

ABSTRACT
PURPOSE OF REVIEW
This article presents an overview of the clinical syndrome of posterior cortical atrophy (PCA), including its pathologic underpinnings, clinical presentation, investigation findings, diagnostic criteria, and management.
RECENT FINDINGS
PCA is usually an atypical form of Alzheimer disease with relatively young age at onset. New diagnostic criteria allow patients to be diagnosed on a syndromic basis as having a primary visual (pure) form or more complex (plus) form of PCA and, when possible, on a disease-specific basis using biomarkers or underlying pathology. Imaging techniques have demonstrated that some pathologic processes are concordant (atrophy, hypometabolism, tau deposition) with clinical symptoms and some are discordant (widespread amyloid deposition). International efforts are under way to establish the genetic underpinnings of this typically sporadic form of Alzheimer disease. In the absence of specific disease-modifying therapies, a number of practical suggestions can be offered to patients and their families to facilitate reading and activities of daily living, promote independence, and improve quality of life
SUMMARY
While rare, PCA is an important diagnostic entity for neurologists, ophthalmologists, and optometrists to recognize to allow for early accurate diagnosis and appropriate patient management. PCA provides an important opportunity to investigate the causes of selective vulnerability in Alzheimer disease.
INTRODUCTION
Posterior cortical atrophy (PCA) is a neurodegenerative syndrome that primarily affects the parietal and occipital lobes.1 While patients with progressive visual impairment with normal acuity had previously been described, the term posterior cortical atrophy was introduced by Benson and colleagues,2 who described a series of patients with deficits in higher-order visual processing and features consistent with aspects of Gerstmann and Balint syndromes but with relatively preserved episodic memory until later in the disease. Subsequent case series determined that the most common underlying pathology was Alzheimer disease (AD),3–5 leading to alternative nomenclature including the visual variant of AD and biparietal AD.4,6 However, as PCA can be due to alternative pathologies, including corticobasal degeneration,7 Lewy body disease, and (very rarely) prion disease, the overarching term posterior cortical atrophy is now preferred to describe the syndrome, with contemporary criteria allowing for subdivisions into PCA-pure, PCA-plus, and pathologic subtypes depending on the clinical presentation and availability of biomarker evidence of underlying pathology.8
EPIDEMIOLOGY
The changing definitions of PCA over recent years and its relative rarity make estimation of incidence and prevalence difficult. However, a striking feature of this syndrome is that the majority of affected individuals have an unusually early age at disease onset, typically presenting between 50 and 65 years of age, although patients with onset in the ninth decade are described.5,9 In the largest series to date studying PCA, of 302 patients, the mean age of onset was 58.9 years (standard deviation 6.9), with 82.5% fulfilling criteria for early-onset dementia (onset before 65 years of age) (figure 3-1).9 The proportion of patients with AD presenting with PCA varies and is likely to depend on clinical context but has been estimated to be about 5% in a specialist cognitive clinic10 and up to 13% in cases of early-onset AD.11 In addition to patients with a clear PCA presentation, a population-based study showed that 14% of patients diagnosed with AD had cognitive profiles consistent with prominent visuospatial problems,12 suggesting that visual problems are underrecognized in individuals diagnosed with “typical” AD and raising questions as to whether PCA is a distinct entity13 or on a phenotypic continuum.14 While some PCA studies have reported a slight overrepresentation of women9 (which may simply reflect that AD is more prevalent in women), others have reported no sex differences.15–17 Few prospective studies have examined disease duration in PCA; while patients with early-onset AD may have faster disease progression than those with later-onset disease, many patients with PCA have a more protracted course extending well over a decade.
FIGURE 3-1.

Age of disease onset in posterior cortical atrophy. Data from an international study9 of 302 patients shows a peak between 50 and 60 years of age, with diminishing incidence with increasing age.
PATHOLOGIC UNDERPINNINGS
Most patients with PCA have underlying AD,4,5,17,18 although cases of PCA can be associated with Lewy body pathology5,17 (either in isolation or, commonly, in combination with AD) and, very rarely, with subcortical gliosis or prion disease.17,19 On postmortem examination, most cases will naturally have end-stage disease, but even at late stages, differences in the distribution of neurofibrillary tangles compared to patients with typical AD have been noted, with particular involvement of primary visual cortices and visual association areas.3,18 Conversely, most studies have not found major differences in amyloid burden across the cortex compared to other forms of AD.5,17
CLINICAL PRESENTATION
The core features of PCA include visuospatial and perceptual deficits as well as features of Gerstmann syndrome (acalculia, left-right disorientation, finger agnosia, and agraphia), Balint syndrome (ocular motor apraxia, optic ataxia, and simultanagnosia), alexia, and apraxia.1,16 In contrast to typical AD, episodic memory is relatively preserved at onset; compared with frontotemporal dementia, aside from the common feature of anxiety,20 personality and behavior are not usually affected in PCA and insight is preserved.
History
Because of its relative rarity, PCA is often diagnosed late, and by the time patients present to neurologists, symptoms will often have been present for many months or years (case 3-1). Very early symptoms can be rather nebulous: patients often describe nonspecific anxiety and a sense that something is wrong, before more concrete problems (usually centered on vision) become apparent. Patients may have a history of repeated visits to optometrists or ophthalmologists and multiple unsuccessful changes in eyeglasses or surgical procedures in an attempt to correct acuity. Patients can often express themselves very clearly and so are not considered as having a “dementia”21 or are diagnosed as having had a stroke before progressive problems emerge.22 Patients often describe causing minor damage to their cars because of problems judging distances while parking. Problems reading analog clocks and digitized pixelated signs are common early features.
