

Abstract
Apicomplexan parasites cause significant morbidity and mortality in humans and livestock, highlighting the need for a deeper understanding of their molecular biology. While techniques for generating targeted gene disruptions have long been available for apicomplexans, such methods are not readily scalable to the entire genome. We recently used CRISPR/Cas9 to disrupt all nuclear protein-coding genes in Toxoplasma gondii using a pooled format. The method relies on transfecting a guide RNA library into parasites constitutively expressing Cas9. Here, we present the complete workflow of such a screen, including preparation of the guide RNA library, growth and testing of the recipient strain, generation of the mutant population, culture conditions for the screen, preparation of genomic DNA libraries, next-generation sequencing of the guide RNA loci, and analysis to detect fitness-conferring genes. This method can be deployed to study how culture conditions affect the parasite’s genetic requirements, which will enable studies of their metabolic needs, host-specificity, and drug-resistance mechanisms. Additionally, by manipulating the background in which the screen is performed, researchers will be able to investigate genetic interactions, which may uncover redundancy or epistasis in the parasite genome. Using this method, a genome-wide screen and its analysis can be completed in three weeks after approximately one month of preparation, making it a powerful tool to uncover functionally important genes in apicomplexan parasites.
Keywords: Toxoplasma, genome-wide screen, CRISPR, Cas9
INTRODUCTION
Apicomplexan parasites cause some of the world’s most prevalent human and veterinary infectious diseases—such as malaria1, cryptosporidiosis2 and toxoplasmosis3,4—and yet research into these pathogens has been limited by our inability to perform saturated genetic screens using targeted mutagenesis. Chemically induced mutagenesis, insertional mutagenesis and genetic crosses have been used to identify loci associated with, for example, virulence5,6, cell cycle progression7, differentiation8 and drug resistance9,10. While effective, these techniques are often technically challenging. Genetic crosses necessitate completing the life cycles of T. gondii or Plasmodium spp. in cats or mosquitos, respectively. Chemical and insertional mutagenesis require large populations of parasites to achieve saturation, and causal mutations can be difficult to distinguish from those that are irrelevant.
CRISPR (Clustered Regularly Interspersed Short Palindromic Repeats) has redefined genetics in many organisms, and apicomplexans are no exception. When used as a genetic tool, CRISPR features an RNA-guided nuclease known as Cas9 that can be directed by 20 base pairs of complementarity between the target genomic region and an initial portion of the RNA guide, called a “protospacer”11-14. Such guide/protospacer combinations are referred to as “single-guide RNAs” or “sgRNAs.” When an sgRNA/Cas9 complex finds a complementary genomic region, Cas9 induces a double-stranded break in the targeted region of the genome, which can be repaired through non-homologous end joining (NHEJ) or homologous recombination15,16. The mechanism underlying CRISPR and its applications to genetics have been reviewed extensively12,17,18. Mammalian geneticists were the first to take advantage of CRISPR’s minimal requirements to design pooled screens in which viral vectors are used to integrate sgRNAs and Cas9 into the genomes of cells in culture, resulting in a population of mutant cells encoding sgRNA barcodes that denote their individual mutations19,20. The relative abundance of these sgRNA barcodes can be quantified using next-generation sequencing after the population undergoes a period of growth or selective pressure, allowing the identification of mutations that increase or decrease fitness under the tested conditions.
We and others have recently demonstrated that CRISPR can be used to induce targeted mutations in Toxoplasma gondii21,22, Plasmodium falciparum23,24 and Cryptosporidium parvum25. Amongst these apicomplexans, Toxoplasma is the only organism that is NHEJ-competent, allowing the double-stranded breaks induced by Cas9 to be efficiently repaired without a homologous DNA template. Compounded with its high transfection rate and the ease with which it can be cultured, this makes T. gondii the ideal apicomplexan species in which to perform pooled CRISPR screens, and we recently reported results obtained using such a system26. Here we elaborate on the mechanics of performing CRISPR screens in T. gondii, and present a detailed protocol.
Overview of T. gondii CRISPR Screens
Our recently published CRISPR data reveals the degree to which each gene from T. gondii contributes to the fitness of parasites grown in human foreskin fibroblasts (HFFs) for seven days, or three passages26. HFF cells are commonly used for in vitro T. gondii studies, and their care is outlined in Box 1. Briefly, a pool of plasmids encoding sgRNAs was transfected into a variant of the RH strain of T. gondii that constitutively expresses the Cas9 nuclease (RH/Cas9). After three passages, we extracted the genomic DNA from these parasites and amplified the integrated sgRNAs by PCR, as described below (Figure 1a). The final PCR amplicon includes adaptors for indexing and next-generation sequencing (Figure 1b). Using next-generation sequencing, we measured the relative abundances of integrated sgRNAs in the passage 3 sample and the transfected plasmid library to identify sgRNAs that were lost over time, which indicates that the mutations they cause reduce parasite fitness. We also treated a pool of CRISPR-generated mutant parasites with 5-fluorodeoxyuridine (FUDR), which is a uracil analog that is toxic to parasites with an active pyrimidine scavenging pathway. Mutations in the gene uracil phosphoribosyltransferase (UPRT) alleviate such toxicity27. We consequently saw a dramatic enrichment in sgRNAs against UPRT when the drug-treated sample was compared to an untreated control, demonstrating that CRISPR screens can also be coupled with positive selection strategies.
BOX 1: HFF Culturing and Infection with T. gondii. TIMING 1 week.
Human foreskin fibroblasts (HFFs) are contact-inhibited primary cells that have been used for decades as a convenient host cell line in which to propagate the lab-adapted RH strain of T. gondii. HFFs cells can be grown in DMEM containing 10% heat-inactivated FBS. We often supplement this media with 10 μg/ml gentamicin to prevent bacterial contamination. Due to their contact-inhibition, HFF cells can be maintained in a flask for up to a month if the media is replaced weekly. This protocol describes splitting a confluent T12.5 flask of HFF cells into four new T12.5 flasks. Researchers may scale this procedure, proportionally with the growth area, to propagate HFF cells onto coverslips or other culture dishes while maintaining a splitting ratio of 1:4.
Wash a confluent monolayer of HFF cells in a T12.5 flask once with 5 ml of PBS.
Remove the PBS and apply 200 μl of 0.05% trypsin to the cell monolayer.
Incubate the trypsin-treated flask at 37°C for 2–10 min, or until the cells have detached from the bottom of the flask, as assessed using an inverted light microscope. CRITICAL STEP Trypsin causes cellular damage after prolonged exposure times. Keep exposure time to a minimum.
Resuspend the trypsinized cells in 10 ml of D10 and divide the cells between four new T12.5 flasks. Incubate these cells at 37°C with 5% CO2.
Once confluent (4–6 days after seeding), researchers may infect the monolayer with T. gondii or replace the media with fresh D10 weekly to maintain uninfected monolayers for up to 1 month.
In order to propagate T. gondii in HFF Cells, exchange the media on a flask of confluent HFF cells for fresh D10, including selective drugs as appropriate.
Add approximately 106 parasites from a freshly-lysed culture (expected to contain 1.5–4 × 107 parasites in total) to a confluent T12.5 flask of HFFs. If using a different format, scale the number of parasites linearly with the area of the confluent monolayer.
Incubate the infected cells at 37°C with 5% CO2 for two days or until the parasites have lysed the host cell monolayer, then return to step 6 to continue propagating the parasites.
Figure 1. A Schematic of the CRISPR Screening Procedure.

(a) RH/Cas9 parasites that have been shown to express active Cas9 are transfected with the linearized CRISPR library. After one week of growth in HFF cells, transfected parasites may be analyzed to identify fitness-conferring genes or subjected to additional selective pressures prior to analysis. Genomic DNA is extracted from the post-growth and post-selection populations and sgRNAs are amplified from this genomic DNA and from the input library through two sequential PCRs. The Illumina sequencing adaptors P5 and P7, as well as indices that facilitate multiplexing, are added during the second PCR. The abundances of sgRNAs in the transfected library relative to all subsequent samples are quantified using next-generation sequencing. Genes that confer fitness will be lower abundance in post-growth or post-selection populations relative to the transfected CRISPR library. (b) Diagram of the amplification of sgRNAs from the CRISPR library prior to sequencing. The sequence of the amplicon is illustrated with N’s denoting the barcode for multiplexing (gray) and the sgRNA (blue). Regions or homology to the vector (orange and green), and Illumina sequencing adaptors (red) are also highlighted.
