments and drafted manuscript; S.-R. J. and I.R.S edited and revised manuscript; all authors approved the final version of the manuscript; I.R.S. conceived and designed research.
References
- 1.McGarry JD, Dobbins RL.. Fatty acids, lipotoxicity and insulin secretion. Diabetologia. 1999;42:128–138. doi: 10.1007/s001250051130. [DOI] [PubMed] [Google Scholar]
- 2.Grill V, Qvigstad E.. Fatty acids and insulin secretion. Br J Nutr. 2000;83:S79–84. [DOI] [PubMed] [Google Scholar]
- 3.Malaisse WJ, Malaisse-Lagae F, Sener A, Hellerstrom C. Participation of endogenous fatty acids in the secretory activity of the pancreatic B-cell. Biochem J. 1985;227:995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Civelek VN, Deeney JT, Shalosky NJ, Tornheim K, Hansford RG, Prentki M, Corkey BE. Regulation of pancreatic beta-cell mitochondrial metabolism: influence of Ca2+, substrate and ADP. Biochem J. 1996;318:615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malaisse WJ, Best L, Kawazu S, Malaisse-Lagae F, Sener A. The stimulus-secretion coupling of glucose-induced insulin release: fuel metabolism in islets deprived of exogenous nutrient. Arch Biochem Biophys. 1983;224:102–110. [DOI] [PubMed] [Google Scholar]
- 6.Doliba NM, Qin W, Vinogradov SA, Wilson DF, Matschinsky FM. Palmitic acid acutely inhibits acetylcholine- but not GLP-1-stimulated insulin secretion in mouse pancreatic islets. Am J Physiol Endocrinol Metab. 2010;299:E475–85. doi: 10.1152/ajpendo.00072.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graciano MF, Valle MM, Kowluru A, Curi R, Carpinelli AR. Regulation of insulin secretion and reactive oxygen species production by free fatty acids in pancreatic islets. Islets. 2011;3:213–223. [DOI] [PubMed] [Google Scholar]
- 8.Berne C. The metabolism of lipids in mouse pancreatic islets. The oxidation of fatty acids and ketone bodies. Biochem J. 1975;152:661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamarit-Rodriguez J, Vara E, Tamarit J. Starvation-induced changes of palmitate metabolism and insulin secretion in isolated rat islets stimulated by glucose. Biochem J. 1984;221:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warnotte C, Gilon P, Nenquin M, Henquin JC. Mechanisms of the stimulation of insulin release by saturated fatty acids. A study of palmitate effects in mouse beta-cells. Diabetes. 1994;43:703–711. [DOI] [PubMed] [Google Scholar]
- 11.Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163–214. [DOI] [PubMed] [Google Scholar]
- 12.Olofsson CS, Salehi A, Holm C, Rorsman P. Palmitate increases L-type Ca2+ currents and the size of the readily releasable granule pool in mouse pancreatic beta-cells. J Physiol. 2004;557:935–948. doi: 10.1113/jphysiol.2004.066258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vara E, Fernandez-Martin O, Garcia C, Tamarit-Rodriguez J. Palmitate dependence of insulin secretion, “de novo” phospholipid synthesis and 45Ca2+-turnover in glucose stimulated rat islets. Diabetologia. 1988;31:687–693. [DOI] [PubMed] [Google Scholar]
- 14.Remizov O, Jakubov R, Dufer M, Drews PK, Drews G, Waring M, Brabant G, Wienbergen A, Rustenbeck I, Schöfl C. Palmitate-induced Ca2+-signaling in pancreatic beta-cells. Mol Cell Endocrinol. 2003;212:1–9. [DOI] [PubMed] [Google Scholar]
- 15.Stein DT, Stevenson BE, Chester MW, Basit M, Daniels MB, Turley SD, McGarry JD. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J Clin Invest. 1997;100:398–403. doi: 10.1172/JCI119546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem. 1992;267:5802–5810. [PubMed] [Google Scholar]
- 17.Thams P, Capito K. Differential mechanisms of glucose and palmitate in augmentation of insulin secretion in mouse pancreatic islets. Diabetologia. 2001;44:738–746. doi: 10.1007/s001250051683. [DOI] [PubMed] [Google Scholar]
- 18.Gravena C, Mathias PC, Ashcroft SJ. Acute effects of fatty acids on insulin secretion from rat and human islets of Langerhans. J Endocrinol. 2002;173:73–80. [DOI] [PubMed] [Google Scholar]
