

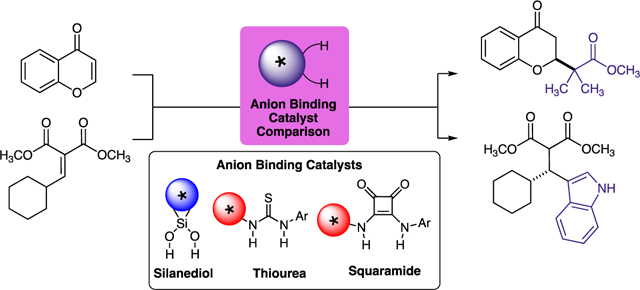
Abstract
Silanediols possess unique and complementary catalytic activity in reactions that are likely to proceed through anion binding. This article directly compares silanediols, thioureas, and squaramides in three separate anion-binding processes. The catalytic abilities of select members of each family are directly correlated to association constant.
Keywords: anion recognition, enantioseletive catalysis, thiourea, silanediol, squaramide
Graphical Abstract
The silanediol functional group, a silicon with two –OH groups bound to it, can host a variety of anions through hydrogen bonding interactions (Scheme 1).1 For instance, seminal work in this area demonstrated that dinaphthyl silanediols host acetate, chloride, and bromide.2 These anion recognition abilities of silanediols can be taken advantage of in other areas of chemistry, such as sensing and catalysis.3,4 Chiral BINOL-based silanediols (e.g., 1a-1c, Scheme 1) have emerged as promising enantioselective anion-binding catalysts for addition reactions to isoquinolinium ions and benzopyrylium ions.5,6
Scheme 1.
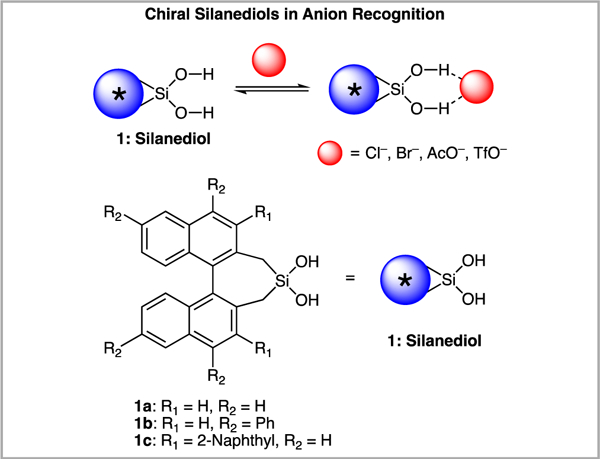
Silanediols (1) can host anionic guests through hydrogen bonding interactions.
In the context of anion-binding catalysis, silanediols can offer an exclusive appeal. From a structural standpoint BINOL-based silanediols stand out as anion-binding catalysts because they are highly aromatic C2-symmetric dual O-H hydrogen bond donors. In comparison, more common anion-binding catalysts, such as (thio)ureas7 and squaramides8, are N-H hydrogen bond donors that typically lack C2-symmetry. Perhaps stemming from their unique structures, BINOL-based silanediols catalyze processes that are complementary to the reactivity patterns observed with the more traditional (thio)urea and squaramide catalysts. Intrigued by the unique catalytic abilities of silanediols, we desired a better understanding of the roles of silanediols in anion-binding catalysis. This article describes the results of the direct comparison of silanediols, thioureas, and squaramides as (i) hosts in anion recognition and (ii) catalysts for enantioselective reactions likely to involve anion-binding catalysis.
The Discovery of Silanediols in Enantioselective Anion-Binding Catalysis
Chiral silanediols as enantioselective anion-binding catalysts first emerged in the literature from our laboratory in 2013 (Scheme 2).6d Inspired by the impressive catalytic abilities of (thio)ureas, in the early stages of our investigations it was hypothesized that silanediols would activate ionic substrates through hydrogen bond recognition of the anionic component (eq 1). As a testing ground, we chose to explore the feasibility of enantioselective silanediol anion-binding catalysis in addition reactions of silyl ketene acetals to N-acyl isoquinolines, reactions that are known to benefit from (thio)urea anion-binding catalysis.7g During our studies we were delighted to find that chiral, enantiopure BINOL-based silanediols influenced the addition of silyl ketene acetals (3) to isoquinolinium ions generated in situ from 2 giving rise to 4 in good yield and high levels of enantiocontrol (eq 2, up to 64% yield and 79% ee). Based upon both prior hypotheses and data collected in our laboratory, it is proposed that the silanediol is operating to hydrogen bond to chloride to create chiral ion pair 5.
Scheme 2.
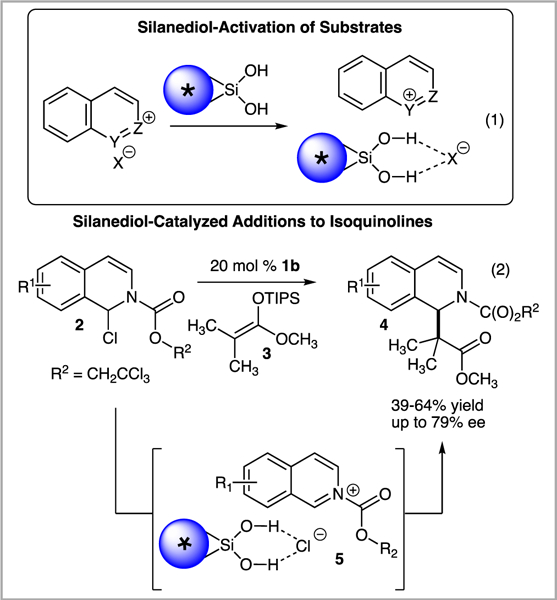
Silanediol-activation of ion pairs (eq 1). The enantioselective functionalization of isoquinolines with silanediol catalysis (eq 2).
Silanediols as Enantioselective Anion-Binding Catalysts
The field of anion-binding catalysis is relatively new. It was only in 2006 that Schreiner described the possibility that hydrogen bond donor catalysts, specifically thioureas, may operate to facilitate ionization.9 In 2007, Jacobsen and coworkers intentionally applied chiral thioureas to influence an enantioselective intramolecular Pictet-Spangler reaction via anion-binding catalysis.10 In the last ten years, further investigations from a number of research teams, have supported the promise of anion-binding catalysis: new families of catalysts are under development and impressive reactivity patterns have been realized.11
Silanediols are a newer and rather unexplored family of hydrogen bond donor anion-binding catalysts in comparison to the more well-known (thio)ureas. Once we had success in the enantioselective addition of silyl ketene acetals to isoquinolinium ions, a known reactivity pattern, we became interested to identify useful, unique reactivity patterns of enantioselective silanediol anion-binding catalysis. Our attention first turned toward the enantioselective functionalization of chromenones to generate 2-alkyl chroman-4-ones, oxygen heterocycles that are frequently found in bioactive secondary metabolites.12 Prior to our studies, no reports were present in the literature describing anion-binding catalysis as a strategy to control the reactions of chromenones via the in situ generation of 4-siloxybenzopyrylium triflates.6b We hypothesized that silanediols could activate benzopyrylium trifates via anion binding to generate a chiral ion pair and allow for the enantioselective alkylation of chromenones.