Comparison with Previous Genetic Techniques
CRISPR allows us to assess the fates of dozens of independently-derived mutants per gene in the course of a single experiment. This provides an advantage over chemically-induced mutagenesis or genetic crosses, which traditionally compare the phenotypes of a few isolated clonal strains, limiting the reliability of these analyses. The ability to control the precise locations of mutations also assures that screens achieve saturation, limits mutations to coding regions, and allows screens to focus on subsets of genes using small populations of parasites. Furthermore, the results of CRISPR screens can be easily analyzed and interpreted by a researcher with basic programming skills.
However, more traditional mutagenesis strategies still hold several advantages over CRISPR screens. Cas9-induced double-strand breaks repaired through NHEJ usually cause frame-shifts that result in loss-of-function mutations. While this simplifies screen interpretation, the more subtle point mutations induced by chemical mutagenesis can be advantageous when researchers wish to enrich for gain-of-function mutations or partial defects in essential genes28. Genetic crosses take advantage of phenotypes that vary naturally between strains: an advantage that CRISPR lacks6,10. Pooled CRISPR screens will also be difficult to implement in other apicomplexans, such as Plasmodium spp. and Cryptosporidium parvum, as these organisms lack NHEJ. However, CRISPR activation and CRISPR inhibition, two techniques that use catalytically inactive Cas9 to modulate transcription without causing DNA breaks, may overcome this hurdle29,30. Therefore, traditional and emerging genetic techniques will continue to be necessary to unravel different aspects of apicomplexan biology.
Potential Applications and Limitations of the Technology
CRISPR screening allows researchers to ask questions on a scale that was previously unattainable in organisms like T. gondii, and it is easy to envision a range of possible applications. For example, screens performed in the presence of immunological assaults could reveal factors involved in host/pathogen interactions, studies of synthetic lethality could elucidate signaling pathways, and positive selection strategies could identify drug targets. Already, cancer researchers have used CRISPR screens to identify genes associated with susceptibility 31,32, and to begin improving treatments 33,34. Immunologists and microbiologists have dissected the response to bacterial infection 35, identified genes necessary for viral production 36, and mapped signaling pathways in bacteria 37. This technology has opened doors across a multitude of disciplines, and will surely lead to further discoveries in parasitology. In addition to holding great promise, CRISPR screening entails technical hurdles and researchers should understand the technology’s limitations before implementing it.
Pooled CRISPR screens present several challenges that are less pronounced when using CRISPR to mutate individual genes. For instance, constitutive expression of Cas9 appears to be necessary to achieve high mutation rates26, yet overcoming Cas9-associated toxicity has been challenging. We circumvented this problem by expressing a “decoy” sgRNA, which, we hypothesize, limits Cas9 toxicity by outcompeting endogenous RNAs that may misdirect Cas9 (see the Experimental Design section for additional details). Although using a decoy sgRNA has allowed us to express Cas9 in the lab-adapted RH strain of T. gondii, the decoy sgRNA appeared to be less effective in other strains (data not shown). Further research into the decoy’s mechanism of action may allow us to create strain-specific decoys. Developing T. gondii strains that express Cas9 inducibly may also help circumvent toxicity. Achieving high rates of transfection and DNA integration present additional challenges to performing CRISPR screens using manageably-sized input populations. The RH strain performs well in these areas; however, low transfection and plasmid integration rates have precluded genome-wide CRISPR screens in other T. gondii strains (data not shown), although screening smaller libraries may still be feasible.
In some cases, the screening results for individual genes may be compromised by problems with sgRNA design. Faulty gene models may result in the design of sgRNAs with targets outside coding regions, due to misannotated start codons, alternative splicing, or erroneous exon/intron calls. Additionally, similarity between potential target sites can lead to off-target cleavage events, resulting, for instance, in sgRNAs against dispensable genes appearing to reduce fitness38,39. High-fidelity Cas9 variants have recently been identified, and these enzymes could reduce off-target effects40,41.Repetitive genomic regions can also result in single sgRNAs causing multiple double-stranded DNA breaks, which may lead to cell death42. Conversely, some sgRNAs may inefficiently target Cas9, possibly due to the inaccessibility of certain genomic regions, inefficient transcription of the sgRNA, or weak interaction with Cas919. In practice, using multiple sgRNAs per gene and eliminating outliers from screening analysis minimizes concerns related to off-target and low-efficiency cleavage.
In summary, despite some limitations, CRISPR screening provides a powerful complement to traditional approaches for unraveling the biology of T. gondii, which will provide insight into the entire apicomplexan phylum. By contributing this protocol, our plasmids and RH/Cas9 to the research community, it is our hope that other groups will continue to develop these methods and apply CRISPR screening to the wide range of fundamental questions that remain in parasitology.
EXPERIMENTAL DESIGN
Developing a Cas9-Expressing T. gondii Strain
Transiently transfecting T. gondii with Cas9 and an sgRNA yields a mutation rate of approximately 20% in the absence of selection21, which is less than what is needed for pooled genetic screens. To increase this mutation rate, we built a strain that constitutively expresses Cas9 (Figure 2), which required overcoming Cas9 toxicity, as described below. We coupled this strain with the plasmid pU6-DHFR, which encodes an sgRNA of choice and confers resistance to pyrimethamine through a resistant version of the dihydrofolate reducatase thymidylate synthase (DHFR-TS) gene. Transfecting RH/Cas9 parasites with pU6-DHFR encoding an sgRNA against the surface antigen gene SAG1 (pU6-SAG1) yielded a population in which approximately 97% of the parasites lacked SAG1 after selection with pyrimethamine, demonstrating the suitability of this strain for pooled screening (Figure 3)26. We showed previously that T. gondii repairs many CRISPR-induced mutations through integration of large portions of the sgRNA-encoding plasmid21, and this is likely why we are able to achieve mutation rates higher than those resulting from the frame-shift mutations typically introduced by NHEJ.
Figure 2. Assessing Cas9 Expression by Immunofluorescence.

Cas9 is visible as a green nuclear dot in RH/Cas9 parasites stained with mouse anti-FLAG followed by Alexa488-conjugated goat anti-mouse. An antibody against a cytoplasmic protein such as actin can be used as a counterstain, and Hoechst can be used to visualize DNA. The parental strain RH lacks the green nuclear staining that is indicative of Cas9 expression. See the Supplementary Methods for a detailed procedure.
Figure 3. Assessing Cas9 Activity Using an sgRNA Targeting SAG1.
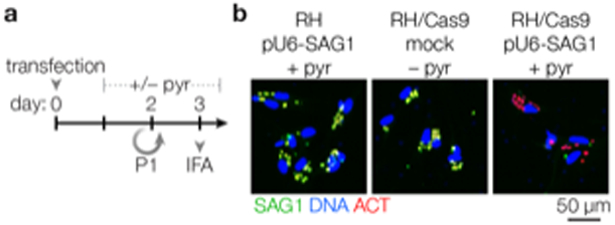
(a) RH/Cas9 and RH are transfected with pU6-SAG1 or mock transfected, then cultured for 3 days. Pyrimethamine is added after 24 hours to select for transfected parasites (or not, in the case of mock transfected parasites). (b) Immunofluorescence assays (IFAs) performed after 3 days reveal loss of SAG1. Active Cas9 is expected to result in loss of SAG1 in 90–97% of parasite vacuoles. See steps 1–25 for a detailed procedure.
Several preliminary attempts to generate a Cas9-expressing strain were unsuccessful, presumably because of toxicity associated with Cas9. We therefore co-expressed Cas9 with the afore-mentioned decoy sgRNA, which targets the 3’ untranslated region of NHE1 and has the sequence AGAATGCAGTTTAGCACCGT. The Cas9-expressing strain was readily obtained by co-transfecting two plasmids encoding Cas9: one also encoding a chloramphenicol acetyltransferase selectable marker (pCas9-CAT) and one encoding the decoy sgRNA (pU6-Decoy). We have since been able to generate similar strains with a single plasmid containing the Cas9, a selectable marker, and the decoy sgRNA. pCas9-CAT and pU6-Decoy are available from Addgene, and RH/Cas9 will soon be available from the ATCC. Investigators wishing to generate their own Cas9-expressing strains may use the plasmids available from Addgene, and should take care to characterize the efficiency of gene disruption in a clonal line of parasites that homogenously expresses Cas9.