- 19.Parker SM, Moore PC, Johnson LM, Poitout V. Palmitate potentiation of glucose-induced insulin release: a study using 2-bromopalmitate. Metabolism. 2003;52:1367–1371. [DOI] [PubMed] [Google Scholar]
- 20.Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD. Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem Biophys Res Commun. 2005;335:97–104. doi: 10.1016/j.bbrc.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 21.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 22.Prentki M. New insights into pancreatic beta-cell metabolic signaling in insulin secretion. Eur J Endocrinol. 1996;134:272–286. [DOI] [PubMed] [Google Scholar]
- 23.Corkey BE, Glennon MC, Chen KS, Deeney JT, Matschinsky FM, Prentki M. A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic beta-cells. J Biol Chem. 1989;264:21608–21612. [PubMed] [Google Scholar]
- 24.Alquier T, Peyot ML, Latour MG, Kebede M, Sorensen CM, Gesta S, Ronald Kahn C, Smith RD, Jetton TL, Metz TO, et al. Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes. 2009;58:2607–2615. doi: 10.2337/db09-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagasumi K, Esaki R, Iwachidow K, Yasuhara Y, Ogi K, Tanaka H, Nakata M, Yano T, Shimakawa K, Taketomi S, et al. Overexpression of GPR40 in pancreatic beta-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes. 2009;58:1067–1076. doi: 10.2337/db08-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. [DOI] [PubMed] [Google Scholar]
- 27.Matschinsky FM. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45:223–241. [DOI] [PubMed] [Google Scholar]
- 28.Gilbert M, Jung SR, Reed BJ, Sweet IR. Islet oxygen consumption and insulin secretion tightly coupled to calcium derived from L-type calcium channels but not from the endoplasmic reticulum. J Biol Chem. 2008;283:24334–24342. doi: 10.1074/jbc.M802097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satin LS, Tavalin SJ, Kinard TA, Teague J. Contribution of L- and non-L-type calcium channels to voltage-gated calcium current and glucose-dependent insulin secretion in HIT-T15 cells. Endocrinology. 1995;136:4589–4601. doi: 10.1210/endo.136.10.7545106. [DOI] [PubMed] [Google Scholar]
- 30.Bokvist K, Eliasson L, Ammala C, Renstrom E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. Embo J. 1995;14:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sweet IR, Cook DL, DeJulio E, Wallen AR, Khalil G, Callis J, Reems J. Regulation of ATP/ADP in pancreatic islets. Diabetes. 2004;53:401–409. [DOI] [PubMed] [Google Scholar]
- 32.Jung SR, Kuok IT, Couron D, Rizzo N, Margineantu DH, Hockenbery DM, Kim F, Sweet IR. Reduced cytochrome C is an essential regulator of sustained insulin secretion by pancreatic islets. J Biol Chem. 2011;286:17422–17434. doi: 10.1074/jbc.M110.202820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung SR, Reed BJ, Sweet IR. A highly energetic process couples calcium influx through L-type calcium channels to insulin secretion in pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2009;297:E717–27. doi: 10.1152/ajpendo.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rountree AM, Neal AS, Lisowski M, Rizzo N, Radtke J, White S, Luciani DS, Kim F, Hampe CS, Sweet IR. Control of insulin secretion by cytochrome C and calcium signaling in islets with impaired metabolism. J Biol Chem. 2014;289:19110–19119. doi: 10.1074/jbc.M114.556050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yaney GC, Corkey BE. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia. 2003;46:1297–1312. doi: 10.1007/s00125-003-1207-4. [DOI] [PubMed] [Google Scholar]
- 36.Chen S, Ogawa A, Ohneda M, Unger RH, Foster DW, McGarry JD. More direct evidence for a malonyl-CoA-carnitine palmitoyltransferase I interaction as a key event in pancreatic beta-cell signaling. Diabetes. 1994;43:878–883. [DOI] [PubMed] [Google Scholar]