The promise of silanediol-enabled control of chromanone functionalization was realized in the addition of silyl ketene acetals to benzopyrylium ions, reactive oxygen heterocycles generated in situ from 6 and a suitable silyl triflate (Scheme 3). Desirable products (8) were isolated in high yield with decent levels of enantiocontrol in this first example of an enantioselective functionalization of 4-siloxybenzopyrylium triflates. In collaboration between the Kondo and Mattson laboratories, data was collected by fluorescence spectroscopy that suggests that the silanediol is able to host a triflate anion through hydrogen bonding interactions.
Scheme 3.
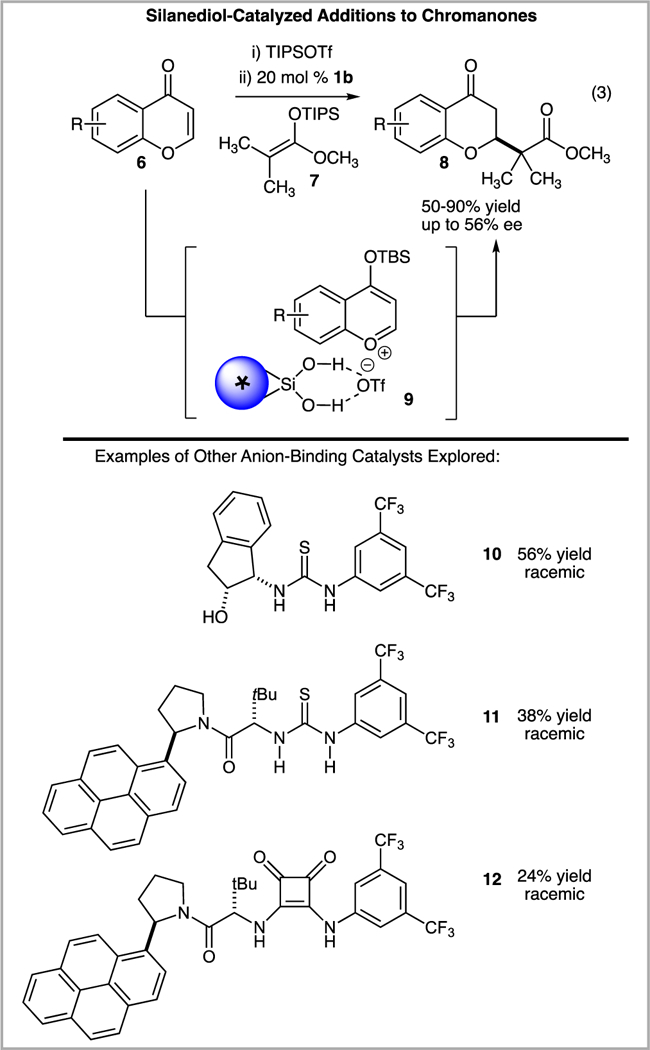
Silanediol 1b as a catalyst for the addition of silyl ketene acetals to 6 via proposed ion pair 9.
An important aspect of the ongoing work in our laboratories has identified the enantioselective functionalization of in situ generated benzopyrylium ions with silanediol anion-binding catalysis. In a direct comparison, it was found that popular (thio)urea catalysts, such as 10 and 11,13 were unable to control the facial selectivity of the addition of 7 to 6. Likewise, squaramide 12,13 a hydrogen bond donor recently reported to participate in triflate binding, provided only racemic product.8b
The distinctive catalytic abilities of silanediols triggered us to consider further (i) additional catalytic processes that may be unique to silanediols and (ii) what specific properties of the silanediol are responsible for its one-of-a-kind catalytic performance.
Silanediols and Lewis Acid Hybrid Anion-Binding Catalysts
Encouraged by the development that silanediol-specific catalytic outcomes are feasible, we sought to branch out from the traditional anion-binding catalyst activation of substrates. We reasoned that if silanediols can activate substrates then they can likely activate other components of a reaction system. For example, we hypothesized that silanediols could active Lewis acids thereby enabling the generation of hybrid anion-binding and Lewis acid catalyst systems that benefit from enhanced activity (13, Scheme 4).
Scheme 4.
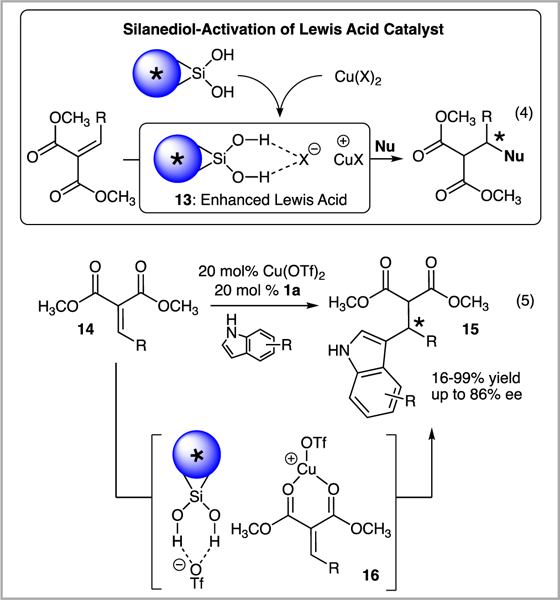
Silanediol-activated Lewis acid (13) as a catalyst for the addition of indoles to 14.
