

ABSTRACT
PURPOSE OF REVIEW
Hippocampal sclerosis, argyrophilic grain disease, and primary age-related tauopathy are common Alzheimer disease mimics that currently lack clinical diagnostic criteria. Increased understanding of these pathologic entities is important for the neurologist who may encounter patients with an unusually slowly progressive degenerative dementia that may appear to meet criteria for Alzheimer disease but who progress to develop symptoms that are unusual for classic Alzheimer disease
RECENT FINDINGS
Hippocampal sclerosis has traditionally been associated with hypoxic/ischemic injury and poorly controlled epilepsy, but it is now recognized that hippocampal sclerosis may also be associated with a unique degenerative disease of aging or may be an associated pathologic finding in many cases of frontotemporal lobar degeneration. Argyrophilic grain disease has been recognized as an enigma in the field of pathology for over 30 years, but recent discoveries suggest that it may overlap with other tau-related disorders within the spectrum of frontotemporal lobar degeneration. Primary age-related tauopathy has long been recognized as a distinct clinical entity that lies on the Alzheimer pathologic spectrum, with the presence of neurofibrillary tangles that lack the coexistent Alzheimer plaque development; thus, it is thought to represent a distinct pathologic entity.
SUMMARY
Despite advances in dementia diagnosis that suggest that we have identified and unlocked the mysteries of the major degenerative disease states responsible for cognitive decline and dementia in the elderly, diseases such as hippocampal sclerosis, argyrophilic grain disease, and primary age-related tauopathy demonstrate that we remain on the frontier of discovery and that our diagnostic repertoire of diseases responsible for such clinical symptoms remains in its infancy. Understanding such diagnostic confounds is important for the neurologist in assigning appropriate diagnoses and selecting appropriate therapeutic management strategies for patients with mild cognitive impairment and dementia.
INTRODUCTION
It is widely recognized that the diagnostic accuracy of the clinical diagnosis of Alzheimer disease (AD) is less than 80% in most centers.1 The important question that emerges from this finding is what is the neuropathologic substrate for the 20% of cases that are misdiagnosed? Is it simply overlap in clinical features with other common neuropathologic conditions? Amnestic variants of dementia with Lewy bodies (DLB), frontotemporal dementia (FTD), or vascular dementia might be considered.2 Or is the underlying disorder instead related to the many other prevalent neuropathologic conditions that remain poorly characterized clinically, such as hippocampal sclerosis of aging, argyrophilic grain disease, or primary age-related tauopathy (PART)? These clinically uncharacterized neuropathologic disease states are found at high frequency in the elderly population and are likely contributors to the observed inaccuracy of clinical diagnostic criteria in clinicopathologic correlation studies.
Recent and ongoing work over the past several decades has brought to light the importance and high prevalence of diseases such as hippocampal sclerosis,3 argyrophilic grain disease,4 and PART5 as common mimics of AD and other dementing disorders, largely because their own clinical phenotype has been poorly elucidated to date. Each of these diseases can be found in isolation at autopsy, but they are more commonly found in association with other comorbid pathologies, further complicating our understanding of the clinical phenotype for the diseases.1,2,6 Yet their prevalence can be as high as 20% of normal controls and higher than 50% in individuals with clinical dementia.1,2,6 This article focuses on our understanding of the pathology and clinical features of these conditions, including signs and symptoms, imaging findings, biomarker discoveries, and familial and genetic predisposition, which have not yet been fully embraced within clinics and medical institutions as part of the standard antemortem diagnostic considerations responsible for cognitive decline and dementia in the aging population today (case 10-1). It should be noted that this article focuses on distinct pathologic disease states that do not yet have established clinical diagnostic criteria. Atypical clinical characteristics of other pathologically defined degenerative diseases that may mimic alternative clinical diagnoses (such as DLB or vascular dementia mimicking AD or vice versa) are discussed in other articles in this issue.
CASE 10-1
An 82-year-old woman presented for evaluation of short-term memory loss. The patient was able to converse normally, and she personally described only mild memory problems described as difficulty remembering the names of casual acquaintances. She denied any difficulties with her daily function. Her husband, however, stated that he had noticed progressive short-term memory decline with frequent repeating behavior over the past 4 years. He stated that this had barely changed over time, although, looking back over the past 4 years, he noted that she was clearly more impaired now than when he first noticed her problems. She frequently misplaced items and forgot dates and appointments unless reminded, but he stated she was otherwise fully engaged in all her daily activities at home, including more complex activities such as cooking and managing the household. She had been driving without incident, and her husband had no concerns about her driving ability. She remained engaged socially with both church activities and a local gardening club. She had no family history of dementia but noted that she had a grandfather who died of amyotrophic lateral sclerosis. Her past medical history was notable only for hypertension that was well-controlled on lisinopril 10 mg/d.
On examination, she scored 22/30 on the Montreal Cognitive Assessment (MoCA), missing three points on orientation and all five points on delayed recall. Her performance was fully intact on the remainder of the examination. Her neurologic examination was otherwise normal, without evidence of focal neurologic deficits, motor neuron disease, or parkinsonism.
Laboratory workup for reversible causes of dementia was unrevealing. MRI of her brain (similar to that presented in figure 10-1) demonstrated focal hippocampal and medial temporal lobe atrophy with a question of increased T2 signal in the hippocampus bilaterally. Amyloid positron emission tomography (PET) was negative for cerebral amyloid. CSF showed normal levels of amyloid β1-42 and phosphorylated tau protein.
FIGURE 10-1.
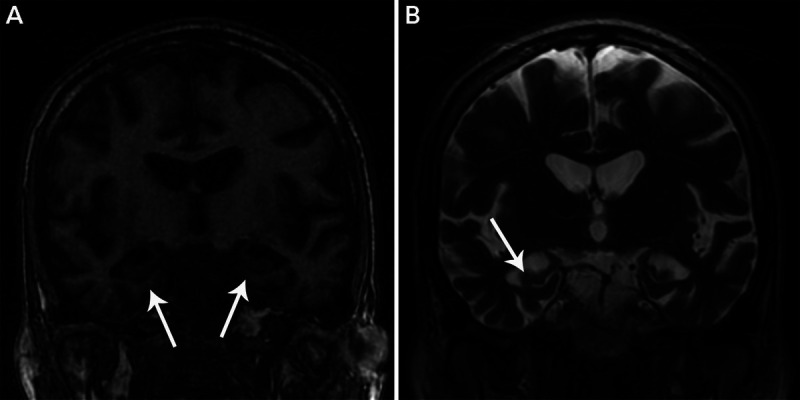
Hippocampal sclerosis of aging. A, Coronal T1-weighted MRI of the medial temporal lobes shows atrophy of the hippocampus and neighboring medial temporal lobe structures (arrows) similar to that seen in the patient in case 10-1. B, Coronal T2-weighted MRI shows ex vacuo enlargement of the adjacent temporal horn of the lateral ventricle (arrow). Note the relative absence of atrophy outside the medial temporal lobe structures.
Other than the negative molecular biomarkers, she met clinical diagnostic criteria for mild cognitive impairment of the Alzheimer type, which was felt to be consistent with the MRI results. She was treated with donepezil for her cognitive decline and had follow-up appointments for another 6 years before she died.
At autopsy, the gross weight of her brain was 1180 g (2.6 lb), and mild global cerebral atrophy and bilateral gross focal medial temporal and hippocampal atrophy were noted on visual inspection. No appreciable vascular lesions were noted on macroscopic or microscopic examination. She was found to have Braak stage I neurofibrillary tangles in the entorhinal cortex and an almost complete absence of even diffuse amyloid plaque deposition. Hematoxylin and eosin (H&E) stains of the hippocampus revealed severe neuronal loss and gliosis throughout the CA1 sector and subiculum. Transactive response DNA-binding protein 43 (TDP-43) antibody staining showed characteristic Type A neuronal inclusions.
COMMENT
The primary pathologic diagnosis given was hippocampal sclerosis of aging despite a clinical presentation consistent with an antemortem diagnosis of Alzheimer disease. This case illustrates the confound inherent in clinical diagnosis of an early amnestic syndrome and highlights the importance of consideration of alternative pathologic diagnoses in such cases.
HIPPOCAMPAL SCLEROSIS
Hippocampal sclerosis is a term that describes two key pathologic features that frequently involve the hippocampus and associated structures: neuronal loss and widespread gliosis.3,7 These pathologies can be seen in association with a variety of potentially causative conditions, including advanced aging, anoxic/hypoxic injury, frontotemporal lobar degeneration (FTLD), temporal lobe epilepsy, and chronic traumatic encephalopathy.7–9 Indeed, the hippocampus and its associated structures appear highly vulnerable to a variety of pathologic insults that can lead to both the neuronal loss and the widespread gliosis that define hippocampal sclerosis generically. Yet, despite the superficially similar pathologic end points, the distinct causes of hippocampal sclerosis may actually be quite different from one another in terms of pathogenic mechanisms.
It is widely appreciated that some of the neuronal populations in the hippocampus are particularly vulnerable to anoxic/hypoxic injury.10 The neuronal degeneration and gliosis associated with hippocampal sclerosis is largely seen within the CA1 hippocampal subfield and subiculum, with relative sparing of the CA2–CA4 hippocampal subfields and other neighboring or anatomically connected regions (figure 10-2).11 The precise causes for the selective vulnerability may include the high degree of plasticity seen in this cortical area, its location in a watershed distribution between the anterior and posterior choroidal arteries, and the high prevalence of glutamatergic synapses in this “feed-forward” hippocampal circuit, leading to excess calcium influx in times of stress and the triggering of the excitotoxic and apoptotic cascade.11–14 These same features may help to explain the high vulnerability of hippocampal neurons to excitotoxic stresses caused by recurrent seizure activity, which may be primarily mediated through N-methyl-d-aspartate (NMDA) receptors in this region.12 Epilepsy and anoxia/hypoxia were considered primary in the development of hippocampal sclerosis until the recent discovery of the association of hippocampal sclerosis with some forms of FTLD seen at autopsy.15,16 The discovery of abnormal accumulations of transactive response DNA-binding protein 43 (TDP-43) in forms of FTLD and the subsequent development of antibodies detecting these pathologic inclusions has greatly enhanced our understanding of hippocampal sclerosis.9,17,18
FIGURE 10-2.