Prior to performing a CRISPR screen, we tested the ability of RH/Cas9 to survive electroporation, integrate pU6-DHFR and survive pyrimethamine selection in an effort to estimate the appropriate size for the initial transfected parasite population. Maintaining a population of parasites that is at least 100 times the size of the library is critical to avoid stochastic loss of library diversity. We concluded that 2–5% of the transfected parasites would survive these manipulations and carry through to the eventual knockout population. For each replicate of the screen, we therefore transfected 4 × 108 parasites to achieve at least 100-fold coverage of the CRISPR library in the manipulated population. Researchers may wish to plaque parasites transfected with empty pU6-DHFR in the presence of pyrimethamine in parallel to their screens to ensure that they obtain similar results.
Parasites transfected with the library will sometimes receive more than one sgRNA plasmid, potentially complicating screen outcomes. In an effort to minimize this problem, we used qPCR to analyze the number of sgRNAs integrated into the genomes of Cas9-expressing parasites after transfection with varying amounts of CRISPR library. After analyzing 9 clonal lineages of transfected parasites, we found that transfecting 50 μg of plasmid DNA in a 400 μl reaction resulted in an average of 1.1 sgRNAs integrating per parasite (data not shown). This may be a slight under-estimate, as parasites that receive multiple guides are more likely to succumb to the effects of detrimental mutations. Increasing the DNA concentration did not yield a greater number of resistant parasites, whereas lowering the concentration reduced the yield. We therefore settled on using 50 μg of DNA per transfection to achieve high transfection efficiency while minimizing the number of parasites that receive more than one sgRNA. In cases where parasites do receive more than one plasmid, these random pairings should affect all genes equally, and are unlikely to alter the ranking of genes in the screen.
Pyrimethamine is the current front-line treatment for toxoplasmosis and researchers should take extra precautions to prevent lab-acquired infections with resistant parasites. In particular, the generation of uncharacterized mutations over the course of the CRISPR screen may lead to unpredictable consequences over the course of an infection. Researchers should be especially cautious when using needles to release parasites from infected host cells. We often use blunt needles—or leave the sheaths of sharp needles in place while clipping the ends of the sheaths off—to avoid needle-stick injuries.
Constructing the Genome-Wide sgRNA Library
We designed a library of sgRNAs encoding ten guides against each of the 8,158 protein-coding genes that are annotated in the GT1 genome (ToxoDB release 28), except for DHFR-TS and HXGPRT, which we excluded to prevent interference with drug-resistance markers. We selected sgRNAs based on previously described criteria19. Linearizing the library with AseI to improve integration caused the loss of 17 sgRNAs, as we neglected to select against AseI sites during the library design. We removed these guides from our downstream analysis. Polynucleotide tracks were avoided as part of the previously published design algorithm, preventing polyT tracks from causing premature transcriptional termination19. CRISPR library design is an evolving field, and researchers who choose to create new T. gondii CRISPR libraries may wish to consider the sgRNA selection criteria used in more recent publications39,43.
Our CRISPR library was generated using massively parallel oligonucleotide synthesis by CustomArray Inc. The pool of sgRNAs was synthesized flanked by priming sequences bearing homology to the U6 promoter and the constant region of the sgRNA found in pU6-DHFR. These flanking regions allow us to clone sgRNAs into pU6-DHFR using Gibson Assembly (Figure 4)44. Our oligonucleotide pool also contained outer priming sequences that defined subsets of genes. By amplifying the pool using primers against these outer sequences prior to amplifying it with primers against the inner sequences, we can clone sub-sets of sgRNAs used to perform smaller, more targeted screens. Our library of sgRNAs cloned into pU6-DHFR is available from Addgene (Lourido Toxoplasma CRISPR Library V1). Ensuring comprehensive sgRNA representation in the transfected library is crucial to achieving reliable results. A protocol for cloning an oligonucleotide pool into pU6-DHFR and transforming the library into E. coli without losing diversity is outlined in the Supplementary Methods.
Figure 4. Cloning a CRISPR library into pU6-DHFR.
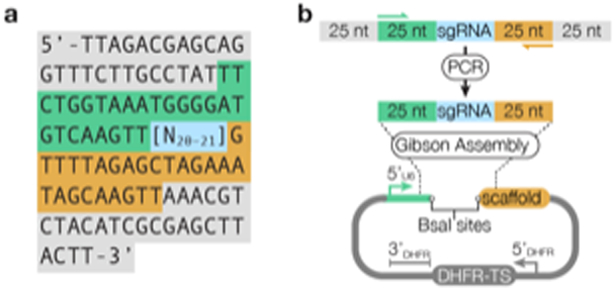
(a) Oligonucleotide arrays contain sgRNAs (highlighted in blue) flanked by regions homologous to pU6-DHFR (green and orange) and sequences for the isolation of sgRNAs targeting subsets of genes (gray). (b) sgRNAs plus regions bearing homology to pU6-SAG1 are amplified using Primer 5 and Primer 6 (Table 1). The resulting DNA fragments are inserted into BsaI-linearized pU6-DHFR using Gibson Assembly. See the Supplementary Methods for a detailed procedure.
Pre-Screen Quality Control
Cas9 expression is occasionally lost in a small portion of the RH/Cas9 population, and during a screen such parasites will quickly out-compete mutants and confound results. It is therefore necessary to confirm by immunofluorescence that 100% of the parasite population is Cas9-positive prior to beginning a screen, as outlined in the Supplementary Methods and illustrated in Figure 2. We also recommend that researchers transfect RH/Cas9 with pU6-SAG1, which they can obtain from Addgene, and ensure that approximately 97% of the post-selection population is SAG1-negative before starting their first CRISPR screen (steps 1–25; Figure 3). Success in this assay confirms that Cas9 is functioning properly and that pyrimethamine is efficiently killing untransfected parasites.
Positive Selection Screens
CRISPR screens can identify genes whose loss is positively selected by a given treatment or genetic precondition. In our recent publication26, we demonstrate this potential by treating the pool of mutant parasites obtained after the third passage of a CRISPR screen with the toxic uracil analog 5-fluoro-deoxyuridine (FUDR) and showing enrichment of sgRNAs targeting UPRT, which is involved in pyrimidine scavenging. After three passages, sgRNAs targeting the most critical genes have been depleted from the population, with 20–30% of the sgRNAs completely absent from the sequencing results. We have seen that continuing to passage the pool of mutant parasites beyond this point results in further purification of mutants for dispensable genes, but does not significantly alter the ranking of genes based on their phenotype scores.26 The third passage is therefore an opportune time to add a selective pressure to the mutant pool and enrich for resistant mutants. If the genes expected to confer resistance cause growth defects, researchers may vary the timing of the selective pressure to avoid losing the mutants from the population prior to selection. However, one must keep in mind that levels of the proteins corresponding to mutated genes will require time to drop, and so adding selection too early may also be detrimental. Recent experience in our lab has shown that even mutants with strong growth defects can be enriched when selection is added after three passages (data not shown). The precise nature of the selection will require optimization to allow for outgrowth of modestly resistant mutants. For example, in the case of FUDR and other small-molecule inhibitors, we have selected the minimum concentration that prevents plaque formation. Because of the scale of the positive selection experiments, it is also possible to try several different conditions in parallel populations, and compare the results of the different selective pressures.
Sequencing and Analysis
CRISPR screening data are distilled to phenotype scores (sometimes also called CRISPR scores) that describe each gene’s contribution to cellular fitness. To arrive at these scores, the relative abundance of each sgRNA is calculated from next-generation sequencing of the sgRNAs present in either the plasmid library or genomic DNA isolated from the various parasite populations. Our previous protocol used two nested PCR steps to isolate the sgRNAs prior to sequencing. In the present method we have streamlined this process, which can now be accomplished in a single reaction, reducing the risk for cross-contamination. By varying a barcode sequence in one of the primers used for the PCR, investigators can multiplex several samples within a single next-generation sequencing reaction to maximize the data obtained from a single sequencing run.
