

Abstract
Purpose:
Head and neck squamous cell carcinoma (HNSCC) is driven largely by the loss of tumor suppressor genes, including NOTCH1, but lacks a biomarker-driven targeted therapy. Although the PI3K/mTOR pathway is frequently altered in HNSCC, the disease has modest clinical response rates to PI3K/mTOR inhibitors and lacks validated biomarkers of response. We tested the hypothesis that an unbiased pharmacogenomics approach to PI3K/mTOR pathway inhibitors would identify novel, clinically relevant molecular vulnerabilities in HNSCC with loss of tumor suppressor function.
Methods:
We assessed the degree to which responses to PI3K/mTOR inhibitors are associated with gene mutations in 59 HNSCC cell lines. Apoptosis in drug-sensitive cell lines was confirmed in vitro and in vivo. NOTCH1 pathway components and PDK1 were manipulated with drugs, gene editing, knockdown, and overexpression.
Results:
PI3K/mTOR inhibition caused apoptosis and decreased colony numbers in HNSCC cell lines harboring NOTCH1 loss-of-function mutations (NOTCH1MUT) and reduced tumor size in subcutaneous and orthotopic xenograft models. In all cell lines, NOTCH1MUT was strongly associated with sensitivity to six PI3K/mTOR inhibitors. NOTCH1 inhibition or knockout increased NOTCH1WT HNSCC sensitivity to PI3K/mTOR inhibition. PDK1 levels dropped following PI3K/mTOR inhibition in NOTCH1MUT but not NOTCH1WT HNSCC, and PDK1 overexpression rescued apoptosis in NOTCH1MUT cells. PDK1 and AKT inhibitors together caused apoptosis in NOTCH1WT HNSCC but had little effect as single agents.
Conclusion:
Our findings suggest that NOTCH1MUT predicts response to PI3K/mTOR inhibitors, which may lead to the first biomarker-driven targeted therapy for HNSCC, and that targeting PDK1 sensitizes NOTCH1WT HNSCC to PI3K/mTOR pathway inhibitors.
Keywords: PDK1, NOTCH1, head and neck squamous cell carcinoma, PI3K
Introduction
Although head and neck squamous cell carcinoma (HNSCC) has a high mutation rate, genomic sequencing has not detected druggable driver mutations in the disease. Rather, HNSCC is dominated by mutations in non-targetable tumor suppressor genes such as TP53 (72%), NOTCH1 (18%), KMT2D (16%), and AJUBA (6%) (1,2). The most frequently altered mitogenic signaling pathway in HNSCC is the phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway, with 54% of patients having mutations or copy number alterations in PIK3CA (35%), PTEN (6%), RICTOR (7%), AKT1 (3%), PIK3R1 (2%), and MTOR (3%) (3). PIK3CA, the third most frequently mutated gene in HNSCC (18%), has frequent hotspot mutations in the helical (E542K or E545K) and kinase (H1047R) domains (3,4).
PI3K signaling and its effectors protein kinase B (AKT), mTOR, and phosphoinositide-dependent kinase 1 (PDK1), which play critical roles in cell proliferation and survival, are validated therapeutic targets in other cancers (5). With the exception of PI3K inhibitors that are approved for the treatment of hematologic malignancies and target of rapamycin complex 1 (TORC1) inhibitors that are approved for renal cell carcinoma, PI3K/AKT/mTOR inhibitors have elicited only modest response rates in solid tumors (5). Strategies guiding the selection of patients for these agents also remain elusive. Whether PIK3CA mutant (PIK3CAMUT) tumors have increased sensitivity to PI3K/AKT/mTOR inhibitors is unclear (6–8). PI3K/AKT/mTOR inhibitors have a low clinical response rate in PIK3CAMUT tumors (~30%) (7) compared with other targeted therapies such as the epidermal growth factor receptor inhibitor osimertinib, which has a response rate of about 80% in EGFRMUT lung cancers (9). Consistent with these clinical findings, PIK3CAMUT HNSCC cell lines and patient-derived xenografts (PDXs) are more sensitive to PI3K/mTOR pathway inhibitors than PIK3CA wild type (PIK3CAWT) HNSCC cells are; however, the drugs cause only cell-cycle arrest with no apoptosis in the mutant cell lines (4,10–13).