CASE 3-1
A 57-year-old woman presented because of progressive visual difficulties. She had initially presented to an optometrist at age 50, having noted difficulties when driving and problems while filling syringes and taking stitches out in her work as a hospital nurse. The optometrist noted a “difficulty with peripheral vision” and referred her to an ophthalmologist, who determined no ophthalmic basis for her difficulties. She subsequently returned to the optometrist, who prescribed multifocal lenses followed by two additional pairs of eyeglasses. The patient moved to a general practice nursing role but noticed further difficulties with visually demanding tasks, such as writing patient notes, reading prescriptions, and transcribing information to a computer. Her husband was also increasingly aware that the patient did not always see everything around her, particularly on the left side; she became lost during barn dances, and on one occasion they became separated when he turned left into the subway station they were walking to, while she continued straight ahead. Over the course of the next 6 years, the patient had five physician visits, including one visit to a memory clinic, which noted “there does not appear to be any evidence of a dementia.” It was only when her husband and son went through the radiologist report from a brain scan and noted the terms posterior, cortex, atrophy, and parietal that diagnostic progress was made. Her husband reported, “I just searched for those words online and what came up was posterior cortical atrophy.”
Neuropsychological assessment revealed significant early visual, visuoperceptual, and visuospatial dysfunction; despite good visual acuity (Snellen equivalent: 6/9), she failed tests of shape discrimination, dot counting, and fragmented letter perception. She had additional deficits in numeracy, praxis, and auditory-verbal short-term memory. These deficits were observed in the context of relatively well-preserved episodic memory on verbal recognition tasks and only occasional errors in the phonologic components of word retrieval. This cognitive profile was consistent with the posterior cortical atrophy (PCA) syndrome, as per the PCA consensus criteria (classification level 1).
Neurologic examination and review of neuropsychiatric symptoms revealed no evidence of symptoms consistent with any additional neurodegenerative syndromes (therefore, consistent with PCA-pure at classification level 2). Standard visual rating of [18F]amyloid imaging yielded results consistent with Alzheimer disease pathology (supportive of PCA-Alzheimer disease at classification level 3).
COMMENT
This case is a typical example of the delayed diagnosis of PCA. The visual symptoms that typify this condition are commonly inaccurately presumed to result from ocular rather than cortical pathology, and, in the absence of “typical” memory problems, a neurodegenerative dementia is often not considered.
Over time, difficulties with reading emerge in the vast majority of patients.15,16 Patients typically get lost while reading text, particularly when moving from line to line or when there is too much competing textual information (so-called crowding)23 or may describe a sense that words are moving or slipping off the page.24 Most patients with PCA will be unable to read within a few years of symptom onset. Occasionally, patients may describe abnormalities with color vision, describing washes of color or prolonged afterimages.25 Patients may describe difficulties in finding items in front of them, particularly in more cluttered environments (simultanagnosia, discussed later in this article) or a sensation that items are moving.26 Patients often become anxious about riding on escalators, particularly when going down; can be cautious when crossing the road because of difficulties in judging the speed of traffic; and can have difficulty with revolving doors or identifying steps or slopes when walking on patterned carpets. Featureless environments, such as white-tiled bathrooms and shiny surfaces, can be particularly disorienting. Patents will often develop impaired facial recognition (prosopagnosia),5,15,16 often while still being able to identify people by voice. Some individuals describe unusual symptoms that can be misconstrued as nonorganic, including finding it easier to read small rather than large text, and difficulties in identifying static objects while being able to track items in motion. An extreme example of this phenomenon is rare patients who can play sports such as tennis or badminton but are unable to locate the ball or shuttlecock when it is on the floor.
Outside of the visual domain, common symptoms are those attributable to impairments of dominant parietal lobe function,5,16 including difficulty with calculation and handling money or spelling. Dyspraxia (acquired deficits in performing complex motor programs) is very common.16,27 Combinations of visual problems and dyspraxia have significant functional consequences, including difficulty in getting dressed (eg, problems buttoning or zipping, tying neckties, or finding the sleeves of coats or putting on clothes backward); cooking; and using cell phones, remote controls, and computers.
Episodic memory and executive impairments are typically not observed in the early stages, although patients may fail certain memory and executive tasks owing to visual, imagery, and other deficits, but emerge as the disease progresses. Language impairment is typically considered a late feature of this disorder, although evidence exists that early anomia is relatively common when specifically looked for.5,28–30 This may reflect that while in their purest forms, the different established symptoms of AD dementia can easily be distinguished, in practice and as expected for a neurodegenerative process that spreads over time, a degree of overlap exists between different syndromic variants; this becomes increasingly apparent as the disease advances.
Aside from dyspraxia, a number of other motor features can be seen in PCA, including limb rigidity, myoclonus, and tremor.31 While these can reflect underlying pathologies other than AD (eg, corticobasal degeneration16 or, when parkinsonism is present, dementia with Lewy bodies), these can also be seen in PCA underpinned by Alzheimer pathology.31
As PCA progresses, symptoms inevitably worsen. The majority of patients will become functionally blind, which has major implications for the level of care that is required and can be the cause of considerable distress to the individual, who will often have good insight into his/her problems. Being unable to read or use other communication devices and becoming increasingly dependent on others often leads individuals to feel disempowered and depressed.32 Patients who already have severe visual problems will become at high risk of falls as other cognitive and motor problems emerge. In its later stages, PCA is often indistinguishable from advanced typical AD.
Atypical Presentations
Considerable debate has existed as to whether the presence of early visual symptoms is essential for a diagnosis of PCA20 or whether presentations with other parietal features (eg, early prominent dyspraxia) with subsequent emergence of more typical visual features should still be classified under the PCA umbrella. This has implications for diagnostic criteria, as patients with early motor features can overlap with other conditions, such as corticobasal syndrome, an issue that is now addressed in the most recent consensus criteria.8 Some patients with a pure PCA phenotype may have corticobasal degeneration or Lewy body pathology rather than AD. However, the presence of features typical for these conditions (eg, alien limb phenomena for corticobasal degeneration or early hallucinations, rapid eye movement [REM] sleep behavior disorder, or parkinsonism for dementia with Lewy bodies) may provide evidence for these alternative pathologies. While the Heidenhain variant of Creutzfeldt-Jakob disease (which presents with early visual impairment) can, in theory, be mistaken for PCA, other clinical features, including the (usual) rapidity of the presentation and the nature of the visual disturbance (which, in prion disease, is often associated with frightening visual distortions), usually mean this is not a major diagnostic challenge. In rare overlap cases, the MRI features and evidence of diffusion changes, in particular in Creutzfeldt-Jakob disease, are particularly valuable.19
BEDSIDE COGNITIVE ASSESSMENT
The bedside cognitive examination can be particularly revealing in PCA. Screening cognitive tests such as the Mini-Mental State Examination (MMSE)33 or Addenbrooke’s Cognitive Examination III (ACE-III)34 will often clearly demonstrate preservation of orientation, repetition, and recall, with severe difficulties in dominant parietal tasks (eg, calculation and spelling) and right parietal function (eg, copying complex figures or clock drawing).