To this end, our investigations led us to probe the effect of BINOL-based silanediol 1a on Cu(OTf)2 in the addition of indoles to alkylidene and arylidene malonates (14, eq 5, Scheme 4).6a We were delighted to find that the silanediol 1a and Cu(OTf)2 cocatalyst system was effective in the addition of indoles to 14, affording desirable products (15) with excellent yields (typically >90%) and up to 86% enantiomeric excess. Although the mechanism of this process is still under investigation, it is feasible that ion pair 16 is operating as a key intermediate.6a
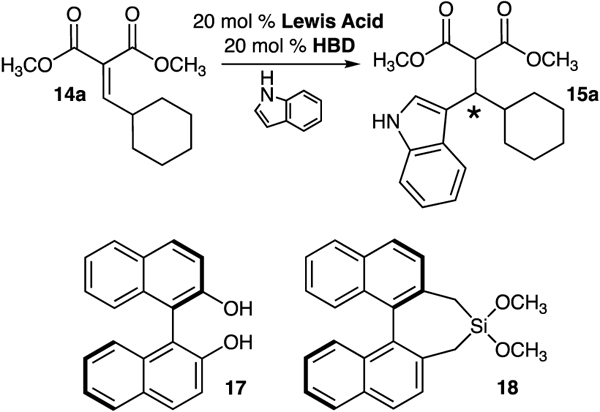
A brief survey of popular Lewis acid and dual hydrogen bond donors led us to conclude that there is something unique about the silanediol-Cu(OTf)2 catalyst system. Inferior yields and enantiomeric excesses were obtained for several other Lewis acids employed in the reaction system (Table 1). Cu(OTf)2 gave rise to 15a in 92% yield and 72% ee (entry 1). While high yields of product were achieved using Sc(OTf)3 and In(OTf)2, the enantiomeric excess never reached beyond 10% (entries 2 and 3). Both the oxidation state of copper and the anion involved have huge influences on the reaction: CuOTf afforded just 10% yield of 15a in 31% ee, while CuCl, CuI, and CuSO4 did not enable the reaction to proceed (entries 4–6). BINOL-based silanediol 1a proved to be the best hydrogen bond donor in the process. The addition of steric bulk to the silanediol scaffold, such as BINOL-based silanediols 1b and 1c, resulted in steep declines in both yield and enantiomeric excess (entries 7 and 8). Attempts to use thiourea catalysts 10 and 11 prevented the formation of product (entries 9 and 10). Low yields and enantiomeric excesses of 15a were observed with squaramide 12 operating as the cocatalyst (entry 11). BINOL (17) was also tested in the transformation and gave rise to racemic 15a in 28% yield, the same yield obtained as the background rate of the reaction (entry 12). The importance of the silanediol functional group was supported with the observation that dimethoxysilane 18 was unable to control the absolute stereochemistry in the synthesis of 15a (entry 13).
Table 1.
On the Unique Nature of Silanediol 1a-Cu(OTf)2 Hybrid Catalysisa
 | ||||
| Entry | HBDb | Lewis Acid | Yield (%) | ee (%) |
| 1 | 1a | Cu(OTf)2 | 92 | 72 |
| 2 | 1a | Sc(OTf)3 | 95 | 9 |
| 3 | 1a | In(OTf)2 | 88 | 10 |
| 4 | 1a | CuOTf | 10 | 31 |
| 5 | 1a | CuCl | 0 | -- |
| 6 | 1a | CuSO4 | 0 | -- |
| 7 | 1b | Cu(OTf)2 | 76 | 13 |
| 8 | 1c | Cu(OTf)2 | 14 | 6 |
| 9 | 10 | Cu(OTf)2 | 0 | -- |
| 10 | 11 | Cu(OTf)2 | 0 | -- |
| 11 | 12 | Cu(OTf)2 | 10 | 10 |
| 12 | 17 | Cu(OTf)2 | 28 | racemic |
| 13 | 18 | Cu(OTf)2 | 25 | racemic |
See the experimental sections for details of the procedures.
HBD = hydrogen bond donor.
Host:Guest Interactions of Silanediols
With the identification of a second reaction unique to silanediols, our curiosity to better understand their anion-binding properties grew stronger. Investigations were initiated to explore the abilities of our chiral, BINOL-based silanediol 1a to recognize chloride and triflate in two solvents (e.g., chloroform and toluene) using UV-Vis spectroscopy and nuclear magnetic resonance spectroscopy. Under identical experimental conditions, the association constants of thiourea 11 and squaramide 12 for both chloride and triflate were also measured so as to be able to compare anion-binding and catalysis of the three different hydrogen bond donor families (Figures 1 and 2).
Figure 1:
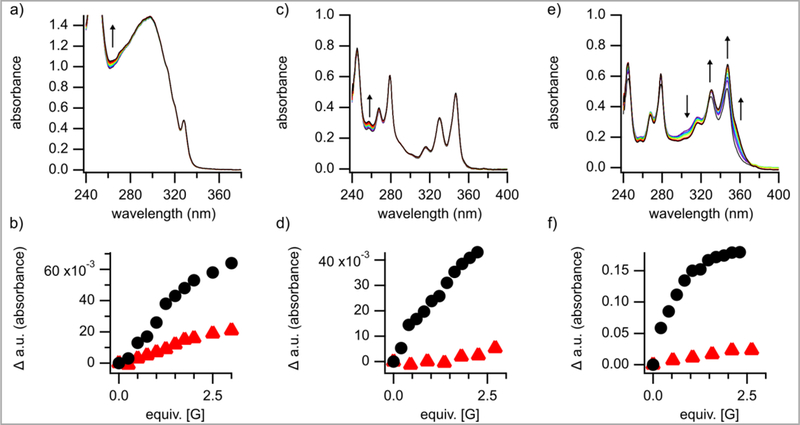
UV-Vis titrations of silanediol 1a (a), thiourea 11 (c), and squaramide 12 (e) with TBACl in CHCl3. b) Change in 262nm when silanediol 1a was titrated with [G] = TBACl ( ) and TBAOTf (
) and TBAOTf ( ). d) Change in 259nm when thiourea 11 was titrated with [G] = TBACl () and TBAOTf (). f) Change in 348nm when squaramide 12 was titrated with [G] = TBACl () and TBAOTf ().
). d) Change in 259nm when thiourea 11 was titrated with [G] = TBACl () and TBAOTf (). f) Change in 348nm when squaramide 12 was titrated with [G] = TBACl () and TBAOTf ().
Figure 2:
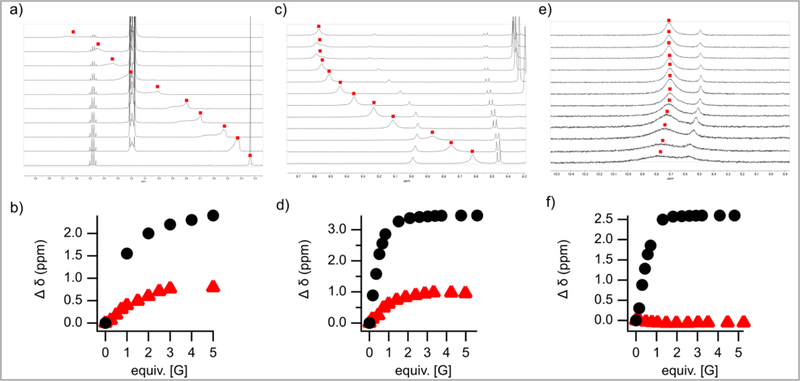
NMR titrations of silanediol 1a (a), thiourea 11 (c), and squaramide 12 (e) with TBAOTf in CDCl3. b) Change in 2.33 ppm (-OH) when silanediol 1a was titrated with [G] = TBACl () and TBAOTf (). d) Change in 8.62 ppm (-NH) when thiourea 11 was titrated with [G] = TBACl () and TBAOTf (). f) Change in 9.78 ppm (-NH) when squaramide 12 was titrated with [G] = TBACl () and TBAOTf ()
The association of silanediol 1a with both chloride and triflate in chloroform and toluene was observed using UV-Vis spectroscopy (Figure 1a and 1b and supporting information). For example, plotting the change in absorbance upon the addition of 0–5 equiv of tetrabutylammonium chloride (TBACl) and tetrabutylammonium triflate (TBAOTf) at 262nm generated the curves depicted in Figure 1b. From these data, the association constant for silanediol:chloride was determined to be 1.9 × 103 M−1 in CHCl3 and 3.8 × 104 M−1 in toluene (Table 2, entry 1). The silanediol:triflate association constant in toluene was measured to be 2.8 × 103 M−1 (entry 2). No association constant was determined for the triflate in CHCl3 by UV-Vis spectroscopy because the change in spectra upon the addition of TBAOTf was too small.