Molecular therapies targeting activated oncogenes are common, but only one such therapy has been approved for cancers driven by the loss of tumor suppressor function: poly (ADP-ribose) polymerase (PARP) inhibitors for BRCA1/BRCA2MUT breast cancer (14). Perhaps because of feedback pathways, targeting pathways activated downstream of tumor suppressors has not been effective for cancer therapy. To date, the new genomic information available for this disease has not been translated into clinical care largely because the genomic landscape is dominated by tumor suppressors. In this study, we tested the hypothesis that an unbiased pharmacogenomics approach would identify novel, translationally applicable molecular vulnerabilities to HNSCC with the loss of tumor suppressor function. To address the lack of effective molecularly targeted therapies for HNSCC, we used an unbiased pharmacogenomics approach to integrate drug sensitivity data for seven diverse PI3K/mTOR pathway inhibitors in a panel of 59 molecularly characterized HNSCC cell lines (15,16). We identified a striking correlation between NOTCH1 loss of function (LOF) mutations and sensitivity to PI3K/mTOR pathway inhibitors in HNSCC that we confirmed with both in vitro and in vivo studies.
To the best of our knowledge, this is the first study to establish a therapeutic vulnerability of NOTCH1MUT HNSCC to any class of drugs. Thus, our findings have the potential to advance the approval of a biomarker-driven targeted therapy for HNSCC. Further, based on the mechanism of sensitivity of NOTCH1MUT HNSCC, we have identified a combination therapy that may be effective against NOTCH1WT HNSCC.
Materials and Methods
Cells and reagents
We obtained and subjected 50 human papilloma virus (HPV)-negative and 9 HPV-positive HNSCC cell lines to whole-exome sequencing, reverse-phase protein array analysis, and gene expression profiling as described previously (15,17,18). All the cell lines were genotyped by short tandem repeat analysis, and all cell lines were mycoplasma-free at the time of testing with a mycoplasma detection kit (Lonza, Walkersville, MD). UMSCC49 parental and NOTCH1 knockout (KO) cells were purchased from Dr. Chad Brenner at the University of Michigan. An erythromycin ribosomal methylase (ERM) plasmid expressing PDK1−green fluorescent protein was a gift from Dr. Gordon Mills. Cells were transfected with the PDK1-expressing plasmid with Lipofectamine 3000 (Life Technologies, Grand Island, NY) for 6 hours and selected with 1–2 mg/ml G418 (Sigma, St. Louis, MO). To create the PJ34, FaDU, and MDA686LN NOTCH1 KO lines, we transfected the parental cell lines with a NOTCH1 CRISPR/Cas9 KO plasmid (sc-421930; Santa Cruz, Dallas, TX) using GenJet DNA transfection reagent (Signagen, Rockville, MD). The transfected cells were sorted based on green fluorescent protein expression, and individual clones were obtained. PQR309 was provided by PIQUR Therapeutics AG (Basel, Switzerland). All other drugs were purchased from Selleck Chemicals (Houston, TX) and prepared as 10 mmol/L stock solutions in dimethyl sulfoxide (DMSO).
Cell viability assays
HNSCC cell lines were treated with DMSO (vehicle) or PI3K/mTOR pathway inhibitors at seven different concentrations (0.018–9.613 µM) for 72 hours. A CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI) was performed as described previously (31, 32). Inhibitory concentration (ICs) and area under the curve (AUC) values were calculated using the drexplorer R package with a best-fit dose-response model (33). The combination indices were calculated using the Chou-Talalay method (34) in CalcuSyn (Biosoft, Cambridge, UK). We tested the reproducibility and robustness of the data generated using three quality control parameters as described previously (29). These parameters were the concordance correlation coefficient, location shift, and maximum standard deviation between two biological replicates for three technical and two biological replicates. Based on heuristics from our previous screening studies in lung cancer (29), the cut-offs for reproducibility were a concordance correlation coefficient greater than 0.8, a location shift less than 0.9, and a standard deviation less than 0.23 based on the normal mixture fit model. Experiments not satisfying these criteria were repeated. The replicate with the smallest experimental variation measured by the median of standard deviation was chosen as a representative of the replicates, and its IC values served as the final values for subsequent analysis.
Western blot analysis
Western blot analysis was performed as described previously (28). In brief, cells were lysed with ice-cold lysis buffer, and the lysates were centrifuged at 20,000×g for 10 minutes at 4°C. Cell samples containing equal amounts of protein were resolved using sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and immunoblotted with different primary antibodies. Protein expression was detected using a horseradish peroxidase–conjugated secondary antibody (Bio-Rad, Hercules, CA) and electrochemiluminescence reagent (Amersham Biosciences, Pittsburg, PA). The antibodies are listed in Table S1.