On additional testing, a marked discrepancy is often seen between patients’ ability to read single letters and short words compared to blocks of texts as they will often get lost moving from word to word or line to line (figure 3-235); this may improve if they are allowed to use a finger to track. Simultanagnosia (the inability to interpret the entirety of a visual scene) can often be demonstrated by asking an individual to describe a complex picture; rather than describing it in its entirety, individuals with PCA will often hone in on specific features and fail to see the picture as a whole. This can be demonstrated on a research basis using eye tracking (figure 3-3). A particularly striking and very common feature of PCA is the presence of an apperceptive agnosia. This can be easily demonstrated by showing patients degraded letters (figure 3-436), numbers, or objects, which they typically cannot make sense of, and then showing them the same letters in completed form, which they can recognize. Right parietal dysfunction can also be demonstrated through difficulties in dot counting, line bisection, or clock drawing.
FIGURE 3-2.

Eye tracking demonstrates preserved reading in typical Alzheimer disease (tAD) (A) and impaired reading in posterior cortical atrophy (PCA) (B, C). Blue arrows outline reading order, red arrows indicate omission of subsequent words through reading later sections of text, and yellow arrows indicate reading of earlier sections of text. Transcripts of the participants' corresponding spoken output are beneath each example (italicized text). Each hyphen in the transcript beneath each example indicates a pause of 3 seconds. Numbers refer to where words were repeated.
Reprinted with permission from Yong KX et al, Neurology.35 © 2015 American Academy of Neurology.
FIGURE 3-3.

Visual dysfunction in posterior cortical atrophy (PCA). Eye tracking studies in healthy individuals (A) and patients with PCA (B) demonstrate simultanagnosia in PCA. Circles represent fixation locations and circle size (and corresponding figure in milliseconds) indicates fixation durations. Individuals with PCA fixate on prominent features initially (eg, the dome on the pier) but subsequently fixate on relatively uninformative aspects of the scene (eg, the sea or sky), missing important contextual details (eg, the beachfront or near the end of the pier).
Reprinted from Crutch SJ, et al, Lancet Neurol.1 © 2012 Elsevier Ltd.
FIGURE 3-4.

Fragmented letters. Individuals with posterior cortical atrophy will often be able to read individual letters, but not when they are presented in degraded form (apperceptive agnosia).
Reprinted with permission from Warrington EK.36 © 2010 National Hospital, Institute of Neurology.
Dominant parietal dysfunction can be demonstrated through bedside tests of calculation and spelling, although these should be administered verbally. Similarly, when testing for apraxia, to exclude problems solely related to visual difficulties, patients should be asked, for example, to put their thumb on their ring finger or to salute, rather than asking them to copy the examiner’s hand movements.
Visual disorientation (likely reflecting a combination of simultanagnosia and optic ataxia), when present, is a striking sign. The patient is seated in front of the examiner and asked to concentrate on the examiner’s nose, as would be done with visual field testing. The patient is instructed to reach out and take the examiner’s moving hand in the patient’s peripheral vision. Typically, a patient with PCA will be able to see the hand and will often copy the hand movements but will have difficulties locating the examiner’s hand in space (video 3-1, links.lww.com/CONT/A266). This phenomenon may be much more apparent in one hemifield than the other or may be present in both and is often present in central vision, distinguishing the misreaching in PCA from classic optic ataxia. In more advanced cases, the patient will reach instead toward the examiner’s tie (the tie sign) or face instead of reaching for the target hand.
NEUROLOGIC EXAMINATION
In patients with typical PCA due to AD, aside from demonstrating the higher-order visual signs and dyspraxia described above, the neurologic examination is typically unremarkable, although, as mentioned, a proportion of individuals will have mild motor signs or myoclonus.31 Specific features that should be assessed include the presence of significant parkinsonism, which may reflect underlying Lewy body pathology, and very asymmetric motor features (dystonia, dyspraxia, myoclonus, and alien limb), which might reflect a corticobasal syndrome. Patients will not typically have upper motor neuron signs or features of amyotrophy. Watching the patient walk may be very informative, as mild balance problems are not infrequently encountered by patients with PCA, and the functional consequences of higher-order visual problems may be demonstrated if patients bump into walls or doorframes while walking.
INVESTIGATIONS
While a syndromic diagnosis of PCA is usually apparent on the basis of the history and bedside examination, investigations can provide additional evidence for both the extent and nature of the cognitive and visual deficits and for the underlying causal pathology.
Visual Fields
While formal ocular assessment performed carefully can demonstrate normal acuity and fundi, visual field testing in PCA often reveals hemifield impairments or constriction,37 including unusual and variable field deficits not confirming to classic cortical lesions,38 which can be mistaken as being nonorganic (figure 3-5).
FIGURE 3-5.

Visual fields in posterior cortical atrophy. In patient 2 and patient 10, visual field deficits are principally limited to a hemifield; in patient 3 and patient 4, both hemifields are affected to some extent.
Reprinted from Millington RS, et al, Neuroimage Clin.38 © 2017 The Authors.
Neuropsychology
Detailed neuropsychological assessment provides objective evidence for the cognitive domains that are both affected and preserved in PCA, with implications for diagnosis and potentially for management. A standard neuropsychometric battery can be employed in patients with PCA but with a few caveats. It is important that the testing psychologist is aware of the individual’s difficulties with vision, ensuring that test material is, whenever possible, presented in verbal rather than visual form (eg, nominal skills may be better determined by naming to description than to a picture). Similarly, assessment of premorbid IQ based on reading words from the National Adult Reading Test39 may or may not be reliable depending on the extent of visual impairment.