Table 2:
Association Constants Determined for 1a, 11, 12 with Chloride and Triflate (M−1)
| Entry | Host : Guest | UV-Vis | |
| CHCl3 | Toluene | ||
| 1 | 1a:Cl– | 1.9±0.22 × 103 | 3.8±0.13 × 104 |
| 2 | 1a:–OTf | ND | 2.8±0.40 × 103 |
| 3 | 11: Cl– | 1.8±0.37 × 104 | 7.9±0.13 × 106 |
| 4 | 11: –OTf | ND | 6.5±0.27 × 104 |
| 5 | 12: Cl– | 7.6±0.16 × 105 | >106 |
| 6 | 12: –OTf | ND | 3.8±0.04 × 105 |
ND = Not Determined: The absorbance change was too small to accurately determine the association constant
Thiourea 11 and squaramide 12 were also found to recognize both chloride and triflate through UV-Vis titration experiments (Figure 1c–1f). A binding constant of 1.8 × 104 M−1 was extrapolated from the change in UV-Vis spectra observed at 259nm upon the addition of TBACl to thiourea 11 in CHCl3. Measuring of the binding constant in toluene (Ka = 7.9 × 106 M−1) indicated a stronger host:guest interaction relative to that observed in CHCl3 (Table 2, entry 3). The association constant of squaramide 12 and chloride was found to be 7.6 × 105 M−1 in CHCl3 and >106 M−1 in toluene (entry 4). In the cases of both the thiourea 11 and squaramide 12, the association constant found for triflate was lower than that found for chloride (entries 3 and 5 v. entries 4 and 6). In toluene, the association constant for 11:–OTF was found to be 6.5 × 104 M−1 and 12: –OTF was found to be 3.8 × 105 M−1 (entries 4 and 6, respectively). Similar to the silanediol, the change in spectra was too small for both thiourea 11 and squaramide 12 to accurately determine a binding constant of triflate in CHCl3.
NMR titration experiments were also used to analyze the association of silanediol 1a, thiourea 11, and squaramide 12 to the host chloride and triflate anions in both chloroform (Figure 2). In all cases, the introduction of TBACl caused larger shifts in the 1H NMR spectra than TBAOTf. This data suggests that silanediol 1a, thioureas 11, and squaramides 12 all operate as hosts of both chloride and triflate, although they bind more strongly to chloride than triflate (Figure 2).
With the collection of the association constant of both chloride and triflate with our silanediol, thiourea, and squaramide catalysts we correlated the association constant to yield and enantiomeric excess in three reactions: (i) additions of silyl ketene acetals to isoquinolinium chlorides; (ii) additions of silyl ketene acetals to benzopyrylium triflates; (iii) additions of indoles to cyclohexylidene malonate in the presence of copper(II) triflate (Scheme 5).
Scheme 5.
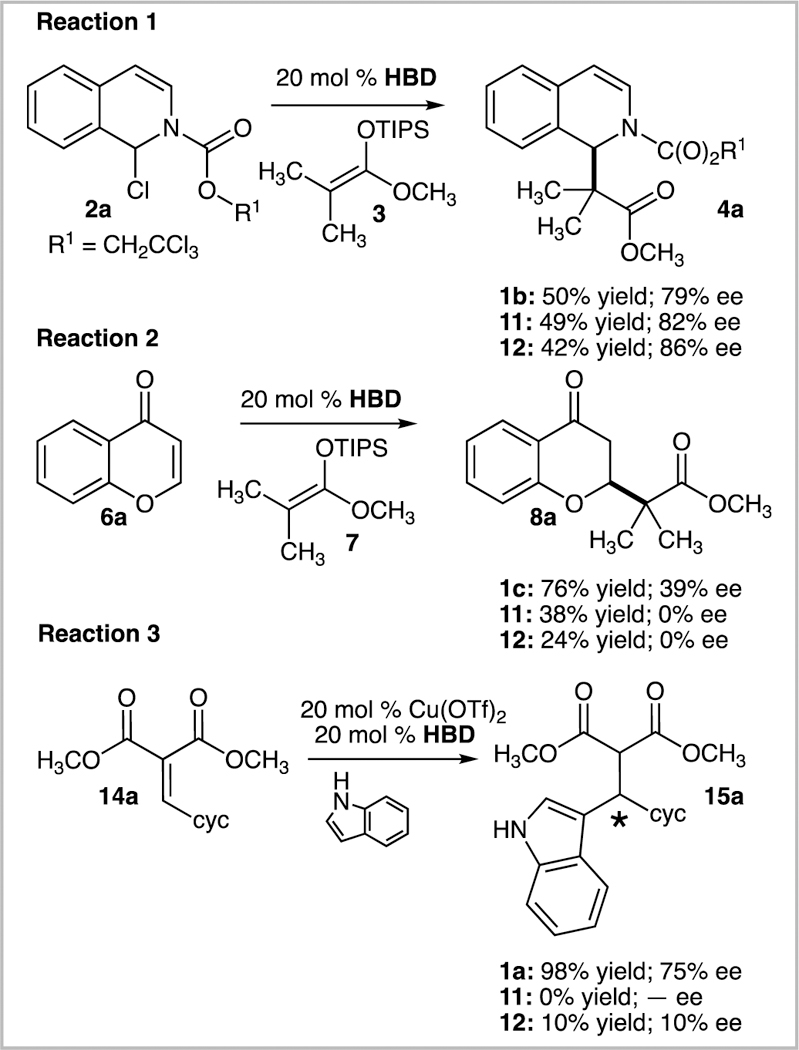
Reactions 1–3 comparing silanediol 1a, thiourea 11, and squaramide 12. Reaction 1: alkylation of isoquinoline with silyl ketene acetal, Reaction 2: conjugate addition of silyl ketene acetal to chromen-4-one, Reaction 3: Lewis acid catalyzed Friedel-Crafts alkylation of indole with alkylidene malonate
In the first reaction, the addition of silyl ketene acetal 3 to 2a to generate 4a, plausibly proceeds through an isoquinolium chloride ion pair (Reaction 1, Scheme 5). This reaction system allows us to directly study the effect of silanediols, thioures, and squaramides on ion pairs containing chloride (Figure 3). There appears to be a clear correlation of the strength of association to enantiomeric excess. Specifically, the more tightly bound thiourea 11 and squaramide 12 generate improved enantiomeric excesses when compared to the silanediol 1a, which has a lower association constant.