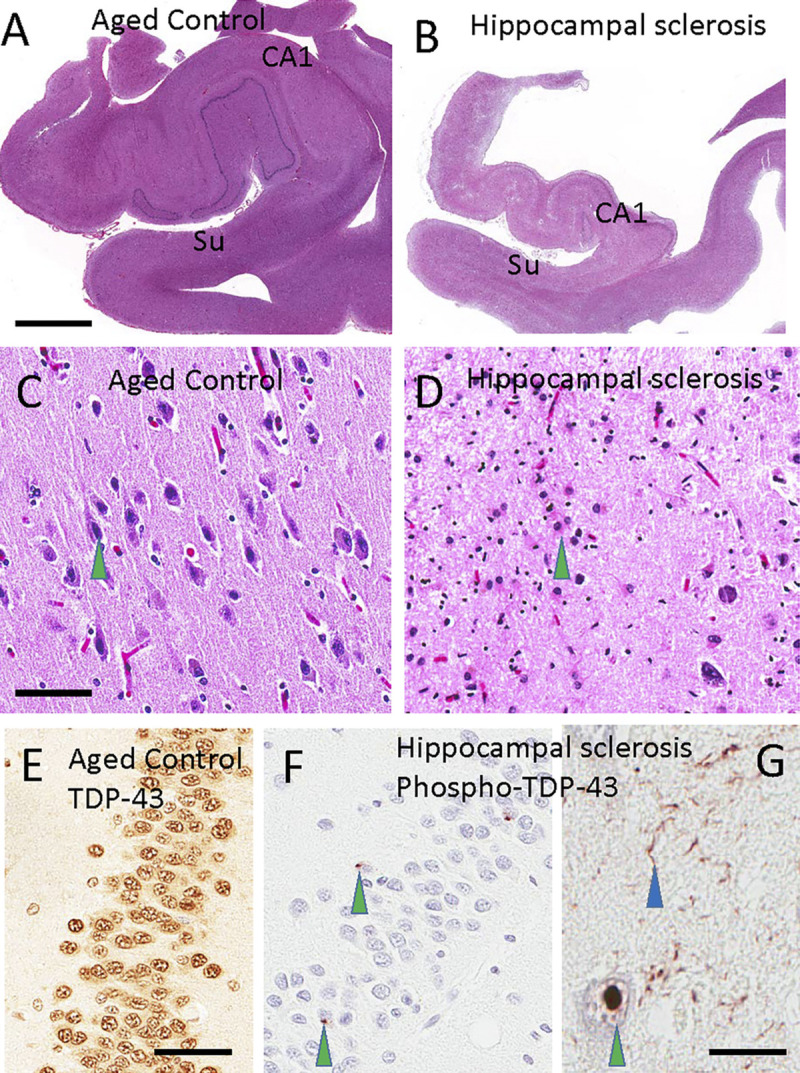
Histopathology of a normal hippocampus compared to hippocampal sclerosis. Hematoxylin and eosin (H&E) staining of a healthy control hippocampus (A) and hippocampus with hippocampal sclerosis (B) highlights selective involvement of the CA1 sector of the hippocampus and subiculum (Su) in hippocampal sclerosis. Note that panels A and B were taken at the same magnification. A normal subiculum stained with H&E, with a pyramidal neuron (C, green arrowhead) and the subiculum of a patient with hippocampal sclerosis of aging, in which the cellular population of neurons has been replaced by reactive astrocytes (D, green arrowhead). Transactive response DNA-binding protein 43 (TDP-43) is normally present in cell nuclei, as shown here by using an antibody against nonphosphorylated TDP-43 in the dentate granule cells (E). By contrast, in hippocampal sclerosis of aging, phosphorylated TDP-43 pathology is seen in cells of the dentate granule neurons (F, green arrowheads) and inside cells (G; green arrowhead indicates intranuclear deposit). Neuritic pathology is seen in the CA1 sector of the hippocampus (G, blue arrowhead). Scale bars: 5 mm in A, B; 300 microns in C, D; 50 microns in E, F; 60 microns in G.
Before our ability to sensitively and specifically detect TDP-43 inclusions, the diagnosis of hippocampal sclerosis was largely made using hematoxylin and eosin (H&E)–stained slides looking for the neuronal dropout and gliosis characteristic of hippocampal sclerosis (figure 10-2).7 This method was relatively nonspecific and failed to detect a large number of autopsy cases in which the pathology was subtle or found in a comorbid state with diseases such as AD that also lead to neuronal dropout and gliosis within the hippocampus.3,9 It is now recognized that hippocampal sclerosis occurs not only in “pure” or isolated forms but is frequently found to be a comorbid process, coexisting with other neurodegenerative pathologies such as AD, DLB, progressive supranuclear palsy (PSP), and amyotrophic lateral sclerosis (ALS).2,18–20 Hippocampal sclerosis also often develops in the course of FTLD. In aged individuals, the pathogenesis of hippocampal sclerosis is now more likely to be associated with a comorbid neurodegenerative process than the prior associations with anoxic/hypoxic insults and temporal lobe epilepsy that were historically assumed to be the primary causes for hippocampal sclerosis in the elderly.9,11,12,21,22 These findings also began to open the door for exploration into genetic contributions to FTLD associations with hippocampal sclerosis.
Frontotemporal Lobar Degeneration–Related Hippocampal Sclerosis
Hippocampal sclerosis has been identified as an associated pathologic finding in both young and old patients with FTLD who are devoid of a history of epilepsy or anoxic/hypoxic injury.15,16,23,24 This finding was of uncertain significance until the discovery of TDP-43 pathologic inclusions in FTLD cases and the subsequent elucidation of familial genetics in FTLD linked to hippocampal sclerosis. FTLD-related mutations in the GRN gene (a hexanucleotide repeat expansion in a region in chromosome 9 locus strongly associated with both ALS and FTLD [C9orf72]) and the TMEM106B gene have been shown to be strongly associated with the development of hippocampal sclerosis.25–27 The known products of these genes may all contribute to hippocampal sclerosis through shared or independent pathways. GRN has been implicated in neuronal survival, inflammatory pathways in the central nervous system, and the modulation of central nervous system angiogenesis.28 The protein product of the C9orf72 gene, influenced by TMEM106B, has been suggested to contribute to actin-mediated cytoskeletal homeostasis, membrane lipid trafficking, and neuronal autophagy, each of which may play a role in the development of hippocampal sclerosis associated with FTLD and ALS.26,27 These proposed mechanisms may all contribute to the development of hippocampal sclerosis in FTLD and highlight the now-accepted premise that hippocampal sclerosis is not just a downstream effect of pathology but is a distinct degenerative process.
PATHOLOGY
On H&E stain, hippocampal sclerosis–FTLD is indistinguishable from the other potential causes of hippocampal sclerosis seen in advanced aging, epilepsy, anoxic/hypoxic injury, and chronic traumatic encephalopathy.11 Selective neuronal loss in the CA1 sector of the hippocampus and subiculum and widespread gliosis in these regions is observed across all these disease states.11 Hippocampal sclerosis–FTLD, however, is strongly associated with TDP-43 inclusions within the affected regions. While TDP-43 immunoreactivity can present in several different forms, the TDP-43 inclusions seen in hippocampal sclerosis–FTLD are exclusively Type A, characterized by widespread TDP-43 reactive neurites with associated intraneuronal cytoplasmic TDP-43 inclusions, irrespective of genetic association with GRN or C9orf72 mutations.3,9,17,29 These pathologic findings are seen in the hippocampal CA1 subfield, subiculum, and amygdala. It is important to note that hippocampal sclerosis–FTLD, as with the more widespread cortical and subcortical pathology seen in FTLD, can frequently be unilateral on gross inspection of the brain.30 The overt signs of hippocampal and amygdala atrophy can frequently be unilateral as well. However, if the apparently unaffected side is evaluated with TDP-43 immunohistochemistry, the Type A TDP-43 neuritic and cytoplasmic inclusions are present bilaterally irrespective of the apparent unilaterality of the gross atrophy and apparent sclerosis visualized on H&E staining.
CLINICAL PRESENTATION
While the clinical presentation of FTLD in general is quite heterogeneous, encompassing both behavioral and a variety of language-predominant clinical syndromes, the specific presence of hippocampal sclerosis pathology in FTD dictates common findings attributed to the loss of hippocampal function. In other words, hippocampal injury leads to a decline in anterograde memory function, detected as a decline in delayed recall on formal cognitive testing. To date, clinical diagnostic criteria to evaluate the presence or absence of hippocampal sclerosis in clinical cases meeting criteria for FTD are lacking. The potential for hippocampal sclerosis should be actively pursued in cases with associated motor neuron disease and in the semantic dementia variant of FTD. However, the presence of hippocampal sclerosis should always be suspected in patients diagnosed with FTD who demonstrate appreciable anterograde amnestic signs and symptoms, irrespective of other associated clinical features or diagnoses.
Hippocampal Sclerosis of Aging
Hippocampal sclerosis has long been recognized as seen at autopsy in a relatively large proportion of the “oldest-old” (older than 85 years of age).3,7 Hippocampal sclerosis can be seen in the absence of TDP-43 inclusions, presumably in these cases due to either anoxic/hypoxic brain injury or the presence of clinically evident, clinically silent, or undiagnosed seizure activity.7 Hippocampal sclerosis without concomitant TDP-43 pathology is relatively uncommon in the aging population.3 Extensive review of the clinical histories of most patients with TDP-43–negative hippocampal sclerosis of aging pathology typically fails to reveal evidence of a prior anoxic/hypoxic event. Nonetheless, it is possible that reduced oxygen, nutrient delivery, or glucose fluxes secondary to systemic or vascular disease may produce a chronic state of insufficient vascular flow to support neuronal health, leading to the pathologic findings of TDP-43–negative hippocampal sclerosis of aging pathology. Other conditions, such as chronic hypoxia from chronic obstructive pulmonary disease, emphysema, or severe obstructive sleep apnea, could also be potential contributors to the development of hippocampal sclerosis in affected individuals. Epilepsy, while commonly associated with hippocampal sclerosis in the young, has not been convincingly demonstrated to be a potential cause of hippocampal sclerosis among elderly individuals. Survival effects in patients with uncontrolled epilepsy who may die prematurely, never achieving the advanced age characteristic of hippocampal sclerosis of aging, may bias the aged population evaluated for hippocampal sclerosis of aging against consideration of this possibility. A counterargument could be made, however, that over 10% of patients with dementia develop seizures that are often atypical clinically as their disease progresses.31,32 Frequently, such seizures remain undiagnosed, and the potential prevalence of unrecognized recurrent seizure activity or a predisposition to seizures determined by routine EEG in the aging population with dementia may approach 38%.32 Thus, the possibility that unrecognized or undiagnosed late-life epilepsy could be a contributor to hippocampal sclerosis in advanced age remains a possibility.
PATHOLOGY
In hippocampal sclerosis of aging, as in hippocampal sclerosis–FTLD, TDP-43 inclusions, when found as they are in the vast majority of cases, are best classified as Type A, with predominant TDP-43 immunoreactive neurites and neuronal cytoplasmic inclusions in the setting of widespread neuronal loss and gliosis in the hippocampal CA1 subfield, subiculum, and amygdala.3,9 Recent confounds identified in the pathologic diagnosis of hippocampal sclerosis of aging include the potential for unilateral rather than bilateral sclerosis to be present and the recently appreciated issue of “segmental” hippocampal sclerosis of aging.9,30,33 In segmental hippocampal sclerosis of aging, the sclerosis is patchy, so the pathologic diagnosis of hippocampal sclerosis of aging may be missed if multiple sections through the hippocampus are not analyzed.33 This phenomenon has implications not only as a confound in the recognition of hippocampal sclerosis of aging but also for research into why specific hippocampal regions may or may not be affected with the disease process. This finding further raises questions about the early stages of hippocampal sclerosis of aging and whether all hippocampal sclerosis of aging will spread to the entire hippocampus and beyond or whether it can remain isolated to segmental regions of the hippocampus.
It is important to note that the extent of TDP-43 pathology seen commonly, if not almost universally, in hippocampal sclerosis of aging can frequently extend well beyond the medial temporal lobe to include neighboring regions of the frontal and temporal cortices as well as subcortical regions,3,29 demonstrating that hippocampal sclerosis of aging is not confined to the medial temporal lobe structures but instead can be associated with more widespread pathology. The pathologic overlap between hippocampal sclerosis of aging and hippocampal sclerosis–FTLD has led some to conclude that hippocampal sclerosis of aging may be more representative of a forme fruste of FTLD than a unique neurodegenerative disease of aging.