Patients with PCA typically show a marked discrepancy between performance IQ (low) and verbal IQ and deficits in parietal tasks with preservation of memory and executive tests, provided that imagery and other deficits are taken into account (eg, recognition memory performance may remain preserved when recall appears impaired) (figure 3-640). On this basis, neuropsychological research criteria for PCA have been described that require, for example, evidence for impairments (performance at less than the 5th percentile) on at least two out of four parietal tests (object perception, space perception, calculation, spelling) with evidence of preservation (greater than the 5th percentile) on a recognition memory test, in addition to fulfilling clinical criteria.41
FIGURE 3-6.

Pattern of cognitive impairment in typical Alzheimer disease (A) and posterior cortical atrophy (B). Increasing impairment in a cognitive domain is represented by retreat of the color in its segment toward the center of the circle. Thus, verbal and nonverbal memory are profoundly affected in panel A but relatively preserved in panel B, while profound posterior cortical impairment is seen in panel B.
Modified with permission from Warren JD, et al, Nat Rev Neurol.40 © 2012.
Structural Imaging
As the name of the condition implies, most individuals with PCA will have prominent volume loss reflecting neuronal tissue loss in posterior brain regions (figure 3-7). On a group level, numerous studies have confirmed reductions in parietal and occipital volume and thinning in these regions in patients with PCA compared with both controls and patients with typical AD.41,42 In practice, posterior volume loss is very clear in some cases, but others with very clear symptoms can have little volume loss, made more difficult to discern by natural variability of parietal lobe anatomy. Volumetric T1-weighted acquisitions in the coronal plane supplemented by sagittal views are often best for appreciating the distribution and extent of atrophy, and visual rating scales such as those introduced by Koedam and colleagues43 may be helpful in providing an objective measure of parietal volume loss (figure 3-8). However, in the presence of a typical history for PCA, the absence of marked parietal volume loss should not exclude the diagnosis. Hippocampal volumes are typically relatively preserved in PCA compared to typical AD dementia, in keeping with the relative preservation of episodic memory. It is also important to exclude vascular disease, inflammation, and mass lesions within the posterior cortices, which can mimic neurodegeneration; when Creutzfeldt-Jakob disease is possible, diffusion-weighted imaging (DWI) may be very helpful, typically showing cortical ribboning in occipital lobes in the Heidenhain variant (figure 3-9).19
FIGURE 3-7.

Typical MRI in posterior cortical atrophy. Sagittal (A) and coronal (B) MRIs show marked parietal lobe volume loss.
FIGURE 3-8.

Visual rating scale for the posterior brain regions. In sagittal, axial, and coronal orientation, this rating scale rates atrophy as follows: 0 = no atrophy, 1 = minimal atrophy, 2 = moderate atrophy, and 3 = severe atrophy.
PAR = parietal lobe; PCS = posterior cingulate sulcus; POS = parietooccipital sulcus; PRE = precuneus.
Reprinted with permission from Koedam EL, et al, Eur Radiol.43 © 2011 The Authors.
FIGURE 3-9.

Cortical ribboning in Heidenhain variant of Creutzfeldt-Jakob disease. Axial diffusion-weighted 1.5T MRI shows right occipital lobe cortical ribboning (arrow) in a patient presenting with the Heidenhain phenotype, which can clinically resemble posterior cortical atrophy.
Reprinted with permission from Townley RA, et al, Neurocase.19 © 2018 Information UK Limited.
Functional Imaging
In cases in which the clinical phenotype of PCA is clear and posterior atrophy is seen on MRI, further imaging is not typically required. However, in equivocal cases, fludeoxyglucose positron emission tomography (FDG-PET) may be extremely valuable in demonstrating hypometabolism within the parietooccipital cortices,44,45 which is typically more extensive than the pattern of atrophy and may be seen earlier than structural changes are apparent (figure 3-10). While not in routine clinical use, arterial spin labeling MRI (which provides a measure of cortical blood flow) may provide similar information, albeit reflecting blood flow rather than metabolism.46
FIGURE 3-10.

Multimodal imaging in posterior cortical atrophy (PCA). Single-participant axial images for one control participant and five patients with posterior cortical atrophy showing cerebral blood flow (CBF) (arterial spin labeling [ASL]), glucose metabolism (fludeoxyglucose positron emission tomography [FDG-PET]), atrophy (structural MRI), and amyloid deposition (florbetapir PET). Amyloid deposition varies between individuals but is distributed throughout the cortex in all. Cerebral blood flow, hypometabolism, and atrophy are, by contrast, all restricted to posterior cortical areas.
f = female; m = male; SUVR = standardized uptake value ratio.
Reprinted with permission from Lehmann M, et al, J Neurol Neurosurg Psychiatry.46 © 2016 British Medical Journal.
Molecular Imaging
As patients with PCA, by definition, have atypical disease and almost all present with early-onset dementia, most will fall within appropriate use criteria for amyloid PET imaging.47 Where available, amyloid PET can provide invaluable evidence for underlying AD. However, in contrast to the striking focality both of the symptom and structural imaging changes, amyloid deposition is usually seen across the cortex, with amyloid PET scans from patients with PCA often being indistinguishable from patients with more typical forms of AD.45,48 Thus, while amyloid PET has a role in defining pathology, it is not useful in defining AD syndromes. By contrast, tau PET scanning, which is currently only available in a research setting, often shows very striking posterior cortical deposition of tau pathology in PCA (figure 3-11).49
FIGURE 3-11.