Once the samples are sequenced, we used a combination of custom-built Perl and R scripts to arrive at this metric, but others may wish to use an online analysis portal such as CRISPRAnalyzeR (http://crispr-analyzer.dkfz.de)45. For each sample, we first counted the sequencing reads that matched each sgRNA in the library, taking only perfect matches into account. To avoid taking the logarithm of zero, we assigned pseudocounts to any sgRNAs that were not found. Pseudocounts corresponded to 90% of the lowest non-zero sgRNA count in a particular sample. Alternatively, missing cases could be avoided by adding one count to every sgRNA. Underrepresented sgRNAs—in the bottom 5% of the transfected library by abundance—were considered unreliable and removed from subsequent analyses across all samples. This corresponded to removing sgRNAs with less than 30X coverage in the input library from the final analysis. We divided these adjusted counts by the total number of perfect matches per sample to determine the proportion of the sample comprised by each sgRNA, and we called these “match-normalized counts.” For negative selection screens, match-normalized counts from populations post-growth were divided by counts from the corresponding sgRNAs in the input library, and the log2 of this fold change was calculated. This metric describes the change in relative abundance of each sgRNA after a growth period. For positive selection experiments, we divided the match-normalized counts from the drug-treated population to those from the untreated population before calculating the log2 of the fold change. Limiting our analysis to the five sgRNAs with the highest phenotype scores per gene reduced variation due to spontaneous sgRNA loss. By calculating the mean of the scores associated with these five sgRNAs, we obtained a single phenotype score for each gene.
Because sgRNAs that target fitness-conferring genes are lost during negative selection, the relative abundance of sgRNAs targeting dispensable genes increases despite their neutral effect on parasite fitness. We therefore compared phenotype scores to the median score of 40 genes that were previously shown to be dispensable rather than to a phenotype score of zero, which would typically indicate no change. For each gene, we compared the phenotype scores from four biological replicates to the median phenotype score for the control genes using one-sided t-tests. We also compared the fold changes of sgRNAs targeting each gene to that of the control-targeting sgRNAs using a one-sided Mann-Whitney U test. For each set of tests, we adjusted the resulting p-values using the Benjamini-Hochberg method. We considered genes fitness-conferring if their adjusted p-values were less than or equal to 0.05 in both tests. The scripts described in steps 62-64 and Supplementary Data can be used to calculate phenotype scores and their significance relative to the list of control genes mentioned above.
MATERIALS
Reagents
Eppendorf tubes (Axygen cat. no. MCT-150-C)
50 ml tubes (Corning cat. no. 430828)
15 ml tubes (Corning cat. no. 430052)
Pipettes (Fisher Scientific cat. no. F144802G and F167300G)
Pipette tips with filters (USA Scientific cat. no. 1120–1810, 1120–8810, 1122–1830 and 1120–3810)
Sterile Acrodisc® Syringe Filters with HT Tuffryn® Membrane 0.2 μm, 25 mm (Pall Life Sciences cat. no. 4192).
Petri dishes (Falcon cat. no. 351029)
Culture tubes (Corning cat. no. 352051)
24-well tissue culture dishes (Costar cat. no. 3524)
Parafilm (VWR cat. no. 291–1214)
T12.5 tissue culture flasks (Falcon cat. no. 353018)
15 cm tissue culture dishes (Falcon cat. no. 353025)
Cell scrapers (Costar cat. no. 3010)
PCR tubes (BioRad cat. no. TF102010)
Cryotubes (Thermo Scientific cat. no. 5000–0012)
27½ -gauge needles (BD cat. no. 305109) CAUTION Exercise extreme care when handling live T. gondii with sharps. Blunt needles (SAI Infusion cat. no. B27–50) can be used as an alternative.
pU6-SAG1 (Addgene cat. no. 80322)
Lourido Toxoplasma CRISPR Library V1 (Addgene cat. no. 80636)
pU6-DHFR (Addgene cat. no. 80329)
pCas9-CAT (Addgene cat. no. 80323)
pU6-Decoy (Addgene cat. no. 80324)
Ampicillin (Sigma-Aldrich-Aldrich cat. no. A0166–25G)
Glycerol (EMD cat. no. GX0185–6)
Methanol-Free Formaldehyde (Thermo Scientific cat. no. 28908)
Prolong Gold (Invitrogen cat. no. P1044)
Molecular biology-grade water (e.g., from an EMD Millipore Milii-Q Integral System, Fisher Scientific cat. no. ZRXQ003US)
Gentamicin (Gibco cat. no. 15710–064)
EGTA (Sigma-Aldrich cat. no. E4378–500G)
HEPES (Sigma-Aldrich cat. no. 3375–1KG)
KH2PO4 (Sigma-Aldrich cat. no. P5655)
K2HPO4 (Sigma-Aldrich cat. no. 1551128)
KCl (Fisher Scientific cat. no. SP138–500)
MgCl2 (Mallinckrodt cat. no. 5958)
Methanol for HPLC gradient analysis (Millipore, cat. No. 67–56-1)
EDTA (Amresco cat. no. 0322–500G)
Tris base (Sigma-Aldrich cat. no. TRIS-RO)
Glacial acetic acid (Spectrum cat. no. A1010) CAUTION Glacial acetic acid is corrosive; avoid contact with eyes, skin, and clothing; wear protective gloves while handling.
Na2HPO4 (Sigma-Aldrich cat. no. 255793)
NaCl (Sigma Aldrich cat. no. S7653)
Glutathione (Sigma-Aldrich cat. no. G6013–5G)
Adenosine Triphosphate (ATP, Sigma-Aldrich cat. no. A6419–1G)
CaCl2 (Sigma-Aldrich cat. no. C5080–1KG)
Chloramphenicol (Sigma-Aldrich cat. no. C3175–100MG)
Pyrimethamine (Sigma-Aldrich cat. no. 46706–250MG)
NaOH (Sigma-Aldrich cat. no. S2770) CAUTION Sodium hydroxide is a strong base that can cause severe skin burns and eye injuries; avoid contact with eyes, skin and clothing; use in a fume hood and wear protective eyewear and gloves during handling.
HCl (Millipore cat. no. HX0603–4) CAUTION Hydrochloric acid is a strong acid that can cause severe skin burns and eye injuries; avoid contact with eyes, skin and clothing; use in a fume hood and wear protective eyewear and gloves during handling.
16% Formaldehyde (w/v), Methanol-free (Life Technologies cat. no. 28908) CAUTION Formaldehyde is toxic; use in a fume hood.
Heat-inactivated Fetal Bovine Serum (FBS, Sigma-Aldrich cat. no. F4135–500ML)
Normal Goat Serum (NGS, Sigma-Aldrich cat. no. G6767–500ML)
Triton X-100 (Sigma-Aldrich cat. no. T8787–100ML)
Hoechst (Santa Cruz cat. no. 33258)
Oligonucleotide pool (CustomArray)
Primers (Sigma-Aldrich)
dNTPs (New England Biolabs cat. no. N0447S)
Agarose (Invitrogen cat. no. 16500–500)
Sybr Safe (Invitrogen cat. no. S33102)
Alexa488-conjugated goat anti-mouse (Life Sciences cat. no. A11029)
Alexa594-conjugated goat anti-rabbit (Life Sciences cat. no. A11037)
Mouse anti-FLAG (Sigma-Aldrich cat. no. F1804–50UG)
Counterstain antibody not made in a mouse
DG-52 mouse anti-SAG146
Alexa-488 labeling kit (Life Technology A20181)
Midiprep kit (Macherey-Nagel cat. no. 740412.50)
Miniprep kit (Zymo cat. no. D4054)
DNeasy DNA extraction kit (Qiagen cat. no. 69504)
QIAquick gel extraction kit (Qiagen cat. no. 28706)
Gibson Assembly (New England Biolabs cat. no. E2611L)
E. cloni 10G Supreme electrocompetent cells (VWR cat. no. 95024–012)
AseI and associated NEBuffer 3.1 (New England Biolabs cat. no. R0526L)
DNaseI (Sigma-Aldrich cat. no. DN25–100MG)
Trypsin (Worthington cat. no. LS003704)
iProof and HF buffer (BioRad cat. no. 422840)
Q5 DNA Polymerase (NEB cat. no. M0491L)
BsaI and associated Buffer G (Thermo Scientific cat. no. ER0291)
Calf Intestinal Phosphatase (CIP, New England Biolabs cat. no. M0290S)
HFF cells (American Type Culture Collection cat. no. SCRC-1041)
RH/Cas9 (American Type Culture Collection or by request) CAUTION Check for Cas9 expression and function before starting a screen by following steps 1–25 in the protocol.