Pathologic studies have also demonstrated an association between TDP-43–positive hippocampal sclerosis of aging and brain arteriolosclerosis, suggesting that a chronic reduction in blood flow to the microvasculature in vulnerable watershed areas such as the hippocampus may play a role in the development of hippocampal sclerosis of aging.13 This is in contrast to a clear association between cerebrovascular risk factors that lead to traditional arteriolosclerosis and other pathologic features of cerebrovascular disease, such as large vessel atherosclerosis, cerebral amyloid angiopathy, lacunar infarcts, and large vessel cardioembolic or atheroembolic stroke.7,13 These data suggest that unique contributions to small arteriolar insufficiency, specifically involving the vasculature of the hippocampus, subiculum, and amygdala, may be part of the pathologic cascade of events leading to hippocampal sclerosis of aging that are not directly related to the conventional mechanisms of cerebrovascular disease.13 This hypothesis has found some support regarding the identified genetic associations with hippocampal sclerosis of aging that are discussed below.
Appreciation of the widespread prevalence of hippocampal sclerosis pathology in late-age sporadic AD (approximately 50%) has added greatly to the complexity of both pathologic and clinical diagnostic criteria regarding mixed-pathology disease states. This further highlights the potential for shared mechanisms of disease progression, wherein the development of TDP-43 and hippocampal sclerosis pathology may represent a later downstream stage confluence of pathologic disease mechanisms that contribute to such shared pathology.
GENETICS
A growing appreciation of the importance of hippocampal sclerosis of aging as a unique or comorbid pathologic condition contributing to cognitive decline and dementia and the recognition of the genetic linkage of hippocampal sclerosis with the GRN, TMEM106B, and C9orf72 familial forms of FTLD have recently led researchers to search for genes that may be related to hippocampal sclerosis of aging.3,9,17,29 Despite extensive genetic investigations, linkages between hippocampal sclerosis of aging and AD-related genetic risk loci have not been found.34,35 These findings suggested to researchers that there may be a unique pathogenetic pathway leading to hippocampal sclerosis, propelling further genetic research in the field of hippocampal sclerosis of aging.
Recent genome-wide association studies identified a linkage between two additional genes, ABCC9 and KCNMB2.26,36 The gene product of ABCC9 encodes potassium channel regulators that can influence vascular responses to anoxia/hypoxia and inflammation, providing a plausible mechanism for its relationship with hippocampal sclerosis of aging.26,34,36 Of note, ABCC9 has also been linked to the development of arteriolosclerosis in advanced age, irrespective of the presence of known cerebrovascular risk factors or the presence of other overt cerebrovascular lesions.13,26 This ties in well with the pathologic findings demonstrating an association of hippocampal sclerosis of aging with arteriolosclerosis as described above.13 KCNMB2, like ABCC9, also encodes for potassium channel regulators that have been specifically linked to normal hippocampal physiologic responses important for memory formation.26,36 In addition, the ABCC9 locus resides on human chromosome 12 near another gene, SLCO1C1, which encodes for a protein that regulates brain levels of thyroid hormone, specifically T4 uptake from blood.37 The association of thyroid hormone dysregulation with hippocampal sclerosis of aging was further supported by the finding of increased thyroid hormone levels in the CSF of subjects with hippocampal sclerosis of aging despite normal serum thyroid hormone and other thyroid function testing, suggesting that hippocampal sclerosis of aging may be related to a brain-specific dysregulation of thyroid function.37 Interestingly, a 2018 study found evidence that mutations in ABCC9 and the paralogous gene ABCC8 may be linked to the progression rate of ALS (another TDP-43 disease).38 Otherwise, ABCC9, SLCO1C1, and KCNMB2 have not been linked to FTLD, suggesting that hippocampal sclerosis of aging is a unique neurodegenerative disease, despite sharing overlapping features with other disease processes such as FTLD, vascular dementia associated with arteriolosclerosis, and AD.
CLINICAL FEATURES
Despite the lack of clinical diagnostic criteria for hippocampal sclerosis of aging, the involvement of medial temporal lobe structures with relative (albeit not absolute) sparing of other cortical and subcortical areas is suggestive of a phenotype that may allow the development of such criteria. Such a phenotype would be expected to specifically impair short-term memory mechanisms, leading to a profound anterograde amnesia in the absence of other associated dementia symptoms. Such a diagnostic feature may distinguish hippocampal sclerosis of aging from other amnestic disorders such as AD in its fulminant state as well as other degenerative or vascular processes contributing to cognitive decline and dementia in the aging population today. A caveat is the confounding clinical phenotype of early mild cognitive impairment (MCI) of the AD type, single domain, in which short-term memory loss and an anterograde amnesia are the only findings.39–41 In such cases, longitudinal observation may eventually distinguish MCI of the AD type and hippocampal sclerosis of aging.
While initial reports of a diagnostic algorithm involving a ratio of delayed recall to verbal fluency performance suggest a statistically significant difference in the ratio of word list delayed recall to verbal fluency in subjects with AD compared to those with hippocampal sclerosis of aging, it remains unclear if this algorithm is useful in determining the presence or absence of the disease in a specific patient during life.9 Despite this caveat, the data are intriguing and suggest that additional work focused on the discriminatory ability of either specific cognitive tests or combinations thereof may move the field of neuropsychology closer to developing clinical criteria useful for individualized diagnosis of hippocampal sclerosis of aging rather than simply descriptive of group differences.
The epidemiology of hippocampal sclerosis of aging may also provide diagnostic clues that could eventually be incorporated into clinical diagnostic criteria. Prior studies have suggested that by 85 years of age, hippocampal sclerosis of aging is seen in approximately 10% of the population, increasing to 30% by the age of 100.3,9 A 2018 study further corroborated this and additionally found that hippocampal sclerosis of aging, when present, could account for 25% of prevalent dementia cases as the third leading cause of degenerative dementia in the aging population after AD and DLB.42 As such, considerations of this diagnosis should be reserved for those older than the age of 85 in whom the pretest probability for hippocampal sclerosis of aging is high enough to make clinical diagnostic criteria useful for diagnosis in specific individuals. Prior studies have not found evidence of a role of race, sex, or educational differences to aid in the development of specific clinical diagnostic criteria.
Several studies have described the clinical features of persons with eventual autopsy-confirmed hippocampal sclerosis of aging. While the number of such cases is low at present, it does appear as though cognitive functions that are reliant on neuroanatomic regions outside the hippocampus are relatively spared. In addition, there is a relative paucity or attenuation of neuropsychiatric and behavioral symptoms that would otherwise be seen in confounding neurodegenerative disease states.3,9,22,29 Social functions and daily activities that do not require short-term memory function may remain preserved for many years, with a very slow progression from subjective and objective short-term memory loss to the state of dementia in which functional impairment is evident in daily activities.9,29
Other clinical findings that are often commonly associated with degenerative disease states, such as focal sensory or motor neurologic deficits, aphasia, parkinsonism, motor neuron disease, and apraxia, have not been described in the setting of “pure” hippocampal sclerosis of aging.9,29 The presence of such findings, however, could be associated with hippocampal sclerosis of aging in the setting of mixed pathologic disease states, including other degenerative disorders such as AD, DLB, FTD, motor neuron disease, and vascular dementia. Thus, as diagnostic criteria for even pure hippocampal sclerosis of aging remain elusive, the likelihood that clinical diagnostic criteria for the presence or absence of hippocampal sclerosis of aging in mixed comorbid disease states could be developed remains uncertain for the foreseeable future.
IMAGING
Given the lack of antemortem clinical diagnostic criteria for hippocampal sclerosis of aging, limited data are available on specific imaging findings to guide clinical antemortem diagnosis. Few autopsy-confirmed studies of antemortem imaging have been performed. The most noteworthy of these, a report by Zarow and colleagues,30 describes the extent of hippocampal atrophy in hippocampal sclerosis of aging as statistically more severe than that seen in AD. Postmortem MRI studies confirm the finding of increased hippocampal atrophy in hippocampal sclerosis of aging over that seen in AD.43 These findings are expected given the neuroanatomic focus of disease-defined and pathologic studies (figure 10-2). It remains unclear whether the relative absence or reduced extent of global or more focal brain atrophy in regions outside the hippocampal formations that differ between persons with hippocampal sclerosis of aging and other pathologic disease states can be used to indicate a lower likelihood of pure hippocampal sclerosis of aging. If so, it should be noted that this approach would not be helpful in defining hippocampal sclerosis of aging in the setting of comorbid AD or other disease states that can contribute to focal or more global cerebral atrophy. Thus, while hippocampal atrophy on MRI may be highly sensitive for the diagnosis of hippocampal sclerosis of aging, it clearly lacks the specificity required for accurate diagnostic determination and usefulness as a biomarker of the underlying pathologic disease state.
Techniques looking at the discrete three-dimensional structure of the hippocampus have been developed that demonstrate specific atrophy within subfields of the hippocampus in AD.44 These techniques have not been tested on or validated in hippocampal sclerosis of aging, yet they show significant promise in detecting patterns of focal atrophy that may differ between AD and hippocampal sclerosis of aging. Such techniques may also be helpful in evaluating for unilateral or segmental hippocampal sclerosis of aging.33 It is clear that further work in the area of MRI neuroimaging is needed before a biomarker for hippocampal sclerosis of aging is proven to meet the thresholds for both sensitivity and specificity required for diagnostic use.
Fludeoxyglucose positron emission tomography (FDG-PET) techniques have been developed to look at hypometabolism in the hippocampus. Such techniques can clearly demonstrate differences in metabolic activities in these areas between controls and patients with AD.45 Unfortunately, such techniques are not regularly used or validated in the area of hippocampal sclerosis of aging, so their diagnostic utility remains unknown. The development of amyloid PET and tau PET imaging enables detection of the (presumed) pathology of AD in the antemortem state. Such methods could be adopted to rule out comorbid diagnoses such as AD that could confound the diagnostic accuracy of measures such as MRI for hippocampal atrophy. Thus, a case with advanced hippocampal atrophy that is amyloid and tau negative by PET scanning would have a high probability of representing a case of pure hippocampal sclerosis of aging. Recently, cases of tau-negative amnestic dementia with significant hippocampal atrophy have been reported, which likely represent hippocampal sclerosis of aging.45,46 Indeed, such cases are widely recognized and have been referred to as suspected non-Alzheimer pathology (SNAP), a disease description based on antemortem biomarker findings that may include degenerative processes other than hippocampal sclerosis of aging, including cerebrovascular disease, argyrophilic grain disease, and primary age-related tauopathy (PART).47,48 The development of molecular imaging techniques to visualize TDP-43 pathology may greatly accelerate work in hippocampal sclerosis of aging. However, the development of such imaging agents remains in its infancy. Further work characterizing the imaging findings of hippocampal sclerosis of aging is needed to define whether such imaging abnormalities may demonstrate specificity.
