

ABSTRACT
Hepatocyte-specific knockout of the essential autophagy gene Autophagy-related 7 (Atg7) is sufficient to cause hepatic carcinogenesis. A recent paper by Lee et al. unveils the molecular pathway accounting for hepatic hypertrophy and hyperplasia followed by malignant transformation. This pathway involves the overactivation of the transcription factor yes-associated protein (YAP), which turns out to be an autophagic substrate. Of note, the transcriptional signature activated in mouse hepatocytes lacking Atg7 resembles that found in non-alcoholic steatohepatitis (NASH), as well as in the steatohepatitic subtype of human hepatocellular carcinomas.
KEYWORDS: Age-related disease, cytotoxic T cells, NK cells, immunosenescence, senescence
Hepatocellular carcinoma constitutes an ever more frequent disease entity that develops as a complication of chronic inflammation, frequently in the context of viral hepatitis, ethanol-induced steatohepatitis and obesity-associated non-alcoholic steatohepatitis (NASH).1
Macroautophagy (hereafter ‘autophagy’) is a bulk lysosomal degradation mechanisms in which cytoplasmic constituents are sequestered in autophagosomes, which then fuse with lysosomes to create autophagolysosomes for the digestion of the luminal content by hydrolases operating at an acidic pH optimum.2 Autophagy is induced by starvation of cells from nutrients and is inhibited in the context of caloric excess leading to obesity. Indeed, autophagy allows starved cells to digest macromolecules to energy-rich metabolites, thus satisfying their bioenergetics demand.3 Beyond this metabolic function, autophagy plays a major role in ridding the cytoplasm from damaged, dysfunctional and potentially noxious organelles, protein aggregates, lipid vesicles or invading pathogens. As such, autophagy plays a major role in organellar quality control (and de facto acts to rejuvenate the cytoplasm) as well as in the suppression of inflammation and malignant transformation.3
It had been known that hepatocyte-specific knockout of essential autophagy-relevant genes such as Autophagy-related 5 (Atg5) and Autophagy-related 7 (Atg7) can stimulate hepatomegaly and the formation of adenomas.4,5 This effect had been attributed to the accumulation of Sequestosome-1 (SQSTM1/also referred to as p62) protein. SQSTM1 is a typical autophagic substrate, meaning that inhibition of autophagic flux increases the abundance of this protein to the extent that some autophagy measurements are based on the quantification of SQSTM1 expression levels.6 Indeed, hepatic tumorigenes induced by Atg7 knockout is attenuated upon Sqstm1 knockout,4 and transgenic overexpression of Sqstm1 in the liver is sufficient to drive carcinogenesis.7 Accumulating SQSTM1 protein then binds and inhibits Kelch-like protein 1 (KEAP1), a negative regulator of nuclear factor (erythroid-derived 2)-like 2 (NFE2L2, best known as NRF2), which acts as a pro-tumorigenic transcription factor.8
In a recent paper published in Nature Communications, Lee et al. demonstrate that yet another transcription factor, yes-associated protein (YAP), may contribute to malignant transformation of hepatocytes lacking Atg7.9 Thus, the protein YAP, which turned out to be an autophagic substrate (like SQSTM1), accumulated upon deletion of Atg7, as demonstrated in a tamoxifen-inducible liver cell-specific Atg7 knockout model. This YAP accumulation occurred quickly after Atg7 knockout (within days), this preceding the accumulation of SQSTM1 (which occurred only several weeks later).9 Liver-specific double knockout of Atg7 and Yap resulted in a phenotype in which much of the alterations induced by single knockout of Atg7 were attenuated. Indeed, in contrast to prior studies using a very similar approach in which Atg7 knockout led to the development of benign tumors (adenomas) only,4 Lee et al. observed that Atg7 deletion caused the induction of malignant lesions (carcinomas).9 Such carcinomas developed after an augmentation in liver size (hepatomegaly) due to an increase in hepatocyte volume (hypertrophy) and number (hyperplasia), coupled to increased proliferative activity, lobular and portal inflammation, ductular reaction, steatosis and fibrosis. All these microscopic and macroscopic signs of pathology were significantly improved upon removal of Yap, indicating a major role for YAP throughout all steps of hepatic carcinogenesis.9
Transcriptional profiling of livers that had undergone the knockout of Atg7 revealed a significant overexpression of YAP target genes. More importantly, the transcriptional changes induced by Atg7 inactivation in mouse livers were very similar to those found in the liver from patients with non-alcoholic steatohepatitis (NASH), as well as in the steatohepatitic subtype of human hepatocellular carcinomas (but not that in other subtypes of liver cancer), supporting the translational relevance of this mouse model.9
Based on the interesting work by Lee et al.,9 a few major questions arise:
Is autophagy inhibition by excessive nutrition with consequent elevations in glucose, free fatty acids and triglycerides as well as obesity-associated growth factors (such as insulin, insulin-like growth factor, leptin, etc.) sufficient to provide a stimulus for hepatic oncogenesis in patients with NASH?