RH (American Type Culture Collection cat. no. 50174)
Hanks’ Balanced Salts (Sigma-Aldrich cat. no. H2387–10X1L)
Dulbecco’s Modified Eagle Medium (DMEM) (Sigma cat. no. D7777–10L)
LB Agar (BD cat. no. 244510)
LB (Bio Basic Inc. cat. no. SD7002)
Equipment
Dialysis membranes (Millipore cat. no. VSWP02500)
0.1 cm gap cuvettes for bacterial transformations (BioRad cat. no. 165–2089)
4 mm gap cuvettes for T. gondii transfections (BTX Harvard Apparatus cat. no. 450126)
Gene Pulser Xcell (BioRad cat. no. 1652660)
Electroporator for T. gondii (BTX Harvard Apparatus ECM 830)
New Brunswick Scientific Excella E10 Platform Shaker in a 37°C room
37°C, 5% CO2 incubator (Thermo Scientific Forma Stericycle)
Coverslips (VWR cat. no. 89015–724)
Slides (Corning cat. no. 2948–75X25)
Nikon Eclipse Ti inverted fluorescent microscope
Nikon TMS inverted light microscope
Clinical centrifuge (Eppendorf 5810R)
Bench top centrifuge (Eppendorf 5424R)
−80°C freezer (Thermo Electron Corporation Forma −86°C ULT)
−20°C freezer (Thermo Electron Corporation Forma Pharmacy)
Hemocytometer (VWR cat. no. 15170–173)
Thermal cycler (BioRad cat. no. 51000)
Gel electrophoresis box (Owl Easycast B1A)
BioRad Powerpac Basic
37°C water bath (Fisher Scientific Isotemp)
Software
R (version 3.2.3 and above) (https://cran.r-project.org/mirrors.html)
R Studio (version 1.0.136 and above) (https://www.rstudio.com)
Countess, CRISPR_Analysis.R scripts (https://github.com/LouridoLab/CRISPR_Analysis)
Sample files (GEO acc. no. GSE98227) (https://www.ncbi.nlm.nih.gov/geo/)
Reagent Setup
D10: DMEM containing 10% heat-inactivated FBS, 10 μg/ml gentamicin. Store at 4°C; can be kept for approximately one month.
HHE: HBSS containing 0.1 mM EGTA and 100 mM HEPES, pH 7.4. Store at 4°C; can be kept for approximately two months.
Cytomix: 10 mM KPO4 (see below), 20 mm KCl, 5 mM MgCl2, 25 mM HEPES, 2 mM EDTA, adjust pH to 7.6. Sterilize using a 0.2 μm filter. Store at 4°C; can be kept for approximately four months.
0.1 M KPO4: 125 mM K2HPO4, 25 mM KH2PO4. Store at room temperature; can be kept for at least six months.
Glutathione (GSH): Dilute to 250 mM in sterile water and sterile filter using a 0.2 μm filter. Store at −20°C in 100 μl aliquots; can be kept for a year, avoiding excessive freeze-thaw cycles.
Adenosine Triphosphate (ATP): Dilute to 100 mM sterile water, adjust pH 7.0 and sterilize using a 0.2 μm filter. Store at −20°C in 100 μl aliquots; can be kept for a year, avoiding excessive freeze-thaw cycles.
CaCl2: Dilute to 7.5 mM in sterile water, sterilize using a 0.2 μm filter. Store at −20°C, in 100 μl aliquots; can be kept for a year, avoiding excessive freeze-thaw cycles.
Fixative: 4% methanol-free formaldehyde diluted molecular biology grade water. Store at 4°C; can be kept for one week.
Blocking solution: 5% heat-inactivated fetal bovine serum, 1% normal goat serum diluted in PBS. Store at 4°C; can be kept for one month.
Wash solution: 1% normal goat serum diluted in PBS. Store at 4°C; can be kept for one month.
50X Tris Acetic acid EDTA (50X TAE): 2 M tris base, 5.7% glacial acetic acid, 50 mM EDTA pH 8.0, dilute 50-fold to make 1X TAE. Store at room temperature; can be kept for a year.
Luria-Bertani Broth (LB): Dissolve 25 g of LB powder in 1 L of water and autoclave for 40 min to sterilize. Store at room temperature; can be kept for six months.
Luria-Bertain Broth plus Agar and ampicillin (LB Agar plus ampicillin): Dissolve 40 g of LB powder containing agar in 1 L of water and autoclave for 40 min to sterilize. Allow to reach room temperature, then add ampicillin to a final concentration of 100 μg/mL and pour into petri dishes. Allow plates to solidify before use. Store at 4°C, can be kept for one month.
10X Phosphate-Buffered Saline (10X PBS): 0.1 M Na2HPO4•7H2O, 1.4 M NaCl, 27 mM KCl, 15 mM KH2PO4, adjust pH to 7.4. Autoclave for 40 min to sterilize, dilute 10-fold to make 1X PBS. Store at room temperature; can be kept for six months.
PROTOCOL
Confirming Efficient Gene Disruption in RH/Cas9 TIMING 1 week
Purify 400 μg of pU6-SAG1 at a minimum concentration of 1.25 μg/μl using a commercial midiprep kit.
- Linearize 400 μg of pU6-SAG1 by digesting it at 37°C for 3 hours in the following reaction:
Component Amount per reaction Final concentration pU6-SAG1 400 μg 1 μg/μl NEB Buffer 3.1 40 μl 1x AseI 400 U 1 U/μl UltraPure water To 400 μl Dialyze the digested DNA against water for 20 min using drop dialysis on a dialysis membrane. Avoid dialyzing more than 100 μl per membrane to avoid excessive loss of the DNA. PAUSE POINT Dialyzed DNA can be stored at −20°C for at least 1 week.
Seed HFFs on 3 glass coverslips in a 24-well plate and grow to confluence (approximately 2 days).
Infect two T12.5 flasks of HFFs each with approximately 106 RH parasites and two T12.5s each with approximately 106 RH/Cas9 parasites two days before transfection.
Aliquot the linearized, dialyzed pU6-SAG1 into two microfuge tubes with 100 μg of DNA per tube. Aliquot the same volume of water (elution buffer) into a microfuge tubes to use for a mock transfection.
On the day of the transfection, heat the DNA and elution buffer to 65°C for 10 min to sterilize it.
In the hood, combine each DNA sample with 8 μl of each 7.5 mM CaCl2, 250 mM GSH, and 100 mM ATP, then bring the total volume to 155 μl using Cytomix.
Two days after infection (step 5), filter the parasites from the completely lysed monolayers. Pellet the parasites, resuspend in 10 ml of HHE, and count them using a haemocytometer. Pellet the parasites again and resuspend them at 5 × 107 parasites per ml in Cytomix. CRITICAL STEP Parasites should always pelleted for 10 min at 500 x g, 4°C, and can be separated from host cell debris by filtering through 3 μm membranes in Whatman filter holders (parasites will be present in the flow-through).
- Combine each DNA sample with 245 μl of parasites re-suspended in Cytomix such that there are three samples:
- RH/Cas9 with pU6-SAG1
- RH/Cas9 with mock
- RH with pU6-SAG1
Transfer the parasite/DNA mixture (400 μl total) into 4mm gap electroporation cuvettes, and electroporate using a BTX Square-Wave electroporator set to 1.7 kV, for two 176 μs pulses in 100 ms intervals. CRITICAL STEP An optimal transfection efficiency is necessary to ensure the success of the subsequent screens. This protocol was designed using the assumption that 2—5% of transfected RH/Cas9 parasites form pyrimethamine-resistant plaques after transfection with pU6-SAG1.
Place 200 μl from each transfection into a separate T12.5 containing a confluent monolayer of HFFs and 2.5 ml of D10. Add 40 μg/ml chloramphenicol to the media for the RH/Cas9 transfections, and return to the incubator.
One day after transfection, change the media on the transfected parasites to D10 containing 3 μg/ml pyrimethamine for RH/Cas9 transfections, and D10 only for mock transfected parasites.
Two days after transfection, scrape the infected HFF monolayers to fully release the parasites. Infect the HFFs on coverslips (step 4) with 10 μl of each sample. Maintain pyrimethamine selection as indicated above (step 13).
One day after infection (three days post transfection), perform the immunofluorescence assay. Prior to starting, prepare the blocking and wash solutions (see Reagent Setup section).
Fix the infected HFF cells by removing the media from all wells, then adding 0.5 ml of methanol and incubating on ice for 2 min.
Wash the fixed cells 3 times for 5 min with 1 ml of wash solution.
Block the cells for 10 min in 1 ml of blocking solution.