FLUID BIOMARKERS
Because of the lack of clinical antemortem diagnostic criteria for hippocampal sclerosis of aging, fluid biomarkers have yet to be discovered. However, fluid biomarkers such as amyloid and phosphorylated tau levels in CSF can be used as a proxy for the presence or, more important, the absence of AD pathology to appropriately guide diagnostic thinking regarding a consideration of hippocampal sclerosis of aging as the cause for any clinical symptoms seen.48 A combination of biomarkers could enable the identification of pure hippocampal sclerosis of aging in the presence of short-term memory loss with relative preservation of other cognitive domain function and advanced isolated hippocampal atrophy on MRI. The identification of elevated T3 CSF levels in hippocampal sclerosis of aging that are not seen in controls or patients with AD could also eventually prove useful as an antemortem biomarker for hippocampal sclerosis of aging.37 Confirmatory analysis in a validation cohort, however, is required before this consideration can move forward. Other biomarkers, including inflammatory and angiogenic profiles, are not likely to yield specific results given that alterations in these nonspecific processes are seen frequently in a variety of disease states. The development of CSF and serum assays for TDP-43 is under way, yet none have shown convincing results to date. The use of proteomic and metabolic approaches to identify yet-undiscovered biomarkers of hippocampal sclerosis of aging should be explored.
IMPLICATIONS FOR THERAPEUTIC INTERVENTION
No therapeutic interventions have been approved for this common, yet understudied, disease. Indeed, to the authors’ knowledge, a clinical trial investigating the utility of any available or experimental agents for hippocampal sclerosis of aging has never been conducted. In terms of symptomatic improvement, the potential for acetylcholinesterase inhibitors to help with symptoms of hippocampal sclerosis of aging is uncertain. Likewise, the potential benefits of NMDA partial antagonists, such as memantine, remain unknown, although the possibility exists for both acetylcholinesterase inhibitors and NMDA partial antagonists to be used, as their effects across disparate disease states such as AD, DLB, and vascular dementia suggest generalized improvement in cognitive function irrespective of underlying pathologic cause.
The development of potential experimental therapeutics for hippocampal sclerosis of aging could rely on strategies that attempt to ameliorate risk factor contributions, implicated genetic mechanisms, or the pathologic features of hippocampal sclerosis of aging. Potential strategies could include a focus on arteriolosclerosis, with a myriad of agents developed to treat the risk factors of small vessel ischemic disease and overt cerebrovascular injury as a result of such disease.13 The potential use of antihypertensive, antiplatelet, and anticoagulant therapies could be helpful in reducing the impact of arteriolosclerosis on hippocampal sclerosis of aging disease progression. Prospective clinical trials are currently limited by the inability to objectively operationalize antemortem criteria for inclusion in such studies. Proangiogenic strategies may also prove fruitful given the association of hippocampal sclerosis of aging with haplotype insufficiency (reduced levels of the gene product as a result of the genetic mutation inactivating one copy of the gene) in progranulin levels that may exert their effect through this mechanism. Anti-inflammatory and immune-modulatory strategies could also prove beneficial given the extreme gliosis seen in hippocampal sclerosis of aging. Anti–TDP-43 strategies that may be developed in the future for use in FTLD could likewise be repurposed for use in hippocampal sclerosis of aging.
While this major disease of aging continues to exert a significant impact on the most rapidly growing segment of our population (the oldest-old), research advances moving us forward to potential treatments are moving at an accelerated rate, leveraged by our discoveries over the past several decades in other neurodegenerative diseases of aging. Potential treatments and the development of clinical diagnostic criteria could be seen in as soon as the next few years.
ARGYROPHILIC GRAIN DISEASE
Argyrophilic grain disease was first described as a disease-defining neuropathologic finding in the brains of aged individuals at autopsy.49,50 The neuropathologic features include small (4 μm to 8 μm), spindle-shaped argyrophilic inclusions (“grains”) within the dendritic harbor of neurons that are preferentially evident in medial temporal and limbic regions, including the ambient gyrus, entorhinal cortex, hippocampus, and amygdala, but can also commonly be seen in the lateral hypothalamus and neighboring temporal and extratemporal cortical regions (figure 10-3).49,51 Originally identified using silver staining techniques and then later with tau protein immunohistochemistry, these grains were often difficult to distinguish from the dystrophic neurites that can be widespread in AD.49,50,52,53 They can, however, be distinguished, even with silver staining techniques, by their unique morphology, which is distinct from that typically seen as neuritic pathology in AD.49 Associated neuropathologic changes with argyrophilic grain disease include the presence of “coiled bodies,” which are oligodendroglial tau inclusions; “ballooned” neurons; and “tufted” and/or “thorny” astrocytes.49,50,52,53 All of these pathologic features involve accumulation of phosphorylated tau protein, suggesting its central role in the pathogenesis of argyrophilic grain disease. At the time of their original description, it was understood that they were associated with an increased frequency of clinical dementia, but their presence is not an absolute determinant of clinical dementia.
FIGURE 10-3.
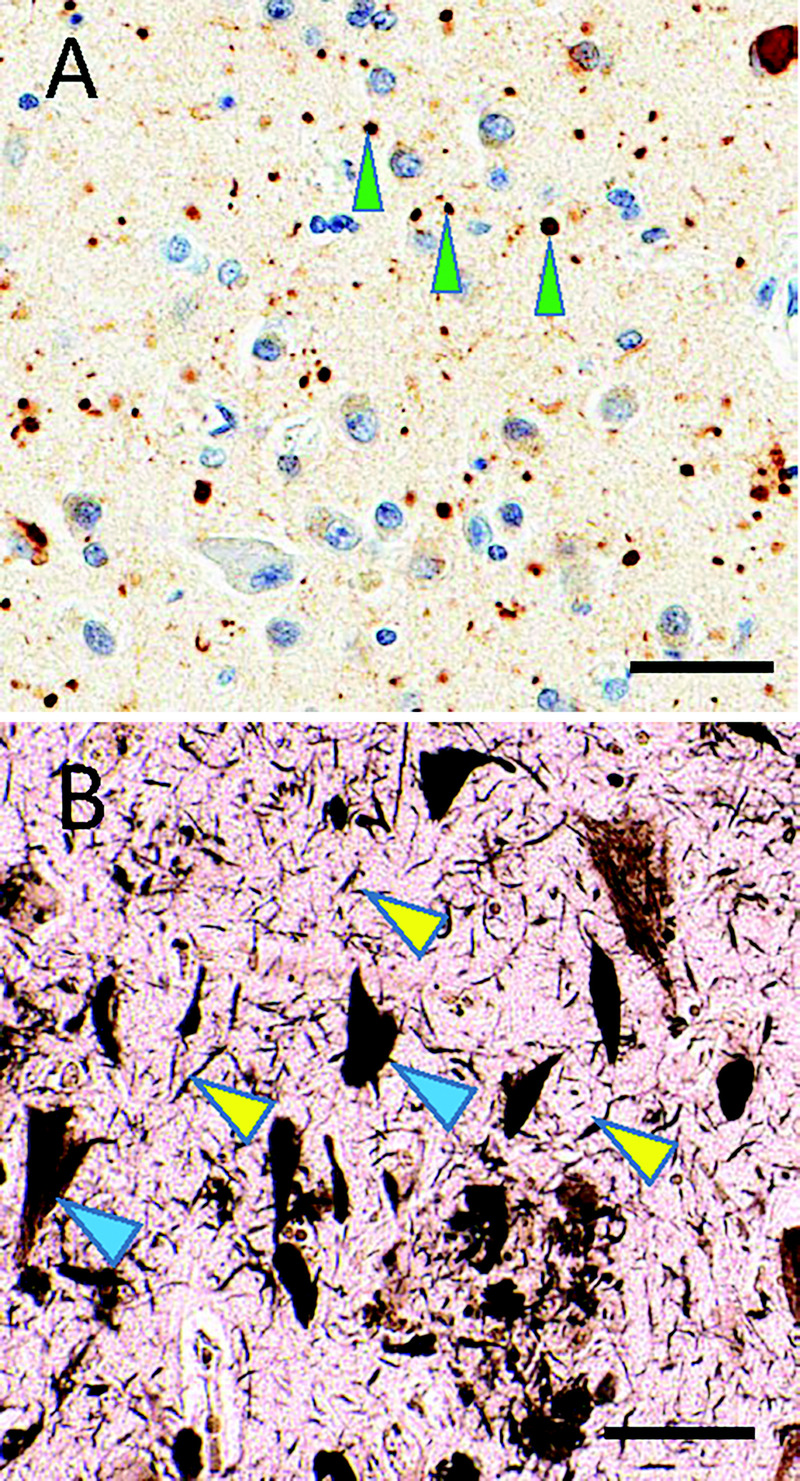
Subtypes of tauopathy. A, Immunostaining shows phosphorylated tau (brown) and counterstaining hematoxylin shows cell nuclei (blue) in argyrophilic grain disease. Note the roundish, approximately 5-micron immunostained structures (green arrowheads), which constitute “grains.” B, In contrast, silver staining highlights Alzheimer disease–type neurofibrillary pathology that includes threadlike structures (yellow arrowheads) and neurofibrillary tangles in neuronal cytoplasm (blue arrowheads). Scale bars: 50 microns in A; 40 microns in B.
Further elucidation of the pathologic features of argyrophilic grain disease revealed that the inclusions were largely composed of 4-repeat tau isoforms that can be visualized using specific immunostains for exon 10 of the tau gene.54–56 Exon 10 of tau encodes the second of four microtubule-binding domains seen in the full-length tau molecule, frequently referred to as “adult” tau, in contrast to 3-repeat isoforms referred to as “fetal” tau. This differs from the dendritic neurites seen in AD, which are composed of both 3-repeat and 4-repeat isoforms of the tau protein. The development of specific monoclonal antibodies recognizing exon 10 greatly enhanced our ability to visualize these specific pathologic features that frequently coexist within a milieu of AD-type pathology.54 Additional biochemical analysis of the tau protein in argyrophilic grain disease has suggested a reduction in or absence of tau acetylation as well as differential phosphorylation patterns that, again, can be used to distinguish these frequently comorbid pathologies.57 In addition, pathologic studies have revealed that argyrophilic grain disease has a predilection for affecting the CA2 sector of the hippocampus, in contrast to AD, hippocampal sclerosis, and other hippocampal insults, which frequently manifest their pathologic features in the CA1 sector of the hippocampus.53 These discoveries suggest that argyrophilic grain disease represents a unique and distinct neuropathologic entity that can be comorbid with AD but is not a manifestation of the primary neuronal degenerative pathologic features of AD.
Argyrophilic Grain Disease of Aging
The initial reports of argyrophilic grain disease pathology from autopsy series demonstrated an increased frequency with advanced age as well as dementia.49 The prevalence of argyrophilic grain disease pathology in cases of dementia with advanced age can be as high as 42%.4 Argyrophilic grain disease has also been reported to affect 30% of centenarians lacking the signs and symptoms of overt dementia.49,58 The clinical features of argyrophilic grain disease have been described as indistinguishable from those of late-onset AD; however, important differences have been noted. Increasing recognition of this entity as a distinct disease state and enhanced immunohistochemical methods to identify this pathologic disease state as a unique contributor to mixed dementia have led to some understanding of the clinical phenotype associated with argyrophilic grain disease.
The relative prevalence of argyrophilic grain disease appears higher in subjects with MCI that in those with overt dementia, suggestive of relatively isolated involvement of medial temporal lobe structures in argyrophilic grain disease as opposed to more widespread cortical progression in cases of late-onset AD.59 In addition, the progression of MCI due to argyrophilic grain disease appeared to be much slower than that seen in MCI related to AD.59 Other than the slower longitudinal course of disease, neuropsychological test findings distinguishing argyrophilic grain disease from MCI related to AD have not been reliably described and validated in the literature. The lack of a diagnostic clinical phenotype and clinical diagnostic criteria for argyrophilic grain disease has limited our ability to advance the field both in antemortem diagnosis and the development of specific treatment modalities that may provide symptomatic relief or slow or halt the progression of argyrophilic grain disease.