- 37.Sweet IR, Gilbert M. Contribution of calcium influx in mediating glucose-stimulated oxygen consumption in pancreatic islets. Diabetes. 2006;55:3509–3519. doi: 10.2337/db06-0400. [DOI] [PubMed] [Google Scholar]
- 38.Fraze E, Donner CC, Swislocki AL, Chiou YA, Chen YD, Reaven GM. Ambient plasma free fatty acid concentrations in noninsulin-dependent diabetes mellitus: evidence for insulin resistance. J Clin Endocrinol Metab. 1985;61:807–811. doi: 10.1210/jcem-61-5-807. [DOI] [PubMed] [Google Scholar]
- 39.Lewis B, Mancini M, Mattock M, Chait A, Fraser TR. Plasma triglyceride and fatty acid metabolism in diabetes mellitus. Eur J Clin Invest. 1972;2:445–453. [DOI] [PubMed] [Google Scholar]
- 40.Park EA, Cook GA. Differential regulation in the heart of mitochondrial carnitine palmitoyltransferase-I muscle and liver isoforms. Mol Cell Biochem. 1998;180:27–32. [PubMed] [Google Scholar]
- 41.Malaisse WJ, Rasschaert J, Villanueva-Penacarrillo ML, Valverde I. Respiratory, ionic, and functional effects of succinate esters in pancreatic islets. Am J Physiol. 1993;264:E428–33. doi: 10.1152/ajpendo.1993.264.3.E428. [DOI] [PubMed] [Google Scholar]
- 42.Erecinska M, Davis JS, Wilson DF. Regulation of respiration in paracoccus denitrificans: the dependence on redox state of cytochrome c and [ATP]/[ADP][Pi]. Arch Biochem Biophys. 1979;197:463–469. [DOI] [PubMed] [Google Scholar]
- 43.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem. 1956;17:65–134. [DOI] [PubMed] [Google Scholar]
- 44.Joseph JW, Koshkin V, Saleh MC, Sivitz WI, Zhang CY, Lowell BB, Chan CB, Wheeler MB. Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004;279:51049–51056. doi: 10.1074/jbc.M409189200. [DOI] [PubMed] [Google Scholar]
- 45.Carlsson C, Borg LA, Welsh N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology. 1999;140:3422–3428. doi: 10.1210/endo.140.8.6908. [DOI] [PubMed] [Google Scholar]
- 46.Cen J, Sargsyan E, Bergsten P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutr Metab (Lond). 2016;13:59. doi: 10.1186/s12986-016-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vara E, Tamarit-Rodriguez J. Glucose stimulation of insulin secretion in islets of fed and starved rats and its dependence on lipid metabolism. Metabolism. 1986;35:266–271. [DOI] [PubMed] [Google Scholar]
- 48.Liang Y, Bai G, Doliba N, Buettger C, Wang L, Berner DK, Matschinsky FM. Glucose metabolism and insulin release in mouse beta HC9 cells, as model for wild-type pancreatic beta-cells. Am J Physiol. 1996;270:E846–57. doi: 10.1152/ajpendo.1996.270.5.E846. [DOI] [PubMed] [Google Scholar]
- 49.Hodson L, Skeaff CM, Fielding BA. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Progress in Lipid Research. 2008;47:348–380. doi: 10.1016/j.plipres.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 50.Basu A, Basu R, Shah P, Vella A, Rizza RA, Jensen MD. Systemic and regional free fatty acid metabolism in type 2 diabetes. Am J Physiol Endocrinol Metab. 2001;280:E1000–6. doi: 10.1152/ajpendo.2001.280.6.E1000. [DOI] [PubMed] [Google Scholar]
- 51.Satin LS. Localized calcium influx in pancreatic beta-cells: its significance for Ca2+-dependent insulin secretion from the islets of Langerhans. Endocrine. 2000;13:251–262. doi: 10.1385/ENDO:13:3:251. [DOI] [PubMed] [Google Scholar]
- 52.Oleson BJ, McGraw JA, Broniowska KA, Annamalai M, Chen J, Bushkofsky JR, Davis DB, Corbett JA, Mathews CE. Distinct differences in the responses of the human pancreatic beta-cell line EndoC-betaH1 and human islets to proinflammatory cytokines. Am J Physiol Regul Integr Comp Physiol. 2015;309:R525–34. doi: 10.1152/ajpregu.00544.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf BA, Colca JR, Turk J, Florholmen J, McDaniel ML. Regulation of Ca2+ homeostasis by islet endoplasmic reticulum and its role in insulin secretion. Am J Physiol. 1988;254:E121–36. doi: 10.1152/ajpendo.1988.254.2.E121. [DOI] [PubMed] [Google Scholar]