Invert the coverslips onto 25 μl drops, spotted on Parafilm, of blocking solution containing an antibody against an abundant parasite protein that was not made in a mouse, such as rabbit anti-TgACT1. Incubate the cells in a humid chamber, such as a 15 cm cell culture dish lined with wet Whatman paper, for 30 min at room temperature.
Transfer the coverslips to a clean 24-well plate and wash as in step 17.
Invert the coverslips onto 25 μl drops of blocking buffer containing a 500-fold dilution of Alexa488-conjugated mouse-anti-SAG1 (DG52), a 1000-fold dilution of Alexa594-conjugated goat-anti-rabbit (or an alternative, as appropriate, depending on the primary antibody used in step 19), and a 2000-fold dilution of Hoechst. Incubate the cells in a humid chamber at room temperature in the dark for 30 min.
Transfer the coverslips back to a 24 well plate and wash as in step 17.
Wash the cells once for 5 min in 1 ml of PBS.
Dip the coverslips in water, blot the edges of the coverslips to remove excess water, then mount on 5 μl drops of Prolong Gold, pressing lightly to remove excess mounting media.
Allow the slides to dry for one hour at 37°C or overnight at room temperature before imaging. Calculate the percentage of vacuoles that express SAG1, as indicated by peripheral green staining, in at least 100 vacuoles from each sample. CRITICAL STEP 90–97% of RH/Cas9 vacuoles should be SAG1 negative after transfection with pU6-SAG1, compared to the control samples, which should consistently express SAG1.
Preparations for the Genome-Wide Screen TIMING 3 weeks
-
26.
Digest 700 μg of CRISPR library for 3 hours using 700 U of AseI as in step 2. Although only 600 μg (600 μl) of digested plasmid will be needed for the transfections, the extra volume accounts for sample loss during drop dialysis. CRITICAL STEP The CRISPR library must be handled in such a way as to prevent loss of diversity, as outlined in the Supplementary Methods.
-
27.
Dialyze the digested DNA using drop dialysis membranes. Avoid dialyzing more than 100 μl per membrane to avoid excessive DNA loss. PAUSE POINT Dialyzed DNA can be stored at −20°C for at least 1 week.
-
28.
Freeze 100 ng of digested, dialyzed CRISPR library DNA to sequence with the post-selection samples.
-
29.
Prepare 26 confluent 15 cm dishes and 3 confluent T12.5 flasks of HFFs (see Box 1 for instructions on standard HFF culture).
-
30.
Two days before transfecting the parasites, infect two 15 cm dishes of HFFs, each with approximately 107 RH/Cas9 parasites. This should yield approximately 5.5 × 108 RH/Cas9 parasites after two days of growth. CRITICAL STEP The RH/Cas9 strain should be maintained under selection with 40 μg/ml chloramphenicol, but this selection may be removed following transfection with the CRISPR library.
Introducing the CRISPR Library into RH/Cas9 TIMING 1 day
-
31.
Divide the digested, dialyzed CRISPR library (step 27) into 10 tubes with 50 μg of DNA per transfection and heat to 65°C for 10 min. Aliquot the same volume of water into one tube to use in a mock transfection. The mock transfection will help confirm the clearance of un-transfected parasites by pyrimethamine.
-
32.
Before starting the transfection, replace the media on eight of the 15 cm dishes from step 29 with 30 ml of D10 per dish. These plates will receive the transfected parasites.
-
33.
Filter the parasites, pellet them and resuspend them in 10 ml of HHE. Count the parasites using a hemocytometer while pelleting them again, then resuspend them in Cytomix at a concentration of 2 × 108 parasites per ml. CRITICAL STEP Parasites should always pelleted for 10 min at 500 x g, 4°C, and can be separated from host cell debris by filtering through 3 μm membranes in Whatman filter holders (parasites will be present in the flow-through).
?TROUBLESHOOTING
-
34.
Combine each DNA sample or mock control with 245 μl of parasites re-suspended in Cytomix and transfer each 400 μl sample into a 4 mm gap electroporation cuvette.
-
35.
Electroporate using a BTX Square-Wave electroporator set to 1.7 kV, for two 176 μs pulses in 100 ms intervals.
?TROUBLESHOOTING
-
36.
Pool all the parasites transfected with the CRISPR library into a tube containing 30 ml of D10. Rinse the cuvettes with D10 to recover the remaining parasites and pool with the others. Bring up the total volume in the tube to 40 ml with D10, then divide equally between the eight 15 cm dishes that were set up in step 32. CAUTION Proceed with extreme caution when generating pyrimethamine-resistant T. gondii strains as this antimicrobial is the current front-line treatment against toxoplasmosis.
-
37.
Transfer 200 μl of mock transfected parasites into a T12.5 containing 2.5 ml of D10.
Culture and Sampling of the Screen Populations TIMING 6 days
-
38.
One day after transfection, gently remove the media from the infected cells. Rinse each dish once with 25 ml of 37°C PBS, then add 30 ml of fresh D10 containing 3 μg/ml pyrimethamine and 10 μg/ml DNaseI to each dish. Replace the media from the mock transfection (step 37) with 2.5 ml of D10 containing 3 μg/ml pyrimethamine and 10 μg/ml DNaseI. CRITICAL STEP DNaseI digests extracellular plasmid DNA and reduces background.
-
39.
Two days post transfection, replace the media on eight of the 15 cm dishes from step 29 with 30 ml of D10 containing 3 μg/ml pyrimethamine per dish.
-
40.
Scrape the partially lysed monolayers from the eight 15 cm dishes infected in step 36.
-
41.
Pellet the parasites and host cell debris for 10 min at 500 x g, 4°C.
-
42.
Resuspend all the parasites and cells in 10 ml of HHE and force them through a 27½-gauge needle to disrupt unruptured host cells. CAUTION To avoid needlestick injuries, use blunt needles (see MATERIALS) or leave the sterile sheath on a hypodermic needle in place and cut off the tip of the sheath.
-
43.
Pellet the parasites, resuspend them once more in 10 ml of HHE, pellet again and resuspend them in a final volume of 40 ml of D10 containing 3 μg/ml pyrimethamine; further treatment with DNaseI is not necessary. CRITICAL STEP Researchers should count the parasites at this point to ensure that the total number of passaged parasites is approximately 1.5 × 108, or sufficient to maintain library diversity.
?TROUBLESHOOTING
-
44.
Divide the parasite suspension by adding 5 ml to each of the eight 15 cm dishes from step 39 and incubate for three days (instead of the typical two needed for lysis) to account for the effects of pyrimethamine selection.
-
45.
Replace the media from a confluent T12.5 of HFFs with D10 containing 3 μg/ml pyrimethamine. Infect the monolayer with 5 × 106 parasites from the mock transfection sample (step 37).
-
46.
Five days post transfection, repeat steps 39–44 to continue the propagation of the mutant population. The mock-transfected parasites are expected to be dead by this stage and may be discarded.
?TROUBLESHOOTING
-
47.
Seven days after transfection, if the parasites have completely lysed the monolayers infected in step 46, proceed to step 48. Otherwise, repeat the process outlined in steps 40–42 to syringe-release the parasites, then resuspend the released parasites in 200 ml HHE.
-
48.
Pass 25 ml of parasite suspension through each of eight 3 μm filters.
-
49.
Pellet the parasites and resuspend them once in 10 ml of HHE.
-
50.
Count the parasites and divide them into aliquots of 108 parasites. CRITICAL STEP We have found that 2 × 108 parasites saturate the columns from Qiagen DNeasy DNA extraction kits, and ensuring that the number of parasites per aliquot remains below this number is important in optimizing the yields of genomic DNA from each sample.
-
51.
Pellet the parasites, remove the media and freeze the pellets at −80°C. PAUSE POINT Parasite pellets may be stored frozen at −80°C for several months.
Positive Selection of Mutant Populations (Optional) TIMING 1 week
-
52.
After three passages, pools of mutant parasites may be subjected to additional selective pressures to identify mutations that cause resistance. An example of such a strategy is outlined in steps 53–56. Continue with step 57 of the protocol to proceed without further enrichment of the parasite population.
-
53.
Replace the media on six 15 cm dishes of confluent HFFs with D10. Add a selective agent to three of the dishes, and an appropriate volume of the vehicle to the three other control dishes. For example, we enriched for sgRNAs targeting UPRT, which is involved in pyrimidine scavenging, by adding the toxic uracil analog 5-fluoro-deoxyuridine (FUDR) at 5 μM26 (see the Experimental Design section). This concentration of FUDR was previously shown to effectively inhibit growth of wild-type T. gondii without affecting a resistant strain28.