Figure 3:
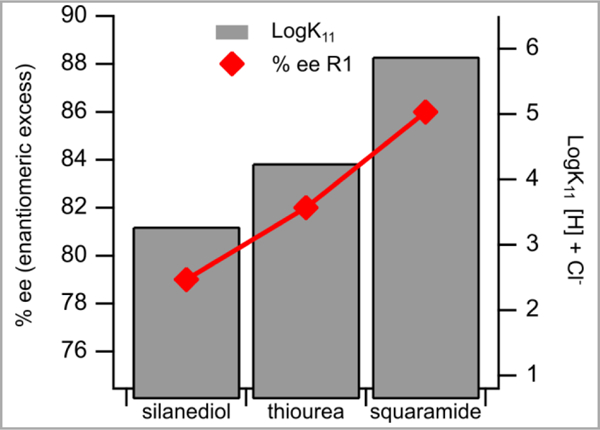
Chloride association constant determined in CHCl3 correlated to enantiomeric excess in the addition of silyl ketene acetals to isoquinolinium ions, reaction 1 (R1).
The influence of silanediols, thioureas, and squaramides on ion pairs containing triflate as the counterion can be studied in Reactions 2 and 3 in Scheme 5. In these cases of triflate ion pairs, the results are less predictable than in the outcomes of the reactions with chloride ion pairs. It was experimentally determined that silanediol 1a recognizes triflates more weakly than either the thiourea 11 or squaramide 12. However, in both the addition of silyl ketene acetal 7 to the benzopyrylium triflate derived from 6a (Reaction 2, Scheme 5) and the addition of indole to cyclohexylidene malonate 14a (Reaction 3, Scheme 5), the silanediol outperformed both thiourea 11 and squaramide 12 in terms of enantiomeric excess and yield (Figure 4).
Figure 4:
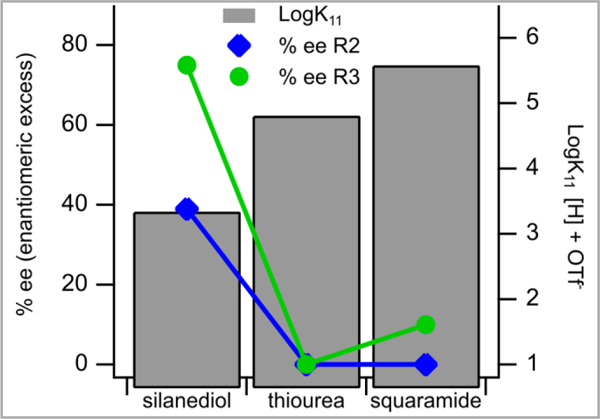
Triflate association constant, determined in toluene, correlated to enantiomeric excess in reactions 2 and 3 (R2 and R3).
Conclusions
Anion-binding catalysis is emerging as an impressive synthetic tool able to catalyze reactions that are inaccessible to more conventional types of catalysis. Several families of enantioselective anion-binding catalysts are now available and it appears that there may be complementary reactivity patterns between them. The identification of parameters able to aid in predicting which anion-binding catalyst to choose to influence a desired reactivity pattern would be an enormous advance in the field.
We have observed that silanediols can offer complementary reactivity patterns when compared to thioureas and squaramides. The origin of the unique catalytic abilities remains unknown and is a point of ongoing study in our research program. This article describes the first direct comparison of silanediols, thioureas, and squaramides in three separate reactions. It has also correlated the association constant of each catalyst to enantiomeric excess in the three processes. In the case of reactions involving chloride ions there appears to be a trend that the stronger the host:guest interaction is, the higher the enantiomeric excess. Alternatively, the silanediol uniquely enables enantioselectivity in the two reactions involving triflate ion pairs that are described herein despite the observation that its binding constant to triflate is lower than both the squaramide and thiourea. Although the reasons for the unique reactivity of silanediols remain uncertain there are additional factors, such as undesired side reactions of the catalysts and non-covalent interactions beyond just anion recognition (e.g., pi-stacking), that may be important to consider. Ongoing investigations in our laboratory are dedicated toward better understanding and capitalizing on the unique role of silanediols in enantioselective anion-binding catalysis.
Toluene was purified by passage through a Pure Process Technology solvent system prior to use. Ethyl acetate and hexanes were used as received. Toluene was dried over 4Å molecular sieves prior to use in the binding constant studies. Chloroform was purified to remove any stabilizer and distilled from CaH2 prior to use in the binding constant studies. Copper (II) triflate was dried at 100°C under vacuum prior to use. Guest compounds TBACl and TBAOTf were dried under vacuum and stored under nitrogen. The silanediol catalyst was prepared according to a literature method.6d The thiourea 11 was prepared according to literature procedures.14 The squaramide 12 was prepared according to literature procedures.8b Indole was recrystallized from hexanes prior to use. All other reagents were used directly as received from the manufacturer unless otherwise noted. Preparative silica gel chromatography was performed using SiliaFlash F60 silica gel (40 – 63 μm). Analytical thin layer chromatography was performed using Analtech 250 μm silica gel HLF plates and visualized under UV 254nm. All 1H NMR spectra were acquired using a Bruker BioSpin 500MHz Avance III Digital NMR spectrometer or JOEL ECA-500 (500MHz) NMR spectrometer and calibrated using the solvent signal (CDCl3 7.26 ppm). Multiplicities were determined using MNova software. All 13C NMR spectra were acquired using a Bruker BioSpin 126MHz Avance III Digital NMR spectrometer or Bruker Avance DPX 400 (100 MHz) and calibrated using the solvent signal (CDCl3 77.16 ppm). Infrared spectra were acquired using a Bruker Vertex 70 with an ATR accessory. High resolution mass spectra were acquired using an Agilent 6520 Q-TOF mass spectrometer. Chiral HPLC analysis was performed using an Agilent 1260 equipped with a diode array detector. Optical rotations were acquired on a Jasco Digital Polarimeter with a 1 dm cell and a sodium lamp. UV-Vis spectrometry experiments were conducted using a Thermo Scientific Evolution 3000 spectrometer or a Shimadzu UV-2500PC spectrometer with 1 cm path length quartz cuvettes.