- 54.Satin LS, Kinard TA. Neurotransmitters and their receptors in the islets of Langerhans of the pancreas: what messages do acetylcholine, glutamate, and GABA transmit? Endocrine. 1998;8:213–223. doi: 10.1385/ENDO:8:3:213. [DOI] [PubMed] [Google Scholar]
- 55.Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51:S405–13. [DOI] [PubMed] [Google Scholar]
- 56.Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 57.Yao CH, Liu GY, Wang R, Moon SH, Gross RW, Patti GJ. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol. 2018;16:e2003782. doi: 10.1371/journal.pbio.2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol. 2000;278:F667–75. doi: 10.1152/ajprenal.2000.278.4.F667. [DOI] [PubMed] [Google Scholar]
- 59.Wolf BA, Easom RA, Hughes JH, McDaniel ML, Turk J. Secretagogue-induced diacylglycerol accumulation in isolated pancreatic islets. Mass spectrometric characterization of the fatty acyl content indicates multiple mechanisms of generation. Biochemistry. 1989;28:4291–4301. [DOI] [PubMed] [Google Scholar]
- 60.Alcazar O, Qiu-Yue Z, Gine E, Tamarit-Rodriguez J. Stimulation of islet protein kinase C translocation by palmitate requires metabolism of the fatty acid. Diabetes. 1997;46:1153–1158. [DOI] [PubMed] [Google Scholar]
- 61.Zawalich W, Zawalich K, Rasmussen H. Insulin secretion: combined tolbutamide, forskolin and TPA mimic action of glucose. Cell Calcium. 1984;5:551–558. [DOI] [PubMed] [Google Scholar]
- 62.Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab. 2009;296:E690–701. doi: 10.1152/ajpendo.90525.2008. [DOI] [PubMed] [Google Scholar]
- 63.Schnell S, Schaefer M, Schofl C. Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from beta-cells through activation of GPR40. Mol Cell Endocrinol. 2007;263:173–180. doi: 10.1016/j.mce.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 64.Roomp K, Kristinsson H, Schvartz D, Ubhayasekera K, Sargsyan E, Manukyan L, Chowdhury A, Manell H, Satagopam V, Groebe K, et al. Combined lipidomic and proteomic analysis of isolated human islets exposed to palmitate reveals time-dependent changes in insulin secretion and lipid metabolism. PLoS One. 2017;12:e0176391. doi: 10.1371/journal.pone.0176391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sargsyan E, Cen J, Roomp K, Schneider R, Bergsten P. Identification of early biological changes in palmitate-treated isolated human islets. BMC Genomics. 2018;19:629. doi: 10.1186/s12864-018-5008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cousin SP, Hugl SR, Wrede CE, Kajio H, Myers MG Jr., Rhodes CJ. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology. 2001;142:229–240. doi: 10.1210/endo.142.1.7863. [DOI] [PubMed] [Google Scholar]
- 67.Richieri GV, Anel A, Kleinfeld AM. Interactions of long-chain fatty acids and albumin: determination of free fatty acid levels using the fluorescent probe ADIFAB. Biochemistry. 1993;32:7574–7580. [DOI] [PubMed] [Google Scholar]
- 68.Matsumoto S, Shibata S, Kirchhof N. Immediate reversal of diabetes in primates following intraportal transplantation of porcine islets purified on a new histidine-lactoioniate-iodixanol gradient. Transplantation. 1999;67:S220. doi: 10.1097/00007890-199904150-00880. [DOI] [Google Scholar]
- 69.Sweet IR, Cook DL, Wiseman RW, Greenbaum CJ, Lernmark A, Matsumoto S, Teague JC, Krohn KA. Dynamic perifusion to maintain and assess isolated pancreatic islets. Diabetes Technol Ther. 2002;4:67–76. doi: 10.1089/15209150252924111. [DOI] [PubMed] [Google Scholar]
- 70.Sweet IR, Khalil G, Wallen AR, Steedman M, Schenkman KA, Reems JA, Kahn SE, Callis JB. Continuous measurement of oxygen consumption by pancreatic islets. Diabetes Technol Ther. 2002;4:661–672. doi: 10.1089/152091502320798303. [DOI] [PubMed] [Google Scholar]
- 71.Kashiwagura T, Wilson DF, Erecinska M. Oxygen dependence of cellular metabolism: the effect of O2 tension on gluconeogenesis and urea synthesis in isolated rat hepatocytes. J Cell Physiol. 1984;120:13–18. doi: 10.1002/jcp.1041200103. [DOI] [PubMed] [Google Scholar]