Comorbid Argyrophilic Grain Disease in Frontotemporal Lobar Degeneration–Tau
The heterogeneous group of diseases classified as FTLD contains a subgroup of disorders linked to abnormal tau accumulations (now termed FTLD-tau), including the familial forms of disease that are linked to specific mutations within the gene encoding the tau protein on chromosome 17 as well as PSP and corticobasal degeneration (CBD). Many of the mutations within the tau gene lie in intronic regions of the gene and appear to influence the relative ratio of 4-repeat to 3-repeat isoforms as a result of transcriptional mRNA processing. In addition, both PSP and CBD are widely recognized as 4-repeat tauopathies. This has led to some speculation of a linkage between all 4-repeat tauopathies. The presence of many of the associated pathologic features of argyrophilic grain disease, including coiled bodies, ballooned neurons, and tufted and/or thorny astrocytes, that are also recognized as core neuropathologic features seen in PSP and CBD lends further credence to this hypothesis. In contrast, pathologic studies to date have not identified argyrophilic grain disease as an invariant feature of FTLD linked to intronic mutations in the tau gene that specifically overexpress 4-repeat tau isoforms, suggesting that a predilection for 4-repeat isoform overexpression of the tau gene is not, in itself, sufficient to produce the neuropathologic features and degeneration characteristic of argyrophilic grain disease. In addition, it should be noted that not all cases of PSP or CBD show the characteristic pathology of argyrophilic grain disease, although many do (case 10-2).60
CASE 10-2
A 72-year-old man presented with a 2-year history of recurrent falls and gait difficulties. Over the past year, he had noticed his voice becoming more monotone and had several instances of swallowing difficulties but had never aspirated. His wife described him as “stiff” and noted that at times he appeared “robotic.” He had previously been diagnosed with Parkinson disease and was started on carbidopa/levodopa, but it failed to improve his motor difficulties and so was discontinued. He had experienced uncontrollable and, at times, inappropriate laughing and crying spells. Over the previous 6 months, his wife had noticed progressive decline in his short-term memory in addition to his progressive motor issues and labile affect.
On examination, he had a mild pseudobulbar affect and impaired downgaze, with prominent axial rigidity. He was also noted to have frequent repeating behavior during history taking. On mental status examination, his delayed recall was completely absent, and he had some difficulties with orientation to time, but otherwise his bedside cognitive testing appeared intact. His brain MRI showed a classic hummingbird sign representing midbrain atrophy. He was diagnosed with progressive supranuclear palsy (PSP) and conservative symptomatic management was initiated.
At autopsy 3 years later, the patient was found to have globose tangles and tufted astrocytes, particularly involving the subthalamic nucleus, substantia nigra, and globus pallidus, which met criteria for a pathologic diagnosis of PSP. Additional findings included widespread argyrophilic grains in the hippocampus particularly affecting the CA2 subfield.
COMMENT
Argyrophilic grain disease in the setting of PSP was considered responsible for the prominent memory deficits seen in this patient. The memory deficit, originally considered a result of PSP pathology or comorbid Alzheimer disease, highlights the need to develop clinical diagnostic criteria for this pathologic entity.
Over recent years, a number of cases with pathologically confirmed argyrophilic grain disease have been described with significant behavioral and psychiatric symptoms in patients with an earlier onset of symptoms than previously described as attributable to argyrophilic grain disease of aging.4,61–65 The coexistence of argyrophilic grain disease pathology with subtypes of FTLD-tau have also been described. All these cases appear to be associated with 4-repeat tauopathies such as PSP and CBD, with the exception of several cases linked to point mutations in the tau gene that are associated with a phosphorylation state rather than isoform expression.61,66,67 These findings have raised speculation that argyrophilic grain disease may exist on the FTLD spectrum rather than as a unique degenerative disease of aging.
Genetics
Some genetic studies in argyrophilic grain disease have suggested an association with the H1 haplotype of the tau gene, which has also been implicated in the development of PSP, but other studies have demonstrated no such association.68,69 It is conceivable that the heterogeneity in the expression of argyrophilic grain disease may be responsible for the varied results in genetic associations between studies that have examined distinct populations, with those enriched for diseases such as PSP demonstrating associations and those excluding such cases failing to find an association. Other studies have demonstrated the linkage between argyrophilic grain disease and the S305I and S305S mutations on chromosome 17 that are linked to PSP and FTLD, respectively.61,66 These genetic findings again support an overlap or shared pathogenesis between FTLD and argyrophilic grain disease. Argyrophilic grain disease in individuals with advanced age has been found to be associated with the APOE ε2 allele.70,71 This allele is typically recognized as protective against AD, again suggesting that late-onset argyrophilic grain disease and AD are distinct entities rather than shared expressions of a single pathogenetic mechanism.
Clinical Features
When argyrophilic grain disease is found in a relatively restricted pathologic distribution involving limbic regions, the clinical phenotype is, again, often a slowly progressive pure amnestic syndrome that can also be seen in association with emotional and personality changes reflecting the limbic predominance of such pathology.4,59,63–65,72,73 Such distributions of argyrophilic grain disease pathology have also been seen in cases of Parkinson disease and are associated with an earlier onset of symptomatic psychotic features such as hallucinations, delusions, and paranoia. Yet other cases with profound behavioral, personality, and psychotic features that appear to exhibit the clinical phenotype of FTD have been shown to have more widespread cortical argyrophilic grain disease pathology in addition to the limbic predominance of this disease.64,65 These findings suggest that the clinical features of argyrophilic grain disease exist on a spectrum, with the clinical presentation mirroring the underlying anatomic involvement of argyrophilic grain disease–specific pathology.
Imaging and Biomarker Studies
Given the lack of antemortem diagnostic criteria for argyrophilic grain disease, the development of fluid and imaging biomarkers of the disease remains in its infancy. It has been suggested that medial temporal lobe atrophy, such as that seen in AD and hippocampal sclerosis of aging, may also be the most prevalent finding in isolated, “pure” argyrophilic grain disease.4 Other imaging findings in argyrophilic grain disease have not been reported, but the development of tau PET ligands may hold promise for detecting argyrophilic grain disease as it is a pure tauopathy, to the best of our knowledge at this time. It remains unclear, however, whether the degree of abnormal tau accumulation within the microscopic grains will be sufficient to allow detection with such methodology. Fluid and structural imaging biomarkers that can distinguish argyrophilic grain disease from other forms of degenerative disease, such as AD and hippocampal sclerosis of aging, have not been developed. Early attempts at identifying argyrophilic grain disease–specific biomarkers are in development based on biochemical analyses that show not only the preponderance of 4-repeat tau but also a lack of tau acetylation in argyrophilic grain disease.54,56,57,74 At present, argyrophilic grain disease remains a diagnosis that can only be made and confirmed at autopsy.
Implications for Treatment
As yet, no specific treatments for argyrophilic grain disease have been developed. The absolute reliance on postmortem examination for determination of the presence or absence of argyrophilic grain disease has precluded clinical trial development and confounded retrospective analysis of any potential benefit for symptomatic treatment. The advent of anti-tau therapies that have been developed for AD and FTLD-tau hold much promise for the treatment of argyrophilic grain disease if antemortem clinical or biomarker-based diagnostic criteria can be developed. Until such time, treatment for argyrophilic grain disease should focus on targeting the symptomatic expression of disease empirically rather than basing treatment paradigms on rational strategies that may be specific for the disease state.
PRIMARY AGE-RELATED TAUOPATHY
PART is a relatively recently recognized pathology that is common in old age.5 In brains with PART pathology, neuritic amyloid plaques are not detected, but tau neurofibrillary tangles are observed. The presence of age-related tauopathy in the absence of substantial amyloid-β (Aβ) plaques has long been appreciated,75–77 but we are now far more aware of the pathology’s prevalence and public health impact. For example, hippocampal neurofibrillary tangles are practically universal in the 20% to 30% of centenarians whose brains lack detectable Aβ plaques.78,79 Interindividual variation is seen in the severity of PART-type pathology,5 but the mechanism(s) responsible for this variation are mostly unknown. By definition, the neurofibrillary tangles of PART are not associated with underlying FTLD or chronic traumatic encephalopathy, differentiating this entity from other degenerative disease states characterized by predominant tau-related pathology.5 In addition, unlike many cases of tau-related FTLD, Pick disease, and argyrophilic grain disease, which are characterized by tau inclusions that may only include either 3-repeat or 4-repeat isoforms of tau, the tangles seen in PART are composed of both isoforms, with comparable phosphorylation patterns and ultrastructural properties (paired helical filaments) that are virtually identical to those seen in AD.5
Although it has become a recognized disease entity, PART has generated some controversy among experts in the field, and divergent opinions have been expressed.80–82 Despite the near-identical molecular and structural nature and anatomic distribution of neurofibrillary tangles between PART and early AD, the neuroanatomic extent of neurofibrillary tangle pathology in PART appears largely restricted to the temporal lobes, unlike in AD, in which the distribution of neurofibrillary tangles can be more widespread cortically (figure 10-4). These areas of overlap and yet restricted distribution have led some to argue that PART is a unique pathologic disease entity,5,82,83 while others have convincingly argued that PART may simply be part of the spectrum of pathology inherent in many cases of AD.76,81 Much remains unknown about the disease, including how it can best be operationalized for research84,85 and how we should define the “border zones” between AD and PART in theory and in diagnostic practice.
FIGURE 10-4.
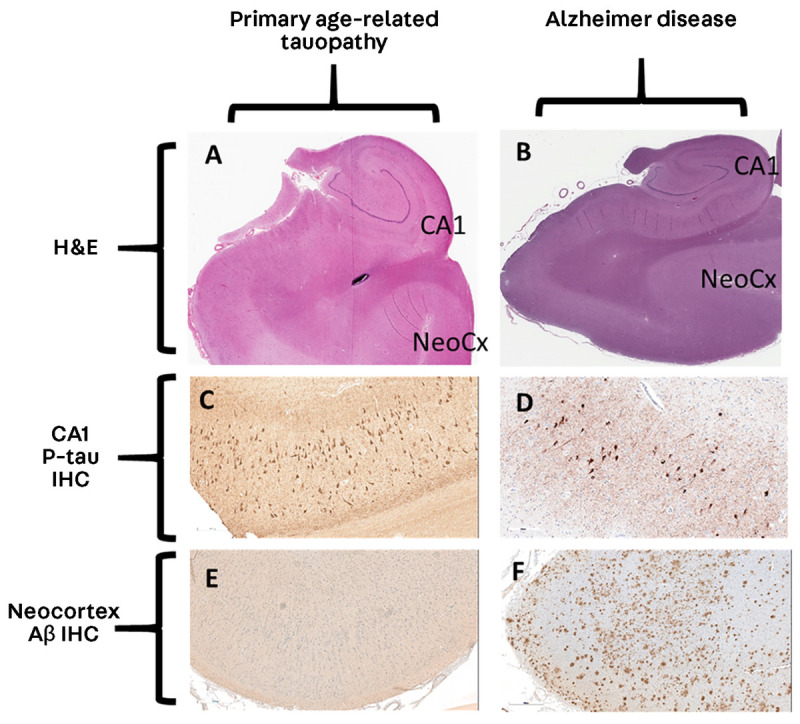
Comparison of primary age-related tauopathy and Alzheimer disease. A, B, Analogous sections from hippocampal-level regions stained with hematoxylin and eosin (H&E) at 1× magnification. C, D, Immunohistochemistry for phosphorylated tau (p-tau) at 20× magnification. E, F, Immunohistochemistry for amyloid-β (Aβ) at 10× magnification. Note that the appearances of the stained sections are very similar for p-tau (C, D); by contrast, the neocortical regions lack Aβ in primary age-related tauopathy (E) but show many Aβ plaques in Alzheimer disease (F).