-
54.
Use the parasites from step 50 to infect each 15 cm dish with 1.2 × 107 parasites, or a sufficient number to maintain library diversity.
-
55.
Continue passaging the treated and untreated populations for the desired period of selection. Control populations will typically lyse the monolayer every two days, and should be passaged as in step 54. Under the appropriate selection, the treated population may not need to be passaged, and it may take up to a week for resistant parasites to form small plaques.
-
56.
Once growth of resistant parasites is observed in the treated populations, isolate the parasites from the treated and control populations following steps 47–51. Note that stringent selection may result in small numbers of resistant parasites, which should not compromise the outcome of the analysis as long as the enriched sgRNAs map to a few genes.
Genomic DNA Extraction and sgRNA Amplification TIMING 2 days
-
57.
Extract the DNA from one pellet containing 108 parasites per sample using a DNeasy Blood & Tissue Kit (Qiagen) following the manufacturer’s instructions, except eluting in 50 μl of molecular biology grade water instead of elution buffer. We advise extracting DNA from the parental strain as a negative control for the PCR. CRITICAL STEP Proceed immediately with the amplification of the CRISPR library from the genomic DNA to avoid any loss of DNA quality that may result from storage.
-
58.Assemble one PCR per sample according to the table below. We recommend using 1 μg of genomic DNA from the transfected populations to provide at least a 100-fold coverage of the CRISPR library as template for the PCR. Primer 2 can be barcoded (Table 1) to allow for sample multiplexing during next-generation sequencing. CRITICAL STEP Use filter tips and perform negative controls using water in place of template DNA to avoid and check for cross contamination.
Component Amount per reaction Final concentration 2X Q5 master mix 50 μl 1x Primer 1 (10 μM) 5 μl 0.5 μM Primer 2 (10 μM) 5 μl 0.5 μM Template DNA 1 μg of genomic DNA or 50 ng of library DNA UltraPure water To 100 μl -
59.Perform PCR following the cycling conditions below and analyze the products using standard gel electrophoresis.
Cycle number Denature Anneal Extend Final 1 98°C, 30 s 2–27 98°C, 10 s 60°C, 20 s 72°C, 15 s 28 72°C, 2 min 29 Hold, 12°C ?TROUBLESHOOTING
-
60.
Purify the resulting 368–369 bp PCR products (Figure 1b) using the PCR purification protocol from a QIAquik gel extraction kit.
TABLE 1:
Primers.
| Primer | Sequence (5’ to 3’) | Notes |
|---|---|---|
| Primer 1 | AATGATACGGCGACCACCGAGATCTACACGAATGACACACAGGAACTACGCG | Contains the P5 Illumina adaptor |
| Primer 2 | CAAGCAGAAGACGGCATACGAGATNNNNNNGATTTTCAAATGGCGACCTGC | Contains the P7 Illumina adaptor and a barcode, written here as “NNNNNN”. We have used the following barcode sequences: ATCACG, CGATGT, TTAGGC, TGACCA, ACAGTG, GCCAAT, CAGATC, ACTTGA, GATCAG, TAGCTT, GGCTAC, CTTGTA, AGTCAA, AGTTCC, ATGTCA, CCGTCC, GTCCGC, GTGAAA, GTGGCC, GTTTCG, CGTACG, GAGTGG, ACTGAT, ATTCCT |
| Primer 3 | CGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC | Used to sequence sgRNAs using Illumina sequencing |
| Primer 4 | GCGCGACAGAAAGCCCCTTCGAAGAGCGCACAGGGAGGAAGCAGGCCTCTGCAGGTCGCCATTTGAAAATC | Used to sequence barcodes using Illumina sequencing |
| Primer 5 | TTCTGGTAAATGGGGATGTCAAGTT | Used to clone the CRISPR library into pU6-DHFR |
| Primer 6 | AACTTGCTATTTCTAGCTCTAAAAC | Used to clone the CRISPR library into pU6-DHFR |
DNA Sequencing and Analysis TIMING 2 weeks
-
61.
Perform next-generation sequencing on the samples isolated in step 60 following your sequencing facility’s instructions. Primer 3 and Primer 4 (Table 1) can be used to sequence the sgRNAs and barcodes, respectively. Samples should be multiplexed such that the number of total reads allows for 200–300X coverage per guide in each sample. Read lengths of 20 bp will ensure sequencing of the entire sgRNA, and PhiX DNA may be added to increase nucleotide diversity and improve sequence quality.
-
62.
The sgRNA sequences obtained through next-generation sequencing should be analyzed as described under Data Analysis in the EXPERIMENTAL DESIGN section. We have provided a custom Perl script called Countess that will identify and tabulate sequences corresponding to individual sgRNAs in FASTQ files containing sequencing reads from an sgRNA library and a single experimental sample. Along with this, we have provided an R script called CRISPR_Analysis.R that will run Countess and calculate phenotype scores for each gene along with a file called sgRNAs_nr_empty that contains sequences for all sgRNAs in the CRISPR library. Links to these scripts are provided under MATERIALS. To start, copy the scripts and their associated files to a local directory. The Supplementary Data provides additional explanation of the commands used in CRISPR_Analysis.R and sample files may be found in the Gene Expression Omnibus repository (acc. no. GSE98227).
-
63.
Download and install R and the work environment R Studio (see MATERIALS). Open the file CRISPR_Analysis.R in R Studio and adjust the user-defined variables as described in the script.
-
64.
Run CRISPR_Analysis.R. A plot illustrating the phenotype scores of each gene and identifying 40 known dispensable and 40 known fitness-conferring genes will appear in the plots quadrant of R studio and a CSV file containing the phenotype score for each gene and its associated statistics will appear in the folder containing CRISPR_Analysis.R. This file can then be analyzed and compared to other experiments using standard spreadsheet software.
?TROUBLESHOOTING
65.
TIMING
Culturing HFF cells prior to beginning a screen (BOX 1 and step 29): 3 weeks
Isolating and preparing CRISPR library DNA (Supplementary Methods and steps 26–28): 2 days
Growing RH/Cas9 parasites in preparation for the screen (step 30): 2 days
Transfecting and culturing parasites to generate mutants (steps 31–51): 1 week divided as follows:
Day 0 (steps 31–37): 6 hours
Day 1 (step 38): 30 min
Day 2 (steps 39–45): 2 hours
Day 5 (step 46): 2 hours
Day 7 (steps 47–51): 2 hours
Optional further enrichment through positive selection (steps 52–56): ~6 days
sgRNA amplification and sequencing (steps 57–60): 2 days
DNA sequencing and analysis (61–64): 2 weeks
ANTICIPATED RESULTS
To achieve full genome coverage, it is necessary to transfect an sgRNA library in which all sgRNAs are present and their abundances are approximately equal. We often use Lorenz curves and their associated Gini indices to describe sgRNA distributions. We obtained Gini indices between 0.35 and 0.40 after analyzing four different sgRNA library preparations using next-generation sequencing. Conversely, sgRNA pools isolated from parasites after three passages had Gini indices ranging from 0.79 to 0.83 (Figure 5a).
Figure 5. Anticipated Screening Results.

(a) Lorenz curves illustrating the abundance disparity in four CRISPR library (orange) and four Passage 3 (blue) samples after Next-Generation Sequencing. (b) Correlation between phenotype scores of two biological replicates of a CRISPR screen for T. gondii genes that confer fitness when parasites are grown in HFF cells. Perfect correlation is indicated by a diagonal line. (c) Phenotype scores associated with 40 known dispensable and 40 known essential genes after a screen for fitness-conferring genes.
The coverage of the CRISPR library will also impact the variability between screen replicates. We typically obtain Pearson correlation coefficients of approximately 0.8 when comparing phenotype scores between replicates of negative selection screens (Figure 5b). Difficulty achieving consistent phenotype scores typically results from a limited population size, and can be addressed by increasing the number of transfected parasites and the number of flasks used in each passage. Transfection efficiency and plasmid integration should also be confirmed by plaquing parasites transfected with pU6-DHFR in the presence of pyrimethamine to ensure that 2–5% of the initial parasite population survives this process.