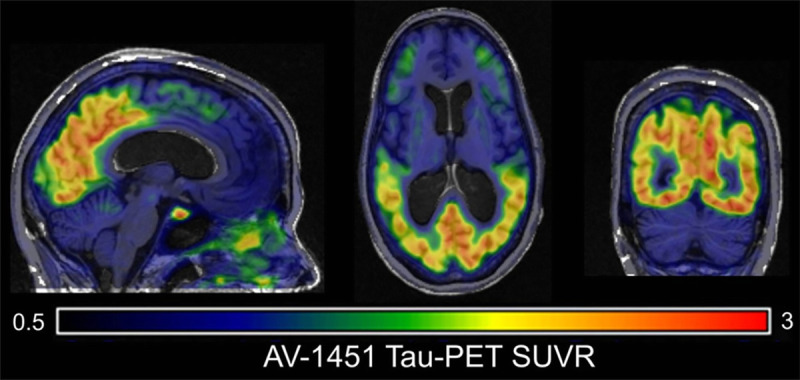
Tau imaging in posterior cortical atrophy. [18F]Flortaucipir positron emission tomography (PET) imaging reveals focal parietooccipital tau pathology.
SUVR = standardized uptake value ratio.
Cerebrospinal Fluid
In keeping with PCA usually being underpinned by AD, CSF amyloid-β (Aβ)1-42 is depressed and concentrations of total tau and phosphorylated tau are increased. However, while levels of Aβ1-42 depression are similar to other forms of AD, some evidence indicates that levels of total tau and phosphorylated tau are not as elevated as in typical AD, perhaps relating to either different intensities or extent of neurodegeneration.50 In practice, therefore, clinicians should be aware that tau to Aβ1-42 ratios may be less elevated in PCA than in typical AD. As with other forms of AD, the total cell count and protein are not elevated; if these are seen, other diagnoses should be considered.
Genetic Testing
PCA due to AD is a sporadic condition, and routine testing for the autosomal dominant forms of the disease is not usually indicated. Testing for APOE genotyping is not recommended as part of the diagnostic workup for any form of AD, and this is particularly the case for PCA, in which evidence indicates that possession of an APOE ε4 allele may be less frequent than in typical cases.9
DIAGNOSTIC CRITERIA
Original diagnostic criteria from Mendez and colleagues15 and Tang-Wai and colleagues5 have been recently updated with the 2017 international consortium criteria (table 3-1).8 These updated criteria describe a three-level classification. Level 1 describes the core clinical, cognitive, and neuroimaging features and exclusions, ie, the clinicoradiologic syndrome. Level 2 distinguishes between a PCA-pure syndrome and, allowing for individuals who also fulfill criteria for another neurodegenerative syndrome, a PCA-plus categorization. Level 3 classifies patients with PCA attributable to a specific underlying disease (eg, PCA due to AD, PCA due to Lewy bodies, PCA due to corticobasal degeneration, or PCA due to prion disease) based on the availability of biomarkers (or histopathologic assessment at autopsy) that can define the underlying pathology. This classification aims also to be complementary to the diagnostic classifications for AD, which increasingly recognize PCA as an atypical form of AD,51,52 providing a framework that can be adapted for different purposes. Thus, for providing appropriate support to individuals, the underlying pathology may be less relevant than the clinical picture, while for research studies aiming to determine the causes of phenotypic heterogeneity, it may be important to classify patients on the basis of both syndrome and pathology.
TABLE 3-1.
Core Features of the Posterior Cortical Atrophy Clinicoradiologic Syndromea,b

MANAGEMENT
While much of the management of PCA is similar to that for typical AD, the dominant visual problems encountered by most patients mean that some specific issues require consideration.
Medical Treatment
Pharmacologic treatments for PCA should be directed to the underlying pathologic substrate. For most patients with PCA due to AD, treatment with acetylcholinesterase inhibitors or memantine, as would be standard treatment for AD, is appropriate. While pivotal clinical trials for the licensing of these drugs focused on typical amnestic AD, individual reports have shown that patients with PCA also benefit.53
Progressive visual impairment and increasing dependence emerging in the face of intact insight often results in significant anxiety, depression, and feelings of guilt.32 While no controlled trials have been conducted, anecdotal evidence suggests that patients may benefit from counseling and other psychological approaches and from standard pharmacologic treatments for mood and anxiety (eg, with selective serotonin reuptake inhibitors [SSRIs]). When myoclonus emerges and becomes problematic, small doses of levetiracetam may be helpful.
Nonpharmacologic Treatments
In the absence of disease-modifying therapies, the mainstay of management of patients with PCA (as with typical AD) is the provision of practical and psychological support to affected patients and their caregivers. Even in the absence of specific therapies, an accurate diagnosis is a vital staging point in an individual patient’s journey, not least as diagnosis is often delayed and symptoms overlooked, misinterpreted, or ignored for many years. Most patients will have stopped, or been stopped from, driving by the time a diagnosis is made, but determining whether patients with PCA are fit to drive is clearly of paramount importance.
The major functional problems in PCA involve everyday skills and self-care.54 Loss of ability to read may be helped with the provision of audio books. Technologic advances such as voice recognition on smartphones, computers, and mobile devices can be invaluable in maintaining an individual’s independence. Specific apps have been developed to help the reading problems in PCA, moving text into the individual’s central vision at an appropriate speed and thus avoiding the need to make saccadic movements from word to word (refer to the Useful Websites section).35 When out of the home, the provision of a white cane is a simple measure that ensures that others are aware of the patient’s potential impairments. Within the house, measures to help with identification of objects and navigation (eg, by putting colored tape on doorframes) or labeling specific items may be helpful. As the disease progresses, home adaptions to minimize stair use and help with bathing and washing may be required. Involving a multidisciplinary team with occupational and physical therapy is often invaluable. Practical tips for patients with PCA and their families are summarized in table 3-2.55 The relative rarity of PCA can lead to isolation. The development of specific support groups for these individuals locally and, increasingly, nationally and internationally can provide a valuable source of support to individuals and their families (for more information, refer to the Useful Websites section).
TABLE 3-2.