CA1 = cornu ammonis 1; IHC = immunohistochemistry; NeoCx = neocortex.
Clinical Features
In large autopsy series, the distribution of PART pathology has been shown to be associated with antemortem cognitive impairment,5,86 and PART is also implicated in subjective memory symptoms87 and MCI.88 PART may be more common in those of advanced age, similar to hippocampal sclerosis of aging and argyrophilic grain disease, differing in this respect from typical AD, which can have an earlier mean age of onset.5,78,82,83,85 In addition, the severity of Braak stage is highly correlated with the degree of cognitive impairment, which is quite similar to what is seen in AD.5,86
A 2017 study using the National Alzheimer’s Coordinating Center cohort compared asymptomatic and symptomatic persons with PART diagnosed at autopsy, based on the classifications of PART-definite (no amyloid deposition, Consortium to Establish a Registry for Alzheimer’s Disease [CERAD] stage 0) and PART-probable (sparse amyloid, CERAD stage 1).89 The symptomatic expression of cognitive decline was more common in those with even mild cerebral amyloidosis (PART-probable, 80% symptomatic) compared to those with a complete absence of cerebral amyloidosis (PART-definite, 58% symptomatic), suggesting a potential exacerbation of PART symptomatology with even the earliest AD-like changes.89 This study also found that PART-definite subjects with cognitive decline were more likely to have higher Braak stages and evidence for comorbid depression than their asymptomatic counterparts.89 In a 2017 study, the cognitive tests showing decline most closely associated with Braak stage were the Wechsler Adult Intelligence Scale Block Design and Trail Making Test Part B, adjusted for age and education using the Mayo Older Americans Normative Studies scores.86 These results would not be predicted on the basis of the anatomic distribution of neurofibrillary tangles in PART but were presumed due to an influence of PART on general cognitive slowing as these are both timed tests.86
Clearly, a more complete understanding of the differences in clinical phenotype between PART, AD, and other AD mimics such as hippocampal sclerosis of aging and argyrophilic grain disease is needed before such antemortem clinical information may be used to accurately identify PART as a distinct clinical entity.
Imaging and Fluid Biomarkers
While further elucidation of the clinical phenotype of PART is awaited, evidence of the ability to detect PART in living individuals using readily available biomarker approaches is mounting.90,91 As understanding the postmortem pathology of PART requires evidence for tau-related neurodegeneration (molecularly similar to that seen in AD and in the same early neuroanatomic distribution) in the absence of cerebral amyloidosis,5 studies of antemortem biomarker detection of PART have focused on using the absence of positive biomarkers of cerebral amyloidosis (amyloid PET and CSF amyloid levels) to rule out AD. Such evidence in combination with markers of tau pathology (tau PET and CSF tau and phosphorylated tau levels) and markers of medial temporal lobe neuronal injury (MRI volumetrics and FDG-PET) may allow a more specific antemortem multimodal characterization of PART.5,48,82,83,85
The cornerstone for such characterization includes the absence of detectable cerebral amyloid levels by amyloid PET or CSF analyses.5,47,48,85,90–93 Such cases, however, may also comprise a host of other degenerative disease states, including vascular dementia, DLB, FTD, hippocampal sclerosis of aging, and argyrophilic grain disease. Exclusion of subjects with DLB or FTD phenotypes and of subjects with vascular dementia based on clinical and imaging evidence of cerebrovascular disease leads to increased specificity for PART. Focusing on medial temporal lobe involvement with the use of MRI volumetrics and FDG-PET further restricts the pathologic entities to PART, FTD-tau (medial temporal lobe predominant), hippocampal sclerosis of aging, and argyrophilic grain disease. Some preliminary evidence indicates that certain tau PET imaging ligands may bind preferentially to tau inclusions with mixed repeat isoforms, which may help to further identify PART-specific tauopathy once AD is excluded.94 Additionally, biochemical analyses of CSF tau levels can help identify phosphorylated species and mixed tau isoform composition that could reliably predict PART.5,45,47,54–57,67,93 Prospective testing of such multimodal use of antemortem biomarkers remains to be confirmed but holds much promise for the antemortem detection of pathologic diseases such as PART.48,90
Genetics
Given the arguable overlap between PART and AD, links between the known genetic associations in AD have been explored in PART. Despite widespread investigations, no associations between common AD genetic risks and PART have been found, and evidence for any association between APOE status and PART is lacking.5,69,82,89,95 A linkage to other isoforms of APOE, such as the ε2 allele that is postulated to play a role in influencing the development of argyrophilic grain disease, has not been found for PART.69–71 This lack of genetic overlap reinforces the current thinking that PART and AD are distinct pathologically described degenerative processes.5,80,82,83,86,93
Given the discovery of mutations in the MAPT gene on chromosome 17 linked to FTLD-tau and the linkages of other tauopathies such as PSP and argyrophilic grain disease to the expression of specific MAPT haplotypes, it is only natural that genetic associations with PART have been explored.61,66–69,74 To date, no specific tau mutations have been linked to PART, although at least one study found a strong association indicating the presence of the MAPT H1 haplotype as a risk factor for PART.95 This association is not unique, however; the haplotype is also a risk factor for the development of both PSP and argyrophilic grain disease (4-repeat tauopathies), suggesting that the H1 haplotype increases the likelihood of developing late-life tau-related neurodegeneration but is not specific regarding genetic influences on any specific tau-related pathway or mechanism.68,69,74,95 Further work confirming this finding and exploring the mechanistic pathways whereby the MAPT haplotype influences the development of distinct tauopathic disease states is needed.
Implications for Treatment
The unique involvement of tau-related pathology in PART makes it an ideal candidate for therapeutic intervention with anti-tau strategies that have been developed for AD, FTLD-tau, and other tau-related disorders such as PSP. The attractiveness of a single molecular target, however, is offset by the current lack of a reliable clinical and biomarker-based rubric for the identification of PART in the antemortem setting. It is hoped that as more data are collected and more validated methods for accurately identifying PART are developed in the antemortem setting, the field will be poised to apply the myriad of anti-tau therapies under development to the treatment of PART.
CONCLUSION
This article focused on three common AD mimics (hippocampal sclerosis of aging, argyrophilic grain disease, and PART) that are frequently found at autopsy in individuals who had been given the clinical diagnosis of AD or other degenerative disorders. Recognition of these entities may help explain the lack of specificity for the diagnosis of AD when based solely on clinical phenotype or anatomic involvement of medial temporal lobe structures. These three diseases, hippocampal sclerosis of aging, argyrophilic grain disease, and PART, all have a predilection for medial temporal lobe and hippocampal involvement, present in a common phenotype as an early amnestic syndrome, and can be associated with medial temporal lobe atrophy. These clinical characteristics make them indistinguishable from AD using routine clinical diagnostic tests and MRI. All three of these entities can be found in “pure” forms that lack associated amyloid deposition, allowing the use of CSF amyloid and amyloid PET measures to at least suggest an underlying disease state distinct from AD.
It is intriguing that both hippocampal sclerosis of aging and argyrophilic grain disease (but not PART) have strong associations with the genetic, biochemical, neuroanatomic, and clinical presentations of FTLD syndromes. These data suggest that FTLD-associated features are common in the aging population. Differences in clinical phenotype between early-onset and late-onset diseases associated with hippocampal sclerosis and argyrophilic grain disease deserve further investigation. Clearly, behavioral and language features predominate in early-onset disease, whereas a stronger amnestic component is seen in late-onset cases. Rethinking conceptions about how the clinical and pathologic features interrelate is a worthwhile endeavor. It is hoped that ongoing research in the field will further contribute to the development of clinical diagnostic criteria for hippocampal sclerosis of aging and argyrophilic grain disease.
Significant overlap with the neuropathologic features of AD and other degenerative disease states has proved problematic in the development of antemortem clinical diagnostic and biomarker criteria specific for these disease states. These challenges have, to date, hampered the development of specific therapeutic strategies. Understanding the presence and significant prevalence of these diseases in the aging population with cognitive decline may help to explain why some patients are less responsive to traditional AD medications than others, as many of the mimics of AD described herein are likely to not show cholinergic deficiency or to have pathology affecting the glutaminergic circuitry as we see in AD. Data acquired over the past decade have advanced our understanding of these diseases and may begin to allow the development of clinical trials for both symptomatic therapeutics and disease-modifying agents.
KEY POINTS
Several clinically uncharacterized neuropathologic disease states are found at high frequency in the elderly population and may contribute to the observed inaccuracy of clinical diagnostic criteria in clinicopathologic correlation studies in dementia.
Recent and ongoing work over the past several decades has brought to light the importance and high prevalence of diseases such as hippocampal sclerosis, argyrophilic grain disease, and primary age-related tauopathy as common mimics of Alzheimer disease and other dementing disorders, largely because their own clinical phenotype has been poorly elucidated to date.
The prevalence of diseases such as hippocampal sclerosis, argyrophilic grain disease, and primary age-related tauopathy can be as high as 20% of normal controls and higher than 50% in individuals with clinical dementia.
It is now recognized that hippocampal sclerosis occurs not only in “pure” or isolated forms but is frequently found to be a comorbid process, coexisting with other neurodegenerative pathologies such as Alzheimer disease, dementia with Lewy bodies, progressive supranuclear palsy, and amyotrophic lateral sclerosis.
The presence of hippocampal sclerosis should always be suspected in patients diagnosed with frontotemporal dementia who demonstrate appreciable anterograde amnestic signs and symptoms, irrespective of other associated clinical features or diagnoses.
The extent of TDP-43 pathology seen in hippocampal sclerosis of aging can frequently extend well beyond the medial temporal lobe to include neighboring regions of the frontal and temporal cortices as well as subcortical regions, demonstrating that hippocampal sclerosis of aging is not confined to the medial temporal lobe structures but instead can be associated with more widespread pathology.
The “coiled bodies,” “ballooned” neurons, and “tufted” or “thorny” astrocytes seen in argyrophilic grain disease all involve accumulation of phosphorylated tau protein, suggesting its central role in the pathogenesis of the disease. At the time of their original description, it was understood that they were associated with an increased frequency of clinical dementia, but their presence is not an absolute determinant of clinical dementia.
Biochemical differences stemming from discoveries in postmortem brain tissue have not yet been translated into antemortem serum or CSF biomarkers for argyrophilic grain disease. At present, argyrophilic grain disease remains a diagnosis that can only be made and confirmed at autopsy.
In brains with primary age-related tauopathy pathology, neuritic amyloid plaques are not detected, but tau neurofibrillary tangles are observed.
By definition, the neurofibrillary tangles of primary age-related tauopathy are not associated with underlying frontotemporal lobar degeneration or chronic traumatic encephalopathy, differentiating this entity from other degenerative disease states characterized by predominant tau-related pathology.