Results from our previous CRISPR screen for fitness-conferring T. gondii genes can be found in the supplement of our recent publication 26 or in ToxoDB, and we recommend that researchers include comparable samples in their screens to ensure that they obtain similar results when applying CRISPR screening to other questions. Our previous publication also provides lists of genes known to be either dispensable or essential for parasite growth in HFF cells, and researchers can check that these genes have high and low phenotype scores, respectively, when performing screens under the conditions that we describe (Figure 5c).
?TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2:
Troubleshooting Table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 33 | Insufficient parasite yield | Unhealthy or insufficient parasites were used to infect HFF monolayers (step 30) | Passage parasites as described in BOX 1 for at least 3 passages prior to infecting HFF monolayers in step 30. Infect each 15 cm dish with 1–2 × 107 parasites |
| Parasites were lost during the filtering and washing that occurs in step 9 | After pelleting, remove the media by decanting rather than aspirating | ||
| 35 | Transformation reactions arc upon electroporation | Excessive salinity in the transformation reaction | Remove excess HHE prior to resuspending the parasites in Cytomix, ensure that Cytomix was made properly, and that DNA was adequately dialysed |
| Electroporated volume exceeded 400 μl | Do not exceed a total volume of 400 μl for the contents of a single cuvette | ||
| Cuvette exterior was wet | Dry cuvette exterior before electroporating | ||
| 43,46 | The parasite yield is insufficient to maintain library diversity | Too few parasites were transfected (step 35) | Ensure that 4 × 108 parasites were transfected in step 35 |
| Parasites were killed during the transfection or were not transfected efficiently | Ensure that electroporator settings are correct Plaque transfected parasites in parallel to screens to determine the rates of parasite survival and pyrimethamine resistance are adequate Assess library DNA quality by running the digested plasmids on a gel, and confirming the presence of a single band at 6.3 kb; Otherwise Isolate and digest new CRISPR library DNA |
||
| 46 | Pyrimethamine did not kill the mock-transfected parasites | RH/Cas9 was contaminated with pyrimethamine-resistant parasites | Thaw a low passage stock of RH/Cas9 strain |
| The pyrimethamine solution was prepared incorrectly | Prepare a new pyrimethamine solution, making sure to dissolve it at 10 mg/ml in ethanol | ||
| 59 | No product observed in the PCR | Genomic DNA was stored too long prior to use | Isolate new genomic DNA and immediately use it in the PCR |
| Samples contained too few parasites for the PCR to produce a strong band. This is especially likely after positive selection screens. | Repeat the PCR using between 30 and 35 cycles instead of 27 cycles. | ||
| Excess genomic DNA inhibited the PCR | Split the requisite 1 μg of genomic DNA template between several reactions | ||
| A product observed in PCR from the parental strain or a negative control | CRISPR library or genomic DNA contaminated the PCR reagents or the DNA extractions | Repeat the DNA extraction and PCR in a UV-treated hood using fresh reagents; take care to avoid cross-contamination | |
| Parasites transfected with pU6-DHFR contaminated the parental RH/Cas9 strain | Repeat the PCR using DNA extracted from a low passage of RH/Cas9 | ||
| 64 | sgRNA abundance shows little change between the plasmid library and selected populations | RH/Cas9 lost Cas9 expression before or during the screen | Repeat the screen using a low passage of RH/Cas9 or derive a homogenous Cas9-expressing population through limited dilution |
| sgRNA abundances are highly variable and do not cluster by gene | The population size was too small leading to genetic bottlenecks | Repeat the screen using larger population sizes Ensure appropriate transfection and plasmids integration |
Supplementary Material
Supplementary Data. R Markdown file showing step-by-step explanations and execution of the analysis scripts used.
Supplementary Methods. Detailed protocols outlining the procedures for (i.) cloning an sgRNA pool into pU6-DHFR, (ii.) library transformation and preparation and (iii.) confirming Cas9 expression.
ACKNOWLEDGEMENTS
We thank Jeroen P. J. Saeij and Tim Wang for helpful advice during the development of the protocol, George Bell and Prathapan Thiru for assistance with bioinformatics, and Emily Shortt for helpful comments during the preparation of this manuscript. Antibodies against SAG1 and ACT were kindly provided by L. David Sibley. This work was supported by the NIH Director’s Early Independence Award (1DP5OD017892) and an NIH Exploratory R21 (1R21AI123746) to S.L.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
REFERENCES
- 1.World Health Organization. World Malaria Report. 2015, World Health Organization; World Malaria Report http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/ Date: (2015). [Google Scholar]
- 2.Checkley W et al. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect Dis 15, 85–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torgerson PR & Mastroiacovo P The global burden of congenital toxoplasmosis: a systematic review. Bull. World Health Organ. 91, 501–508 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agriculture, U. S. D. O. Toxoplasma on U.S. Sheep Operations. [Google Scholar]
- 5.Garrison E et al. A Forward Genetic Screen Reveals that Calcium-dependent Protein Kinase 3 Regulates Egress in Toxoplasma. PLoS Pathog 8, e1003049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su C, Howe DK, Dubey JP, Ajioka JW & Sibley LD Identification of quantitative trait loci controlling acute virulence in Toxoplasma gondii. PNAS 99, 10753–10758 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gubbels M-J et al. Forward genetic analysis of the apicomplexan cell division cycle in Toxoplasma gondii. PLoS Pathog 4, e36 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh U, Brewer JL & Boothroyd JC Genetic analysis of tachyzoite to bradyzoite differentiation mutants in Toxoplasma gondii reveals a hierarchy of gene induction. Molecular Microbiology 44, 721–733 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Pfefferkorn ER, Borotz SE & Nothnagel RF Toxoplasma gondii: characterization of a mutant resistant to sulfonamides. Exp. Parasitol. 74, 261–270 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Pfefferkorn ER & Kasper LH Toxoplasma gondii: genetic crosses reveal phenotypic suppression of hydroxyurea resistance by fluorodeoxyuridine resistance. Exp. Parasitol. 55, 207–218 (1983). [DOI] [PubMed] [Google Scholar]
- 11.Mojica FJM, Díez-Villaseñor C, García-Martínez J & Soria E Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60, 174–182 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasiunas G, Barrangou R, Horvath P & Siksnys V Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 109, E2579–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolotin A, Quinquis B, Sorokin A & Ehrlich SD Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology (Reading, Engl.) 151, 2551–2561 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Marraffini LA & Sontheimer EJ CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doudna JA & Charpentier E Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096–1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Wang T, Wei JJ, Sabatini DM & Lander ES Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sidik SM, Hackett CG, Tran F, Westwood NJ & Lourido S Efficient Genome Engineering of Toxoplasma gondii Using CRISPR/Cas9. PLoS ONE 9, e100450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen B, Brown KM, Lee TD & Sibley LD Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. MBio 5, e01114–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner JC, Platt RJ, Goldfless SJ, Zhang F & Niles JC Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nat. Methods 11, 915–918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghorbal M et al. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol. 32, 819–821 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Vinayak S et al. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature 523, 477–480 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidik SM et al. A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166, 1423–1435.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donald RG & Roos DS Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. PNAS 92, 5749–5753 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfefferkorn ER & Pfefferkorn LC Toxoplasma gondii: characterization of a mutant resistant to 5-fluorodeoxyuridine. Exp. Parasitol. 42, 44–55 (1977). [DOI] [PubMed] [Google Scholar]
- 29.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert LA et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen S et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell 160, 1246–1260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart T et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Pettitt S, Krastev DB, Song F, Ashworth A & Lord CJ Abstract 2743: Finding determinants of PARP inhibitor sensitivity using genome-wide and focused CRISPR screens. Cancer Res 76, 2743–2743 (2016).26964622 [Google Scholar]
- 34.Steinhart Z et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signalling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 23, 60–68 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Parnas O et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marceau CD et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535, 159–163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters JM et al. A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 165, 1493–1506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slaymaker IM et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang T et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tzelepis K et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep 17, 1193–1205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Winter J et al. CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. bioRxiv 109967 (2017). doi: 10.1101/109967 [DOI] [Google Scholar]
- 46.Burg JL, Perelman D, Kasper LH, Ware PL & Boothroyd JC Molecular analysis of the gene encoding the major surface antigen of Toxoplasma gondii. J Immunol 141, 3584–3591 (1988). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data. R Markdown file showing step-by-step explanations and execution of the analysis scripts used.
Supplementary Methods. Detailed protocols outlining the procedures for (i.) cloning an sgRNA pool into pU6-DHFR, (ii.) library transformation and preparation and (iii.) confirming Cas9 expression.