Home Safety Tips and Recommendations for Patients With Posterior Cortical Atrophy and Other Dementias With Visual Dysfunctiona

FUTURE DIRECTIONS
PCA has the potential to provide invaluable insights into the factors underpinning the development of AD generally and the factors influencing phenotypic diversity.56 It is unknown, for example, why patients develop these symptoms on a sporadic basis and at a considerably younger age than patients with typical AD. The single biggest genetic study in AD confirmed that possession of an APOE ε4 allele was a risk factor for PCA but conferred less risk than for typical AD. This study highlighted a number of genes that may potentially influence risk, some of which are implicated in neurodevelopment.9 A 2018 study has also provided evidence that patients with PCA are more likely to report nonlanguage mathematical and visuospatial learning disabilities compared to patients with typical AD and controls.57 From a pathologic perspective, emerging evidence shows different strains of Aβ in PCA compared to typical AD, raising the suggestion that different forms of Aβ may propagate through different brain networks and associate with different forms of the disease.58 These findings, while in their infancy, may in time provide a more detailed understanding of the mechanisms underlying the development of both PCA and typical AD and lead to the development of new and more specific treatments of these conditions.
CONCLUSION
PCA is a rare but important neurodegeneration syndrome typically underpinned by AD. Patients with this condition are often diagnosed late or are misdiagnosed as having a primary ocular or psychologically mediated illness. Recognition of PCA as a distinct syndrome and determination of its underlying cause allow for appropriate nonpharmacologic and pharmacologic treatments to be instigated and for appropriate support to be provided for patients and families. PCA provides a valuable paradigm for exploring the causes of phenotypic heterogeneity in AD.
KEY POINTS
A striking feature of posterior cortical atrophy is that the majority of affected individuals have an unusually early age at disease onset, typically presenting between 50 and 65 years of age.
Most patients with posterior cortical atrophy have underlying Alzheimer disease.
The core features of posterior cortical atrophy include visuospatial and perceptual deficits as well as features of Gerstmann syndrome (acalculia, left-right disorientation, finger agnosia, and agraphia), Balint syndrome (ocular motor apraxia, optic ataxia, and simultanagnosia), alexia, and apraxia.
Patients with posterior cortical atrophy may have a history of repeated visits to optometrists and ophthalmologists and multiple unsuccessful changes in eyeglasses or surgical procedures in an attempt to correct acuity.
Over time, difficulties with reading emerge in the vast majority of patients with posterior cortical atrophy.
Patients with posterior cortical atrophy often become anxious about riding on escalators, particularly when going down; can be cautious when crossing the road because of difficulties in judging the speed of traffic; and can have difficulty with revolving doors.
Combinations of visual problems and dyspraxia in patients with posterior cortical atrophy have significant functional consequences, including difficulty in getting dressed; cooking; and using cell phones, remote controls, and computers.
Simultanagnosia (the inability to interpret the entirety of a visual scene) can often be demonstrated by asking an individual to describe a complex picture; rather than describing it in its entirety, individuals with posterior cortical atrophy will often hone in on specific features and fail to see the picture as a whole.
A particularly striking and very common feature of posterior cortical atrophy is the presence of an apperceptive agnosia.
Visual disorientation (likely reflecting combinations of simultanagnosia and optic ataxia), when present, is a striking sign in patients with posterior cortical atrophy.
When performing neuropsychological testing in posterior cortical atrophy, it is important that the testing psychologist is aware of the patient’s difficulties with vision, ensuring that test material is, whenever possible, presented in verbal rather than visual form.
In the presence of a typical history for posterior cortical atrophy, the absence of marked parietooccipital volume loss should not exclude the diagnosis.
Fludeoxyglucose positron emission tomography may be extremely valuable in demonstrating hypometabolism within the parietooccipital cortices.
While amyloid positron emission tomography has a role in confirming the presence or absence of amyloid pathology, it is not useful in distinguishing between Alzheimer disease syndromes.
Tau positron emission tomography, which is currently only available in a research setting, often shows very striking posterior cortical deposition of tau pathology.
Posterior cortical atrophy due to Alzheimer disease is a sporadic condition, and routine testing for the autosomal dominant forms of the disease is not usually indicated.
For most patients with posterior cortical atrophy due to Alzheimer disease, treatment with acetylcholinesterase inhibitors or memantine, as would be standard treatment for Alzheimer disease, is appropriate.
The mainstay of management of patients with posterior cortical atrophy (as with typical Alzheimer disease) is the provision of practical and psychological support to affected patients and their caregivers.
Most patients with posterior cortical atrophy will not be fit to drive. Establishing driving safety is of paramount importance.
ACKNOWLEDGMENT
This work was supported by a grant from the Economic and Social Research Council-National Institute for Health Research (ES/L001810/1).
VIDEO LEGEND
VIDEO 3-1
Visual disorientation in posterior cortical atrophy. A 77-year-old man with posterior cortical atrophy. The patient is asked to fixate on the examiner’s face and to reach out and take the examiner’s moving hand. The patient is able to see the moving hand (which he imitates) but has major difficulties in locating it in space.
© 2019 American Academy of Neurology.
USEFUL WEBSITES
POSTERIOR CORTICAL ATROPHY SUPPORT USA
Posterior Cortical Atrophy Support USA is a website run by caregivers for and supporters of patients with posterior cortical atrophy.
RARE DEMENTIA SUPPORT
Rare Dementia Support is a UK website that provides a rich support of information for patients with posterior cortical atrophy and their caregivers.
READ-CLEAR
Read-Clear is an app designed to help reading in patients with posterior cortical atrophy and related disorders.