4a:
An oven dried 2-dram vial with screw top cap and septa was equipped with a stir bar and flushed with nitrogen. The vial was sealed and covered further with parafilm. Isoquinoline (11.8 mL, 0.1 mmol, 1.0 equiv) was added via syringe, PhMe (2 mL) was added and the solution was cooled to 0 °C. 2,2,2-Trichloroethyl chloroformate (15.0 mL, 0.22 mmol, 1.1 equiv) was added, the ice bat was removed and the solution was warmed to room temperature while stirring for 30 minutes. The cloudy suspension was cooled to –78 °C. Anion-binding catalyst (0.2 mmol, 0.2 equiv) was added as a solution in PhMe followed by the silyl ketene acetal (0.15 mmols, 1.5 equiv). The reaction vessel was transferred to a –78 °C acetone bath equipped with immersion cooling coil and stirred for 40 hours. The reaction was quenched at –78 °C by the addition of NaOMe (0.2 mL, 0.5 M in Me OH, 1.0 equiv) and then warmed to room temperature before filtration through a short silica gel plug with EtOAc as the eluent. Removal of the solvent in vacuo and subsequent purification via flash column chromatography on silica gel (0:100 EtOAc:Hexanes to 4:96 EtOAc:Hexanes) yielded the title compound as a colorless oil (32.4 mg, 0.80 mmols, 80% yield).
IR (neat) 2991, 2924, 2357, 2343, 1724, 1717, 1627, 1448, 1374, 1322, 1225, 1128, 1046, 941 cm−1
1H NMR (400 MHz, CDCl3): the compound exists as a 3:1 mixture of carbamate rotamers. Signals corresponding to the major rotamer: δ 7.28−7.19(m, 2H), 7.10−6.95 (m, 2H), 6.96 (d, J = 7.6 Hz, 1H), 5.95 (d, J = 7.6 Hz, 1 H), 5.74 (s, 1H),4.97 (d, J = 12.0 Hz, 1H), 4.70 (d, J = 12.0 Hz, 1H), 3.64 (s, 3H), 1.20 (s, 3H), 1.12 (s, 3H).
Representative signals of the minor rotamer: δ 6.05 (d, J = 7.6 Hz, 1H), 5.79 (s, 1H), 4.86 (s,2H), 3.61 (s, 3H), 1.29 (s, 3H), 1.26 (s, 3H).
13C NMR (100 MHz, CDCl3) Signals correspond to major rotamer: δ 175.9, 152.3, 131.3, 128.4, 128.0, 127.2, 125.6, 124.9, 112.0, 95.2, 75.7, 60.9, 52.2, 50.3, 22.6, 21.5.
HRMS (ESI): Mass Calculated for C17H18Cl3NO4 [M+Na]+, 428.0199. Found [M+Na]+, 428.0189.
HPLC (Chiralpak OD-H, 1% isopropanol/99% hexane 0.7 mL/min, tr (minor): 12.8 min, tr (major): 15.7 min.)
[α]24D = –24.5 (c 1.11, CHCl3)
8a:
An 8 mL vial with stir bar was flame dried under vacuum, cooled to room temperature under vacuum, and backfilled with argon gas. Chromone (14.6 mg, 0.1 mmol, 1 eq) and 2,6-di-tert-butyl-4-methylpyridine (6.2 mg, 0.03 mmol, 0.3 eq) was weighed out and placed in the vial. The vial was then placed under vacuum again and backfilled with argon. 200 μL of dry toluene (0.5 M) was added to the vial. Freshly distilled triisopropylsilyl trifluoromethanesulfonate (29.5 μL, 0.11 mmol, 1.1 eq) was added via microliter syringe to the solution and the vial was placed in a 60 °C oil bath for one hour. After the reaction time, the vial was cooled to room temperature and further diluted with 1.3 mL of toluene. The vial was then cooled to –78 °C in an acetone/dry ice bath. After an appropriate amount of time to allow the reaction to come to temperature had passed, a solution of silanediol catalyst in 0.5 mL toluene (12.6 mg, 0.02 mmol, 0.2 eq) was added slowly down the side of the vial. The reaction mixture was stirred for 10 minutes before addition of the silyl ketene acetal (125 μL of a 1 M solution in toluene, 0.125 mmol, 1.25 eq) slowly down the side of the vial. After 4 hours at –78 °C, the reaction was quenched with 200 μL of 3 M HCl (aqueous) (6 eq) at –78 °C. The solution was allowed to warm to room temperature overnight. Then, the crude reaction mixture was extracted with ethyl acetate (5 mL), washed with water (5 mL), dried with Na2SO4, and solvent removed under vacuum. The crude mixture was then dissolved in CDCl3 and 1,3,5-trimethoxybenzene was added as an internal standard for 1H NMR yields. The product was then isolated via silica gel flash column chromatography (100% hexanes to 80/20 hexanes/ethyl acetate) or preparative TLC plates for HPLC analysis (80/20 hexanes/ethyl acetate solvent system). HPLC samples are occasionally filtered through an alumnia plug to remove any undesired silanol by-products. The desired product 8a was prepared 76% by 1H NMR yield.
IR (neat): 2981, 2889, 1729, 1687, 1607, 1463, 1392, 1303, 1221, 1133, 1115, 1078, 990, 870, 764 cm-1.
1H NMR (CDCl3, 400 MHz): δ 7.88–7.86 (m, 1H), 7.48–7.44 (m, 1H), 7.01 (t, J = 7.2 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 4.64 (dd, J = 14, 2.4 Hz, 1H), 3.73 (s, 3H), 2.82–2.75 (m, 1H), 2.62–2.58 (m, 1H), 1.37 (s, 1H), 1.28 (s, 1H).
13C NMR (CDCl3, 100 MHz): δ 192.3, 175.6, 161.7, 136.1, 127.1, 121.6, 120.9, 118.0, 81.8, 52.3, 46.3, 38.5, 20.9, 20.7.
HRMS (ESI): Mass calculated for C14H16NaO4+ [M+Na]+ 271.0941, Found [M+Na]+ 271.0934
HPLC: 30.46:69.54 e.r., Chiralpak AD-H column, 98:2 (Hexanes: isopropanol), 1 mL/min, 254 nm, tr (minor): 11.4 min, tr (major): 13.8 min.
[α]23D = 13.0 (c 0.135, CHCl3).