Despite the near-identical molecular and structural nature and anatomic distribution of neurofibrillary tangles between primary age-related tauopathy and early Alzheimer disease, the neuroanatomic extent of neurofibrillary tangle pathology in primary age-related tauopathy appears largely restricted to the temporal lobes, unlike in Alzheimer disease, in which the distribution of neurofibrillary tangles can be more widespread cortically.
In large autopsy series, the distribution of primary age-related tauopathy pathology has been shown to be associated with antemortem cognitive impairment, and primary age-related tauopathy is also implicated in subjective memory symptoms and mild cognitive impairment.
Prospective testing of multimodal use of antemortem biomarkers remains to be confirmed but holds much promise for the antemortem detection of pathologic diseases such as primary age-related tauopathy.
Hippocampal sclerosis of aging, argyrophilic grain disease, and primary age-related tauopathy all have a predilection for medial temporal lobe and hippocampal involvement, present in a common phenotype as an early amnestic syndrome, and can be associated with medial temporal lobe atrophy. These clinical characteristics make them indistinguishable from Alzheimer disease using routine clinical diagnostic tests and MRI.
Hippocampal sclerosis of aging and argyrophilic grain disease (but not primary age-related tauopathy) have strong associations with the genetic, biochemical, neuroanatomic, and clinical presentations of frontotemporal lobar degeneration syndromes. These data suggest that frontotemporal lobar degeneration–associated features are common in the aging population.
Data acquired over the past decade have advanced our understanding of hippocampal sclerosis of aging, argyrophilic grain disease, and primary age-related tauopathy and may begin to allow the development of clinical trials for both symptomatic therapeutics and disease-modifying agents.
ACKNOWLEDGMENT
This work was supported by a grant from the National Institutes of Health (P30 AG028383).
REFERENCES
- 1. Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005-2010. J Neuropathol Exp Neurol 2012; 71(4): 266– 273. doi:10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009; 66(2): 200– 208. doi:10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nelson PT, Smith CD, Abner EL, et al. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol 2013; 126(2): 161– 177. doi:10.1007/s00401-013-1154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rodriguez RD, Suemoto CK, Molina M, et al. Argyrophilic grain disease: demographics, clinical, and neuropathological features from a large autopsy study. J Neuropathol Exp Neurol 2016; 75(7): 628– 635. doi:10.1093/jnen/nlw034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128(6): 755– 766. doi:10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kovacs GG, Milenkovic I, Wöhrer A, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 2013; 126(3): 365– 384. doi:10.1007/s00401-013-1157-y. [DOI] [PubMed] [Google Scholar]
- 7. Dickson DW, Davies P, Bevona C, et al. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol 1994; 88(3): 212– 221. doi:10.1007/s004010050152. [DOI] [PubMed] [Google Scholar]
- 8. Tai XY, Bernhardt B, Thom M, et al. Review: neurodegenerative processes in temporal lobe epilepsy with hippocampal sclerosis: clinical, pathological and neuroimaging evidence. Neuropathol Appl Neurobiol 2018; 44(1): 70– 90. doi:10.1111/nan.12458. [DOI] [PubMed] [Google Scholar]
- 9. Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain 2011; 134(pt 5): 1506– 1518. doi:10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu H, Yoshimoto T, Imajo-Ohmi S, et al. Why are hippocampal CA1 neurons vulnerable but motor cortex neurons resistant to transient ischemia? J Neurochem 2012; 120(4): 574– 585. doi:10.1111/j.1471-4159.2011.07550.x. [DOI] [PubMed] [Google Scholar]
- 11. Hatanpaa KJ, Raisanen JM, Herndon E, et al. Hippocampal sclerosis in dementia, epilepsy, and ischemic injury: differential vulnerability of hippocampal subfields. J Neuropathol Exp Neurol 2014; 73(2): 136– 142. doi:10.1097/OPX.0000000000000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walker MC. Hippocampal sclerosis: causes and prevention. Semin Neurol 2015; 35(3): 193– 200. doi:10.1055/s-0035-1552618. [DOI] [PubMed] [Google Scholar]
- 13. Neltner JH, Abner EL, Baker S, et al. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 2014; 137(pt 1): 255– 267. doi:10.1093/brain/awt318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cole AJ. Hippocampal sclerosis: an inflammatory hypothesis. Neurology 2007; 69(12): 1204– 1205. doi:10.1212/01.wnl.0000279585.36404.d0. [DOI] [PubMed] [Google Scholar]
- 15. Hatanpaa KJ, Blass DM, Pletnikova O, et al. Most cases of dementia with hippocampal sclerosis may represent frontotemporal dementia. Neurology 2004; 63(3): 538– 542. doi:10.1212/01.WNL.0000129543.46734.C0. [DOI] [PubMed] [Google Scholar]
- 16. Blass DM, Hatanpaa KJ, Brandt J, et al. Dementia in hippocampal sclerosis resembles frontotemporal dementia more than Alzheimer disease. Neurology 2004; 63: 492– 497. doi:10.1212/01.WNL.0000133008.89613.82. [DOI] [PubMed] [Google Scholar]
- 17. Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol 2015; 77(6): 942– 952. doi:10.1002/ana.24388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amador-Ortiz C, Lin WL, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 2007; 61(5): 435– 445. doi:10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yokota O, Davidson Y, Bigio EH, et al. Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 2010; 120(1): 55– 66. doi:10.1007/s00401-010-0702-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takeda T, Uchihara T, Arai N, et al. Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment: distinction from Alzheimer disease. Acta Neuropathol 2009; 117(1): 35– 44. doi:10.1007/s00401-008-0447-2. [DOI] [PubMed] [Google Scholar]
- 21. Cykowski MD, Takei H, Van Eldik LJ, et al. Hippocampal sclerosis but not normal aging or Alzheimer disease is associated with TDP-43 pathology in the basal forebrain of aged persons. J Neuropathol Exp Neurol 2016; 75(5): 397– 407. doi:10.1093/jnen/nlw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brenowitz WD, Monsell SE, Schmitt FA, et al. Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both Alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimers Dis 2014; 39(3): 691– 702. doi:10.3233/JAD-131880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Onyike CU, Pletnikova O, Sloane KL, et al. Hippocampal sclerosis dementia: an amnesic variant of frontotemporal degeneration. Dement Neuropsychol 2013; 7(1): 83– 87. doi:10.1590/S1980-57642013DN70100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Josephs KA, Dickson DW. Hippocampal sclerosis in tau-negative frontotemporal lobar degeneration. Neurobiol Aging 2007; 28(11): 1718– 1722. doi:10.1016/j.neurobiolaging.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 25. Katsumata Y, Nelson PT, Ellingson SR, Fardo DW. Gene-based association study of genes linked to hippocampal sclerosis of aging neuropathology: GRN, TMEM106B, ABCC9, and KCNMB2. Neurobiol Aging 2017; 53: 193.e117– 193.e125. doi:10.1016/j.neurobiolaging.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nelson PT, Wang WX, Partch AB, et al. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol 2015; 74(1): 75– 84. doi:10.1097/NEN.0000000000000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pletnikova O, Sloane KL, Renton AE, et al. Hippocampal sclerosis dementia with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 2014; 35(10): 2419.e2417– 2419.e2421. doi:10.1016/j.neurobiolaging.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (Berl) 2003; 81(10): 600– 612. doi:10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- 29. Cykowski MD, Powell SZ, Schulz PE, et al. Hippocampal sclerosis in older patients: practical examples and guidance with a focus on cerebral age-related TDP-43 with sclerosis. Arch Pathol Lab Med 2017; 141(8): 1113– 1126. doi:10.5858/arpa.2016-0469-SA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zarow C, Weiner MW, Ellis WG, Chui HC. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav 2012; 2(4): 435– 442. doi:10.1002/brb3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cook M, Baker N, Lanes S, et al. Incidence of stroke and seizure in Alzheimer’s disease dementia. Age Ageing 2015; 44(4): 695– 699. doi:10.1093/ageing/afv061. [DOI] [PubMed] [Google Scholar]
- 32. Rao SC, Dove G, Cascino GD, Petersen RC. Recurrent seizures in patients with dementia: frequency, seizure types, and treatment outcome. Epilepsy Behav 2009; 14(1): 118– 120. doi:10.1016/j.yebeh.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ighodaro ET, Jicha GA, Schmitt FA, et al. Hippocampal sclerosis of aging can be segmental: two cases and review of the literature. J Neuropathol Exp Neurol 2015; 74(7): 642– 652. doi:10.1097/NEN.0000000000000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelson PT, Estus S, Abner EL, et al. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol 2014; 127(6): 825– 843. doi:10.1007/s00401-014-1282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Troncoso JC, Kawas CH, Chang CK, et al. Lack of association of the apoE4 allele with hippocampal sclerosis dementia. Neurosci Lett 1996; 204(1–2): 138– 140. doi:10.1016/0304-3940(96)12331-4. [DOI] [PubMed] [Google Scholar]
- 36. Nelson PT, Jicha GA, Wang WX, et al. ABCC9/SUR2 in the brain: Implications for hippocampal sclerosis of aging and a potential therapeutic target. Ageing Res Rev 2015; 24(pt B): 111– 125. doi:10.1016/j.arr.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nelson PT, Katsumata Y, Nho K, et al. Genomics and CSF analyses implicate thyroid hormone in hippocampal sclerosis of aging. Acta Neuropathol 2016; 132(6): 841– 858. doi:10.1007/s00401-016-1641-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vidal-Taboada JM, Pugliese M, Salvado M, et al. KATP channel expression and genetic polymorphisms associated with progression and survival in amyotrophic lateral sclerosis. Mol Neurobiol 2018; 55(1): 7962– 7972. doi:10.1007/s12035-018-0970-7. [DOI] [PubMed] [Google Scholar]
- 39. Sepe-Monti M, De Carolis A, Bomboi G, et al. MRI evidence of bilateral hippocampal sclerosis in amnestic mild cognitive impairment. Eur J Neurol 2006; 13(9): 1031– 1032. doi:10.1111/j.1468-1331.2006.01457.x. [DOI] [PubMed] [Google Scholar]