readclear.co.uk
REFERENCES
- 1.Crutch SJ, Lehmann M, Schott JM, et al. Posterior cortical atrophy. Lancet Neurol 2012;11(2):170–178. doi:10.1016/S1474-4422(11)70289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson DF, Davis RJ, Snyder BD. Posterior cortical atrophy. Arch Neurol 1988;45(7):789–793. doi:10.1001/archneur.1988.00520310107024. [DOI] [PubMed] [Google Scholar]
- 3.Hof PR, Vogt BA, Bouras C, Morrison JH. Atypical form of Alzheimer’s disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Res 1997;37(24):3609–3625. doi:10.1016/S0042-6989(96)00240-4. [DOI] [PubMed] [Google Scholar]
- 4.Galton CJ, Patterson K, Xuereb JH, Hodges JR. Atypical and typical presentations of Alzheimer’s disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain 2000;123(pt 3):484–498. doi:10.1093/brain/123.3.484. [DOI] [PubMed] [Google Scholar]
- 5.Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004;63(7):1168–1174. doi:10.1212/01.WNL.0000140289.18472.15. [DOI] [PubMed] [Google Scholar]
- 6.Ross SJ, Graham N, Stuart-Green L, et al. Progressive biparietal atrophy: an atypical presentation of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 1996;61(4):388–395. doi:10.1136/jnnp.61.4.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang-Wai DF, Josephs KA, Boeve BF, et al. Pathologically confirmed corticobasal degeneration presenting with visuospatial dysfunction. Neurology 2003;61(8):1134–1135. doi:10.1212/01.WNL.0000086814.35352.B3. [DOI] [PubMed] [Google Scholar]
- 8.Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement 2017;13(8):870–884. doi:10.1016/j.jalz.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schott JM, Crutch SJ, Carrasquillo MM, et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimers Dement 2016;12(8):862–871. doi:10.1016/j.jalz.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snowden JS, Stopford CL, Julien CL, et al. Cognitive phenotypes in Alzheimer’s disease and genetic risk. Cortex 2007;43(7):835–845. doi:10.1016/S0010-9452(08)70683-X. [DOI] [PubMed] [Google Scholar]
- 11.Koedam EL, Lauffer V, van der Vlies AE, et al. Early-versus late-onset Alzheimer’s disease: more than age alone. J Alzheimers Dis 2010;19(4):1401–1408. doi:10.3233/JAD-2010-1337. [DOI] [PubMed] [Google Scholar]
- 12.Crane PK, Trittschuh E, Mukherjee S, et al. Incidence of cognitively defined late-onset Alzheimer’s dementia subgroups from a prospective cohort study. Alzheimers Dement 2017;13(12):1307–1316. doi:10.1016/j.jalz.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang-Wai D, Mapstone M. What are we seeing? Is posterior cortical atrophy just Alzheimer disease? Neurology 2006;66(3):300–301. doi:10.1212/01.wnl.0000202093.81603.d8. [DOI] [PubMed] [Google Scholar]
- 14.Stopford CL, Snowden JS, Thompson JC, Neary D. Variability in cognitive presentation of Alzheimer’s disease. Cortex 2008;44(2):185–195. doi:10.1016/j.cortex.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Mendez MF, Ghajarania M, Perryman KM. Posterior cortical atrophy: clinical characteristics and differences compared to Alzheimer’s disease. Dement Geriatr Cogn Disord 2002;14(1):33–40. doi:10.1159/000058331. [DOI] [PubMed] [Google Scholar]
- 16.McMonagle P, Deering F, Berliner Y, Kertesz A. The cognitive profile of posterior cortical atrophy. Neurology 2006;66(3):331–338. doi:10.1212/01.wnl.0000196477.78548.db. [DOI] [PubMed] [Google Scholar]
- 17.Renner JA, Burns JM, Hou CE, et al. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology 2004;63(7):1175–1180. doi:10.1212/01.WNL.0000140290.80962.BF. [DOI] [PubMed] [Google Scholar]
- 18.Hof PR, Bouras C, Constantinidis J, Morrison JH. Selective disconnection of specific visual association pathways in cases of Alzheimer’s disease presenting with Balint's syndrome. J Neuropathol Exp Neurol 1990;49(2):168–184. doi:10.1097/00005072-199003000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Townley RA, Dawson ET, Drubach DA. Heterozygous genotype at codon 129 correlates with prolonged disease course in Heidenhain variant sporadic CJD: case report. Neurocase 2018;24(1):54–58. doi:10.1080/13554794.2018.1439067. [DOI] [PubMed] [Google Scholar]
- 20.Suárez-González A, Crutch SJ, Franco-Macías E, Gil-Néciga E. Neuropsychiatric symptoms in posterior cortical atrophy and Alzheimer disease. J Geriatr Psychiatry Neurol 2016;29(2):65–71. doi:10.1177/0891988715606229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Documentary Channel 112. Terry Pratchett—living with dementia. youtube.com/watch?v=H0HIqfMV2cU. Published October 19, 2011. Accessed November 29, 2018.
- 22.Alzheimer’s Research UK. Trina and Graeme—living with a rare form of dementia. youtube.com/watch?v=rAVUApnsk4M. Published October 2, 2017. Accessed November 29, 2018.
- 23.Yong KX, Shakespeare TJ, Cash D, et al. Prominent effects and neural correlates of visual crowding in a neurodegenerative disease population. Brain 2014;137(pt 12):3284–3299. doi:10.1093/brain/awu293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crutch SJ, Lehmann M, Gorgoraptis N, et al. Abnormal visual phenomena in posterior cortical atrophy. Neurocase 2011;17(2):160–177. doi:10.1080/13554794.2010.504729. [DOI] [PubMed] [Google Scholar]
- 25.Chan D, Crutch SJ, Warrington EK. A disorder of colour perception associated with abnormal colour after-images: a defect of the primary visual cortex. J Neurol Neurosurg Psychiatry 2001;71(4):515–517. doi:10.1136/jnnp.71.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alzheimer’s Research UK. Posterior cortical atrophy animation. youtube.com/watch?v=a4eTGmejkRM. Published May 16, 2016. Accessed November 29, 2018.