15a:
Dimethyl cyclohexylidene malonate (113 mg, 0.5 mmol, 1.0 eq), Cu(OTf)2 (36 mg, 0.1 mmol, 0.2 eq), trifluoroisopropanol (22.6μL, 0.25 mmol, 0.5 eq) and toluene (5mL) were added into a 20 mL screw top reaction vial with a teflon coated septum. The flask was purged with dry N2 and the reaction mixture stirred for 15 minutes or until a homogenous slurry was obtained. The reaction vial was then cooled to –78°C in a dry ice/acetone bath. 2.4 mL of 0.05 M silanediol stock solution (82 mg15, 0.24 mmol, 0.2 eq) in toluene and 2.6 mL of indole solution (88 mg, 0.75 mmol, 1.5 eq) in toluene were added into the reaction vial dropwise. The reaction vial was transferred into a lab freezer (–28°C) and stirred overnight. The reaction was quenched with 2 mL of DI water, stirred for 10 minutes then extracted with EtOAc 3×10mL and dried over Na2SO4. Solvent was removed from the combined organic layer under vacuum to obtain crude product. The crude product was purified by silica gel column chromatography (eluent = 4:1, Hexanes:EtOAc). The resulting material was purified further by silica gel column chromatography (eluent = 100% dichloromethane). After removal of the solvent under vacuum, dimethyl 2-(cyclohexyl(1H-indol-3-yl)methyl)malonate was obtained as an off white solid (159mg, 0.46 mmol, 93% yield).
IR (neat): 3413, 2926, 2853, 1755, 1726, 1457, 1431 cm-1.
1H NMR (CDCl3, 500 MHz): δ = 8.03 (s, 1H), 7.66 (ddt, J = 8.0, 1.5, 0.8 Hz, 1H), 7.32 (dt, J = 8.0, 0.9 Hz, 1H), 7.16 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.10 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 7.03 (d, J = 2.4 Hz, 1H), 4.03 (d, J = 10.8 Hz, 1H), 3.78 (dd, J = 10.8, 4.9 Hz, 1H), 3.73 (s, 3H), 3.35 (s, 3H), 1.78 – 1.55 (m, 6H), 1.31 – 1.08 (m, 2H), 1.02 – 0.81 (m, 3H).
13C NMR (CDCl3, 126 MHz): δ = 169.62, 168.96, 135.76, 128.42, 122.87, 121.85, 119.72, 119.41, 113.90, 111.02, 55.69, 52.64, 52.26, 42.04, 41.16, 32.33, 28.79, 26.68, 26.48, 26.32.
HPLC: 86.0:14.0 e.r., 72% ee, Chiralpak AS-H column (10% iPrOH/Hexanes, 1 mL/min, 225nm); tR (minor) = 8.55 min, tR (major) = 23.80 min. Rf = 0.25 (4:1, Hexanes:EtOAc).
[α]23D = −9.1 (c = 4.0, CH2Cl2)
Determination of Association Constants:
The association constant of the hosts (silanediol 1a, thiourea 11, and squaramide 12) and guests (TBAOTf and TBACl) were determined with UV-Vis spectroscopy. Chloroform was purified to remove any stabilizers and distilled from CaH2 prior to use. Toluene was dried over 4Å molecular sieves prior to use. Commercially available TBAOTf and TBACl were dried under reduced pressure for 1-day prior to use. The titration experiments were carried out with a host solution (3 mL, 1×10−5 M in CHCl3) in a quartz cell and UV-Vis spectra recorded upon the addition of aliquots of the stock solution of guest ion in CHCl3 or toluene with a microsyringe. The association constant was then calculated using a self written non-linear regression analysis program (see supporting information for details). Each titration was repeated in triplicate and the mean K11 was reported.
NMR Titrations:
The NMR titration experiments were carried out with a host solution (1 × 10−2 M) in CDCl3. NMR spectra were recorded upon the addition of guest compound into the host solution. The guest compound was dissolved in the host working solution so as to maintain the concentration of host during the titration.
Supplementary Material
Acknowledgments
Funding Information
The National Science Foundation (1362030), the National Institutes of Health (1R35GM12480401), and Worcester Polytechnic Institute (WPI) are gratefully acknowledged for providing support for our studies. K.O. was supported by a fellowship from Yamagata University to study in the Mattson laboratory to complete these investigations. J. W. A. was supported by a WPI global scholars fellowship to complete investigations in the Kondo laboratory. J. H. is grateful for a WPI Summer Undergraduate Research Fellowship that supported her contribution to this project.
Biosketches

Jonathan Attard obtained his BA in Chemistry and Physics from Whittier college in 2010. He worked in industry for 6 years before beginning his PhD program, in 2016, at Worcester Polytechnic Institute under the guidance of Professor Anita Mattson. His current research involves enantioselective copper catalyzed reactions for the synthesis of chromanone natural product analogues.

Kohei Osawa obtained his BA in Engineering from Yamagata University in 2014. He began his PhD program in 2014 and he is expecting to complete his doctoral research on anion recognition behavior and chiral induction of biaryl-based bis-urea derivatives with Professor Shin-ichi Kondo in 2019 to obtain PhD in Engineering from Yamagata University. He joined Professor Mattson’s current research group from Nov 2017 to Jan 2018 as an international researcher and worked on enantioselective silanediol catalysis.

Yong Guan obtained his BS and MS from Wuhan University in 2004 and 2006. He completed doctoral research on construction of a vaulted biaryl ligand library for the aziridination reaction with Prof. William Wulff and earned a PhD in 2012 from Michigan State University. He performed his postdoctoral research with Prof. Christopher Douglas at University of Minnesota on one-pot Sonogashira coupling and regioselective tetradehydro-Diels-Alder reaction to synthesis rubicenes (2013–2014), and with Prof. Steven Townsend at Vanderbilt University on metal-free synthesis of unsymmetrical organoselenides and selenoglycosides (2015–2016). In 2017, he joined Mattson group at Worcester Polytechnic Institute as a postdoctoral fellow. His current projects are on asymmetric copper catalysis and drug discovery.

Jessica Hatt is expecting to obtain her BS in Chemistry from Worcester Polytechnic Institute in 2020. She joined Professor Mattson’s current research group as an undergraduate researcher in the fall of 2017

Shin-ichi Kondo received his Ph.D. Degree from Kyoto University in 1995 under the supervision of Professor Atsuyoshi Ohno. He became an assistant professor at Ritsumeikan University in 1995 and moved to Gunma University in 1996. In 2009, he became an associate professor at Yamagata University as PI. Since 2012, he has been a full professor at Yamagata University. His research interests include molecular recognition, in particular anion recognition chemistry, fluorescence materials, and organosilicon chemistry

Anita Mattson obtained her BS from Northern Michigan University in 2002 where she conducted undergraduate research with Frankie Ann McCormick. She completed doctoral research on thiazolium-based N-heterocyclic carbene catalysis with Prof. Karl Scheidt and earned a PhD in 2007 from Northwestern University. After working on a synthesis of hemibrevetoxin B as an NIH NRSA postdoctoral fellow in the Crimmins group at the University of North Carolina at Chapel Hill, Anita began her independent career in 2009 at the Ohio State University. After being promoted to Associate Professor with tenure at OSU in 2015, she moved to Worcester Polytechnic Institute in 2016. Anita’s current research program blends her love of organic catalyst design and complex molecule synthesis with drug discovery.