- 40. Abner EL, Kryscio RJ, Schmitt FA, et al. Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 2017; 81(4): 549– 559. doi:10.1002/ana.24903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jicha GA, Parisi JE, Dickson DW, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol 2006; 63(5): 674– 681. doi:10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- 42. Boyle PA, Yu L, Wilson RS, et al. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018; 83(1): 74– 83. doi:10.1002/ana.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dawe RJ, Bennett DA, Schneider JA, Arfanakis K. Neuropathologic correlates of hippocampal atrophy in the elderly: a clinical, pathologic, postmortem MRI study. PLoS One 2011; 6(10): e26286 doi:10.1371/journal.pone.0026286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zarow C, Wang L, Chui HC, et al. MRI shows more severe hippocampal atrophy and shape deformation in hippocampal sclerosis than in Alzheimer’s disease. Int J Alzheimers Dis 2011; 2011: 483972 doi:10.4061/2011/483972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Botha H, Mantyh WG, Murray ME, et al. FDG-PET in tau-negative amnestic dementia resembles that of autopsy-proven hippocampal sclerosis. Brain 2018; 141(4): 1201– 1217. doi:10.1093/brain/awy049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Botha H, Mantyh WG, Graff-Radford J, et al. Tau-negative amnestic dementia masquerading as Alzheimer disease dementia. Neurology 2018; 90(11): e940– e946. doi:10.1212/WNL.0000000000005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Caroli A, Prestia A, Galluzzi S, et al. Mild cognitive impairment with suspected nonamyloid pathology (SNAP): prediction of progression. Neurology 2015; 84(5): 508– 515. doi:10.1212/WNL.0000000000001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jack CR, Jr, Knopman DS, Chetelat G, et al. Suspected non-Alzheimer disease pathophysiology–concept and controversy. Nat Rev Neurol 2016; 12(2): 117– 124. doi:10.1038/nrneurol.2015.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Braak H, Braak E. Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm (Vienna) 1998; 105(8–9): 801–819. doi:10.1007/s007020050096. [DOI] [PubMed] [Google Scholar]
- 50. Tolnay M, Spillantini MG, Goedert M, et al. Argyrophilic grain disease: widespread hyperphosphorylation of tau protein in limbic neurons. Acta Neuropathol 1997; 93(5): 477– 484. doi:10.1007/s004010050642. [DOI] [PubMed] [Google Scholar]
- 51. Tolnay M, Probst A. Argyrophilic grain disease. Handb Clin Neurol 2008; 89: 553– 563. doi:10.1016/S0072-9752(07)01251-1. [DOI] [PubMed] [Google Scholar]
- 52. Jellinger KA. Dementia with grains (argyrophilic grain disease). Brain Pathol 1998; 8(2): 377– 386. doi:10.1111/j.1750-3639.1998.tb00161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tolnay M, Schwietert M, Monsch AU, et al. Argyrophilic grain disease: distribution of grains in patients with and without dementia. Acta Neuropathol 1997; 94(4): 353– 358. doi:10.1007/s004010050718. [DOI] [PubMed] [Google Scholar]
- 54. Fujino Y, Wang DS, Thomas N, et al. Increased frequency of argyrophilic grain disease in Alzheimer disease with 4R tau-specific immunohistochemistry. J Neuropathol Exp Neurol 2005; 64(3): 209– 214. doi:10.1093/jnen/64.3.209. [DOI] [PubMed] [Google Scholar]
- 55. Tolnay M, Sergeant N, Ghestem A, et al. Argyrophilic grain disease and Alzheimer’s disease are distinguished by their different distribution of tau protein isoforms. Acta Neuropathol 2002; 104(4): 425– 434. doi:10.1007/s00401-002-0591-z. [DOI] [PubMed] [Google Scholar]
- 56. Togo T, Sahara N, Yen SH, et al. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol 2002; 61(6): 547– 556. doi:10.1093/jnen/61.6.547. [DOI] [PubMed] [Google Scholar]
- 57. Grinberg LT, Wang X, Wang C, et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol 2013; 125(4): 581– 593. doi:10.1007/s00401-013-1080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ding ZT, Wang Y, Jiang YP, et al. Argyrophilic grain disease: frequency and neuropathology in centenarians. Acta Neuropathol 2006; 111(4): 320– 328. doi:10.1007/s00401-006-0043-2. [DOI] [PubMed] [Google Scholar]
- 59. Jicha GA, Petersen RC, Knopman DS, et al. Argyrophilic grain disease in demented subjects presenting initially with amnestic mild cognitive impairment. J Neuropathol Exp Neurol 2006; 65(6): 602– 609. doi:10.1097/01.jnen.0000225312.11858.57. [DOI] [PubMed] [Google Scholar]
- 60. Yokota O, Miki T, Ikeda C, et al. Neuropathological comorbidity associated with argyrophilic grain disease. Neuropathology 2018; 38(1): 82– 97. doi:10.1111/neup.12429. [DOI] [PubMed] [Google Scholar]
- 61. Rönnbäck A, Nennesmo I, Tuominen H, et al. Neuropathological characterization of two siblings carrying the MAPT S305S mutation demonstrates features resembling argyrophilic grain disease. Acta Neuropathol 2014; 127(2): 297– 298. doi:10.1007/s00401-013-1229-z. [DOI] [PubMed] [Google Scholar]
- 62. Nagao S, Yokota O, Ikeda C, et al. Argyrophilic grain disease as a neurodegenerative substrate in late-onset schizophrenia and delusional disorders. Eur Arch Psychiatry Clin Neurosci 2014; 264(4): 317– 331. doi:10.1007/s00406-013-0472-6. [DOI] [PubMed] [Google Scholar]
- 63. Grau-Rivera O, Gelpi E, Rey MJ, et al. Prominent psychiatric symptoms in patients with Parkinson’s disease and concomitant argyrophilic grain disease. J Neurol 2013; 260(12): 3002– 3009. doi:10.1007/s00415-013-7101-1. [DOI] [PubMed] [Google Scholar]
- 64. Asaoka T, Tsuchiya K, Fujishiro H, et al. Argyrophilic grain disease with delusions and hallucinations: a pathological study. Psychogeriatrics 2010; 10(2): 69– 76. doi:10.1111/j.1479-8301.2010.00318.x. [DOI] [PubMed] [Google Scholar]
- 65. Togo T, Isojima D, Akatsu H, et al. Clinical features of argyrophilic grain disease: a retrospective survey of cases with neuropsychiatric symptoms. Am J Geriatr Psychiatry 2005; 13(12): 1083– 1091. doi:10.1176/appi.ajgp.13.12.1083. [DOI] [PubMed] [Google Scholar]
- 66. Kovacs GG, Pittman A, Revesz T, et al. MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol 2008; 116(1): 103– 118. doi:10.1007/s00401-007-0322-6. [DOI] [PubMed] [Google Scholar]
- 67. Rippon GA, Boeve BF, Parisi JE, et al. Late-onset frontotemporal dementia associated with progressive supranuclear palsy/argyrophilic grain disease/Alzheimer’s disease pathology. Neurocase 2005; 11: 204– 211. doi:10.1080/13554790590944753. [DOI] [PubMed] [Google Scholar]
- 68. Conrad C, Vianna C, Schultz C, et al. Molecular evolution and genetics of the Saitohin gene and tau haplotype in Alzheimer’s disease and argyrophilic grain disease. J Neurochem 2004; 89(1): 179– 188. doi:10.1046/j.1471-4159.2004.02320.x. [DOI] [PubMed] [Google Scholar]
- 69. Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta 2005; 1739(2–3): 240– 250. doi:10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 70. Tolnay M, Probst A, Monsch AU, et al. Apolipoprotein E allele frequencies in argyrophilic grain disease. Acta Neuropathol 1998; 96(3): 225– 227. doi:10.1007/s004010050887. [DOI] [PubMed] [Google Scholar]
- 71. Ghebremedhin E, Schultz C, Botez G, et al. Argyrophilic grain disease is associated with apolipoprotein E epsilon 2 allele. Acta Neuropathol 1998; 96(3): 222– 224. doi:10.1007/s004010050886. [DOI] [PubMed] [Google Scholar]
- 72. Homma T, Mochizuki Y, Takahashi K, Komori T. Medial temporal regional argyrophilic grain as a possible important factor affecting dementia in Parkinson’s disease. Neuropathology 2015; 35(5): 441– 451. doi:10.1111/neup.12208. [DOI] [PubMed] [Google Scholar]
- 73. Ikeda K, Akiyama H, Arai T, et al. Clinical aspects of argyrophilic grain disease. Clin Neuropathol 2000; 19(6): 278– 284. [PubMed] [Google Scholar]
- 74. Miserez AR, Clavaguera F, Monsch AU, et al. Argyrophilic grain disease: molecular genetic difference to other four-repeat tauopathies. Acta Neuropathol 2003; 106(4): 363– 366. doi:10.1007/s00401-003-0742-x. [DOI] [PubMed] [Google Scholar]
- 75. Bouras C, Hof PR, Morrison JH. Neurofibrillary tangle densities in the hippocampal formation in a non-demented population define subgroups of patients with differential early pathologic changes. Neurosci Lett 1993; 153(2): 131– 135. doi:10.1016/0304-3940(93)90305-5. [DOI] [PubMed] [Google Scholar]
- 76. Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 1992; 42(9): 1681– 1688. doi:10.1212/WNL.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 77. Dickson DW, Crystal HA, Mattiace LA, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 1992; 13(1): 179– 189. doi:10.1016/0197-4580(92)90027-U. [DOI] [PubMed] [Google Scholar]
- 78. Neltner JH, Abner EL, Jicha GA, et al. Brain pathologies in extreme old age. Neurobiol Aging 2016; 37: 1– 11. doi:10.1016/j.neurobiolaging.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011; 70(11): 960– 969. doi:10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 80. Braak H, Del Tredici K. Are cases with tau pathology occurring in the absence of Abeta deposits part of the AD-related pathological process? Acta Neuropathol 2014; 128(6): 767– 772. doi:10.1007/s00401-014-1356-1. [DOI] [PubMed] [Google Scholar]
- 81. Duyckaerts C, Braak H, Brion JP, et al. PART is part of Alzheimer disease. Acta Neuropathol 2015; 129(5): 749– 756. doi:10.1007/s00401-015-1390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jellinger KA, Alafuzoff I, Attems J, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 2015; 129(5): 757– 762. doi:10.1007/s00401-015-1407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Giaccone G. The existence of primary age-related tauopathy suggests that not all the cases with early Braak stages of neurofibrillary pathology are Alzheimer’s disease. J Alzheimers Dis 2015; 48(4): 919– 921. doi:10.3233/JAD-150435. [DOI] [PubMed] [Google Scholar]
- 84. Crary JF. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromedicine 2016; 1(6): 53– 57. doi:10.29245/2572.942X/2016/6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nelson PT, Trojanowski JQ, Abner EL, et al. “New old pathologies”: AD, PART, and cerebral age-related TDP-43 with sclerosis (CARTS). J Neuropathol Exp Neurol 2016; 75(6): 482– 498. doi:10.1093/jnen/nlw033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Josephs KA, Murray ME, Tosakulwong N, et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol 2017; 133(5): 705– 715. doi:10.1007/s00401-017-1681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kryscio RJ, Abner EL, Jicha GA, et al. Self-reported memory complaints: a comparison of demented and unimpaired outcomes. J Prev Alzheimers Dis 2016; 3(1): 13– 19. doi:10.14283/jpad.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Abner EL, Kryscio RJ, Schmitt FA, et al. Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 2017; 81(4): 549– 559. doi:10.1002/ana.24903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Besser LM, Crary JF, Mock C, Kukull WA. Comparison of symptomatic and asymptomatic persons with primary age-related tauopathy. Neurology 2017; 89(16): 1707– 1715. doi:10.1212/WNL.0000000000004521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jack CR., Jr PART and SNAP. Acta Neuropathol 2014; 128(6): 773– 776. doi:10.1007/s00401-014-1362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jack CR, Jr, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol 2017; 16(6): 435– 444. doi:10.1016/S1474-4422(17)30077-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wisse LEM, Butala N, Das SR, et al. Suspected non-AD pathology in mild cognitive impairment. Neurobiol Aging 2015; 36(12): 3152– 3162. doi:10.1016/j.neurobiolaging.2015.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dani M, Brooks DJ, Edison P. Suspected non-Alzheimer’s pathology - is it non-Alzheimer’s or non-amyloid? Ageing Res Rev 2017; 36: 20– 31. doi:10.1016/j.arr.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 94. Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun 2016; 4(1): 58 doi:10.1186/s40478-016-0315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Santa-Maria I, Haggiagi A, Liu X, et al. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol 2012; 124(5): 693– 704. doi:10.1007/s00401-012-1017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]