- 27.Kas A, de Souza LC, Samri D, et al. Neural correlates of cognitive impairment in posterior cortical atrophy. Brain 2011;134(pt 5):1464–1478. doi:10.1093/brain/awr055. [DOI] [PubMed] [Google Scholar]
- 28.Migliaccio R, Agosta F, Rascovsky K, et al. Clinical syndromes associated with posterior atrophy: early age at onset AD spectrum. Neurology 2009;73(19):1571–1578. doi:10.1212/WNL.0b013e3181c0d427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magnin E, Sylvestre G, Lenoir F, et al. Logopenic syndrome in posterior cortical atrophy. J Neurol 2013;260(2):528–533. doi:10.1007/s00415-012-6671-7. [DOI] [PubMed] [Google Scholar]
- 30.Crutch SJ, Lehmann M, Warren JD, Rohrer JD. The language profile of posterior cortical atrophy. J Neurol Neurosurg Psychiatry 2013;84:460–466. doi:10.1136/jnnp-2012-303309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan NS, Shakespeare TJ, Lehmann M, et al. Motor features in posterior cortical atrophy and their imaging correlates. Neurobiol Aging 2014;35(12):2845–2857. doi:10.1016/j.neurobiolaging.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suárez-González A, Henley SM, Walton J, Crutch SJ. Posterior cortical atrophy: an atypical variant of Alzheimer disease. Psychiatr Clin North Am 2015;38(2):211–220. doi:10.1016/j.psc.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189–198. doi:10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh S, Schubert S, Hoon C, et al. Validation of the Addenbrooke’s cognitive examination III in frontotemporal dementia and Alzheimer’s disease. Dement Geriatr Cogn Disord 2013;36(3–4):242–250. doi:10.1159/000351671. [DOI] [PubMed] [Google Scholar]
- 35.Yong KX, Rajdev K, Shakespeare TJ, et al. Facilitating text reading in posterior cortical atrophy. Neurology 2015;85(4):339–348. doi:10.1212/WNL.0000000000001782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warrington EK. The Queen Square screening test for cognitive deficits. London, UK: National Hospital, Institute of Neurology, 2010. [Google Scholar]
- 37.Pelak VS, Smyth SF, Boyer PJ, Filley CM. Computerized visual field defects in posterior cortical atrophy. Neurology 2011;77(24):2119–2122. doi:10.1212/WNL.0b013e31823e9f2a. [DOI] [PubMed] [Google Scholar]
- 38.Millington RS, James-Galton M, Maia Da Silva MN, et al. Lateralized occipital degeneration in posterior cortical atrophy predicts visual field deficits. Neuroimage Clin 2017;14:242–249. doi:10.1016/j.nicl.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson HE, Willison J. The National Adult Reading Test (NART) test manual (part 1). Windsor, UK: NFER-Nelson, 1991. [Google Scholar]
- 40.Warren JD, Fletcher PD, Golden HL. The paradox of syndromic diversity in Alzheimer disease. Nat Rev Neurol 2012;8(8):451–464. doi:10.1038/nrneurol.2012.135. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann M, Crutch SJ, Ridgway GR, et al. Cortical thickness and voxel-based morphometry in posterior cortical atrophy and typical Alzheimer's disease. Neurobiol Aging 2011;32(8):1466–1476. doi:10.1016/j.neurobiolaging.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 42.Whitwell JL, Jack CR, Jr, Kantarci K, et al. Imaging correlates of posterior cortical atrophy. Neurobiol Aging 2007;28(7):1051–1061. doi:10.1016/j.neurobiolaging.2006.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koedam EL, Lehmann M, van der Flier WM, et al. Visual assessment of posterior atrophy development of a MRI rating scale. Eur Radiol 2011;21(12):2618–2625. doi:10.1007/s00330-011-2205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nestor PJ, Caine D, Fryer TD, et al. The topography of metabolic deficits in posterior cortical atrophy (the visual variant of Alzheimer's disease) with FDG-PET. J Neurol Neurosurg Psychiatry 2003;74(11):1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenbloom MH, Alkalay A, Agarwal N, et al. Distinct clinical and metabolic deficits in PCA and AD are not related to amyloid distribution. Neurology 2011;76(21):1789–1796. doi:10.1212/WNL.0b013e31821cccad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehmann M, Melbourne A, Dickson JC, et al. A novel use of arterial spin labelling MRI to demonstrate focal hypoperfusion in individuals with posterior cortical atrophy: a multimodal imaging study. J Neurol Neurosurg Psychiatry 2016;87(9):1032–1034. doi:10.1136/jnnp-2015-312782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. J Nucl Med 2013;54(3):476–490. doi:10.1016/j.jalz.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain 2013;136(pt 3):844–858. doi:10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ossenkoppele R, Schonhaut DR, Baker SL, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol 2015;77(2):338–342. doi:10.1002/ana.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paterson RW, Toombs J, Slattery CF, et al. Dissecting IWG-2 typical and atypical Alzheimer’s disease: insights from cerebrospinal fluid analysis. J Neurol 2015;262(12):2722–2730. [DOI] [PubMed] [Google Scholar]
- 51.Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 2014;13(6):614–629. doi:10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 52.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7(3):263–239. doi:10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim E, Lee Y, Lee J, Han SH. A case with cholinesterase inhibitor responsive asymmetric posterior cortical atrophy. Clin Neurol Neurosurg 2005;108(1):97–101. doi:10.1016/j.clineuro.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 54.Shakespeare TJ, Yong KX, Foxe D, et al. Pronounced impairment of everyday skills and self-care in posterior cortical atrophy. J Alzheimers Dis 2015;43(2):381–384. doi:10.3233/JAD-141071. [DOI] [PubMed] [Google Scholar]
- 55.Lake A, Martinez M, Tang-Wai DF. Visual dysfunction in dementia: home safety tips & recommendations. wiki.library.ucsf.edu/download/attachments/320407539/PCA%20Tip%20Sheet%20for%20Patients.pdf?version=1&modificationDate=1386708011000&api=v2. Revised July 2012. Accessed November 29, 2018.
- 56.Mattsson N, Schott JM, Hardy J, et al. Selective vulnerability in neurodegeneration: insights from clinical variants of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2016;87(9):1000–1004. doi:10.1136/jnnp-2015-311321. [DOI] [PubMed] [Google Scholar]
- 57.Miller ZA, Rosenberg L, Santos-Santos MA, et al. Prevalence of mathematical and visuospatial learning disabilities in patients with posterior cortical atrophy. JAMA Neurol 2018;75(6):728–737. doi:10.1001/jamaneurol.2018.0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasmussen J, Mahler J, Beschorner N, et al. Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc Natl Acad Sci U S A 2017;114(49):13018–13023. doi:10.1073/pnas.1713215114. [DOI] [PMC free article] [PubMed] [Google Scholar]