What is the articulation between SQSTM1 and YAP downstream of autophagy inhibition in hepatic carcinogenesis? Are both transcription factors involved in distinct molecular subtypes of liver cancer? Or do they cooperate within the same subtype and, if so, how is this cooperation articulated in mechanistic terms? Is there some crosstalk among the signaling modules or do the effects of transcription factors synergize for the activation of an oncogenic program (Figure 1)?
Is it possible to interrupt the molecular cascades involving autophagy inhibition as well as the activation of SQSTM1/NRF2 and YAP by pharmacological strategies? Would autophagy induction or rather the inhibition of the downstream effectors (SQSTM1/NRF2, YAP) yield a superior result for the chemoprevention of liver cancer? Considering that autophagy modulation, and especially inhibition here, has already been associated with other pathologies like some autoimmune and metabolic disorders, such clinical interventions should be handled with caution.10
Figure 1.
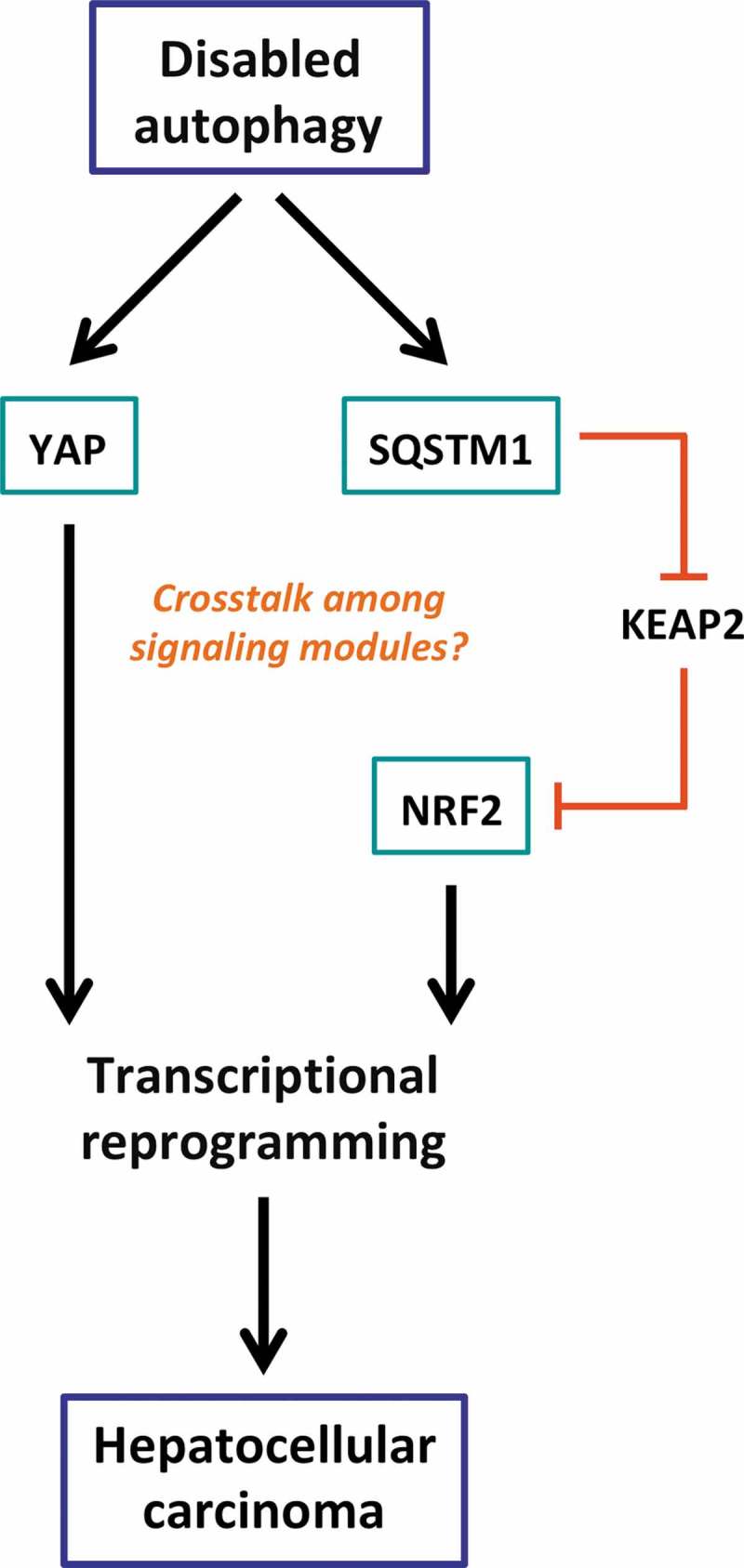
Relationship between autophagy and hepatic carcinogenesis supposing two independent pathways (that may somehow crosstalk to each other) that link autophagy inhibition to the transactivation of oncogenic gene products.
On the one hand, impaired autophagy leading to YAP accumulation in cells is associated with significant overexpression of its target genes and oncogenesis in hepatocytes. On the other hand, SQSTM1 that accumulates upon autophagy deficiency binds to KEAP1 thus preventing its inhibitory interaction with a pro-tumorigenic transcription factor NRF2. KEAP2, Kelch-like protein 1; NRF2, nuclear factor (erythroid-derived 2)-like 2; SQSTM1, Sequestosome-1; YAP, yes-associated protein.
Funding Statement
GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Inserm Transfert; Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM).
Disclosure of Potential Conflicts of Interest
GK is a scientific co-founder of Samsara Therapeutics.
References
- 1.Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouzé E, Blanc J-F, Laurent C, Hajji Y, Azoulay D, Bioulac-Sage P, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol. 2017;67(4):727–738. doi: 10.1016/j.jhep.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Esteban-Martínez L, Sierra-Filardi E, Boya P.. Mitophagy, metabolism, and cell fate. Mol Cell Oncol. 2017;4(5):e1353854. doi: 10.1080/23723556.2017.1353854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015;34(7):856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N.. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu K, Lee J, Ou J-HJ. Autophagy and mitophagy in hepatocarcinogenesis. Mol Cell Oncol. 2018;5(2):e1405142. doi: 10.1080/23723556.2017.1405142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36(13):1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, Zhong Z, Subramaniam S, Raghunandan S, Duran A, et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell. 2016;29(6):935–948. doi: 10.1016/j.ccell.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T, et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun. 2016;7:12030. doi: 10.1038/ncomms12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee YA, Noon LA, Akat KM, Ybanez MD, Lee T-F, Berres M-L, Fujiwara N, Goossens N, Chou H-I, Parvin-Nejad FP, et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat Commun. 2018;9(1):4962. doi: 10.1038/s41467-018-07338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16(7):487–511. doi: 10.1038/nrd.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]