Footnotes
Supporting Information
Is there Supporting Information to be published? Click here to indicate YES or NO (text and links will be updated prior to publication).
References
- (1).For recent reviews on silanediols, see: [Google Scholar]; (a) Wieting JM; Hardman-Baldwin AM; Visco MD; Mattson AE Aldrichimica Acta 2016, 49, 15–20; [Google Scholar]; (b) Franz AK; Wilson SO J. Med. Chem. 2013, 56, 388; [DOI] [PubMed] [Google Scholar]; (c) Sieburth SM, Chen C-A Eur. J. Org. Chem. 2006, 311; [Google Scholar]; (d) Min GK; Heranandez D; Skryidstrup T Acc. Chem. Res. 2013, 46, 457. [DOI] [PubMed] [Google Scholar]
- (2).Kondo S; Harada T; Tanaka R; Unno M Org. Lett. 2006, 8, 4621–4624. [DOI] [PubMed] [Google Scholar]
- (3).For a recent example of silanediols involved in sensing, see: Kondo S; Hie Y; Yamaura M Org. Lett. 2013, 15, 520–523. [DOI] [PubMed] [Google Scholar]
- (4).For selected examples of silanediols involved in achiral catalysis, see: [Google Scholar]; (a) Tran NT; Min T; Franz AK Chem. Eur. J. 2011, 17, 9897; [DOI] [PubMed] [Google Scholar]; (b) Schafer AG; Wieting JM; Mattson AE Org. Lett. 2011, 13, 5228–5232; [DOI] [PubMed] [Google Scholar]; (c) Hardman-Baldwin AM; Mattson AE ChemSusChem 2014, 7, 3275–3278. [DOI] [PubMed] [Google Scholar]
- (5).For reviews on anion-binding catalysis, see: [Google Scholar]; (a) Visco MD; Attard J; Guan Y; Mattson AE Tetrahedron Lett. 2017, 58, 2623–2428; [Google Scholar]; (b) Busschaert N; Caltagirone C; Van Rossom W; Gale PA Chem. Rev. 2015, 115, 8038–8155; [DOI] [PubMed] [Google Scholar]; (c) Brak K; Jacobsen EN Angew. Chem. Int. Ed. 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For examples of silanediols plausibly involved in enantioselective anion-binding catalysis, see: [Google Scholar]; (a) Guan Y; Attard JW; Visco MD; Fisher TJ; Mattson AE Chem. Eur. J. 2018, 24, 7123; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hardman-Baldwin AM; Visco MD; Wieting JM; Stern C; Kondo S; Mattson AE Org. Lett. 2016, 18, 2883–2885; [DOI] [PubMed] [Google Scholar]; (c) Wieting JM; Fisher TJ; Schafer AG; Visco MD; Galluci JC; Mattson AE Eur. J. Org. Chem. 2015, 525–533; [Google Scholar]; (d) Schafer AG; Wieting JM; Fisher TJ; Mattson AE Angew. Chem. Int. Ed. 2013, 52, 11321–11324. [DOI] [PubMed] [Google Scholar]
- (7).For select examples of processes plausibly proceeding through thiourea anion binding catalysis, see: [Google Scholar]; (a) Park Y; Harper KC; Kuhl N; Kwan EE; Liu RY; Jacobsen EN Science 2017, 355, 1620166; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jarvis CL; Hirschi JS; Vetticatt MJ; Seidel D Angew. Chem. Int. Ed. 2017, 56, 2670–2674; [DOI] [PubMed] [Google Scholar]; (c) Kennedy CR; Kehnerr D; Rajapaksa NS; Ford DD; Park Y; Jacobsen EN J. Am. Chem. Soc. 2016, 138, 13525–13528; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhao C; Chen SB; Seidel DJ Am. Chem. Soc. 2016, 138, 9053–9056; [DOI] [PubMed] [Google Scholar]; (e) Reisman SE; Doyle AG; Jacobsen EN J. Am. Chem. Soc. 2008, 130, 7198–7199; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) De CK; Clauber EG; Seidel D J. Am. Chem. Soc. 2009, 131, 17060–17061; [DOI] [PubMed] [Google Scholar]; (g) Taylor MS; Tokunaga N; Jacobsen EN Angew. Chem. Int. Ed. 2005, 44, 6700–6704. [DOI] [PubMed] [Google Scholar]
- (8).For examples of plausible enantioselective squaramide anion binding catalysis, see: [Google Scholar]; (a) Wendlandt AE; Vangal P; Jacobsen EN Nature 2018, 556, 447–451; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Banik SM, Levina A, Hyde AM, Jacobsen Science EN 2017, 358, 761–764; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu RY; Wasa M; Jacobsen EN Tetrahedron Lett. 2015, 56, 3428–3430; [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kotke M; Schreiner PR Tetrahedron 2006, 62, 434. [Google Scholar]
- (10).Raheem IT; Thiara PV; Peterson EA; Jacobsen EN J. Am. Chem. Soc. 2007, 129, 13404–13405. [DOI] [PubMed] [Google Scholar]
- (11).(a) Fischer T; Bamberger J; Gomez-Martinez M; Piekarski DG; Mancheno O Angew. Chem. Int. Ed. 2018, 57, early view; [Google Scholar]; (b) Fischer T; Duang Q-N; Mancheno OG Chem. Eur. J. 2017, 23, 5983–5987; [DOI] [PubMed] [Google Scholar]; (c) Mancheno OG; Asmus S; Zurro M; Fischer T Angew. Chem. Int. Ed. 2015, 54, 8823–8827; [DOI] [PubMed] [Google Scholar]; (d) Zurro M Asmus S Beckendorf S; Muck-Lichtenfeld C; Mancheno OG J. Am. Chem. Soc. 2014, 136, 13999–14002; [DOI] [PubMed] [Google Scholar]; (e) Ohmatus K; Kiyokawa M; Ooi TJ Am. Chem. Soc. 2011, 133, 1307; [DOI] [PubMed] [Google Scholar]; (f) Ohmatsu J; Ando Y; Ooi TJ Am. Chem. Soc 2013, 135, 18706. [DOI] [PubMed] [Google Scholar]
- (12).For reviews including naturally-occurring bioactive chromanones and tetrahydroxanthones, see; [Google Scholar]; (a) Masters KS; Brase S Chem. Rev. 2012, 112, 3717–3776; [DOI] [PubMed] [Google Scholar]; (b) Wezeman T; Brase S; Masters KS Nat. Prod. Rep. 2015, 32, 6–28. [DOI] [PubMed] [Google Scholar]
- (13).Thiourea 11 and squaramide 12 were employed in these studies so that a stronger UV-Vis response may be observed.
- (14).Knowles RR; Lin S; Jacobsen EN J. Am. Chem. Soc. 2010, 132, 5030–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Corrected for 5:1 silanediol:Et2O content
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.