Apoptosis and cell cycle assays
To measure apoptosis, we performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining with an APO-BRDU Kit (BD Biosciences, San Jose, CA) and Annexin V/propidium iodide staining with an FITC Annexin V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA) as described previously (36). For the cell cycle analysis, cells were harvested, fixed, incorporated with bromodeoxyuridine (BrdU), and stained with 7-aminoactinomycin D using a BrdU Flow Kit (BD Biosciences, San Jose, CA). Data were acquired with a three-laser, 10-color Gallios flow cytometer (Beckman Coulter, Brea, CA) and analyzed using Kaluza software (Beckman Coulter, Brea, CA). All apoptosis assays were performed in triplicate, and each test was completed twice on different days.
Colony formation assays
HNSSC cells were seeded in 60-mm plates. One day later, the cells were treated with DMSO or the indicated drugs for 48 hours. The medium was changed, and the cells were incubated in drug-free medium for 14–21 days. The cell colonies were then washed, fixed in 10% formaldehyde, and stained with crystal violet (0.5% w/v). Colony images were taken with a GelCount Tumour Colony Counter (Oxford Optronix Ltd., San Francisco, CA). The total colony number and area were counted and analyzed using the ImageJ software program (National Institutes of Health, Bethesda, MD). Assays were performed in triplicate, and each test was completed twice on different days.
Mouse models
This study was performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by MD Anderson’s Institutional Animal Care and Use Committee. Subcutaneous xenograft and orthotopic nude mouse tongue models were created as described previously (19). GSK2126458 was administered by oral gavage 5 days per week at 1 mg/kg (orthotopic model) or 3 mg/kg (subcutaneous xenograft model). PQR309 was administered by oral gavage 5 days per week at 30 mg/kg (orthotopic model).
TUNEL tissue staining
Harvested tumor tissues were fixed in 10% formalin, embedded in paraffin, cut into 5-μm sections, and stored until further use. Deparaffinized and rehydrated tissue sections were processed for DNA labeling by terminal deoxynucleotidyl transferase, and then streptavidin-conjugated horseradish peroxidase/diaminobenzidine was detected using a TUNEL apoptosis detection kit (Trevigen, Gaithersburg, MD, USA). Images were taken at 40X magnification, and Image J software was used to calculate the percentage of TUNEL-positive cells.
Statistical Analysis
We used the beta-uniform mixture model to control the false discovery rate (17). To identify differentially expressed features between groups, we applied modified two-sample t-tests using the Limma package in R. We used the Fisher exact test and Wilcoxon rank-sum test to evaluate associations between molecular characteristics and drug sensitivities. In vitro experiment results were compared using a two-sample t-test and two-way analysis of variance corrected for multiple comparisons with the Tukey method in GraphPad Prism 7 (La Jolla, CA). In vivo experiment results were analyzed using a linear mixed model in R.
Results
HNSCC cell lines with LOF NOTCH1 mutations are sensitive to PI3K/mTOR pathway inhibitors in vitro
To identify novel predictive therapeutic vulnerabilities linked to PI3K/mTOR pathway inhibition, we treated 59 HNSCC cell lines with seven different PI3K/mTOR pathway inhibitors that were in clinical development at the start of this study (20–27). Of the drug–cell line combinations, 95% met our predefined quality control cutoff. The HNSCC cell lines exhibited diverse sensitivity to PI3K/mTOR pathway inhibitors (Fig. S1A and B and Table S2). Because the dose-response curves for PI3K/mTOR pathway inhibitors often plateau near the IC50 values (10), we used the more robust IC70 and AUC values as parameters for drug potency as described previously (28). Cell lines were classified as sensitive based on IC70 values less than the peak plasma concentration for each drug. To determine if cross-comparison of the PI3K/mTOR inhibitors as a class was feasible, we compared their drug sensitivity patterns with those of three cell cycle kinase inhibitors we tested previously (18). The PI3K/mTOR pathway inhibitors clustered separately from the cell cycle inhibitors (Fig. S1C).
We then compared drug sensitivity to common driver genes identified in HNSCC tumors from The Cancer Genome Atlas (TCGA) (18). Our cell lines did not have any mutations in 11 of the top 50 mutated genes, and 6 genes were mutated in only one cell line, precluding comparison. Of the 515 sequenced TCGA HNSCC tumors, 92% had a mutation in at least one of the remaining 33 genes, demonstrating that our cell lines were genomically representative of HNSCC patients. Of the 33 genes, only mutated NOTCH1 and KRTAP5–5 were significantly correlated with sensitivity to six of the seven drugs (Fig. 1A). We did not study KRTAP5–5 further because it is mutated in only two cell lines and 6% of HNSCC patients (1,2).
Figure 1.

NOTCH1 loss-of-function (LOF) mutations and NOTCH1 signaling in head and neck squamous cell carcinoma (HNSCC) are correlated with sensitivity to PI3K/mTOR pathway inhibitors. A. HNSCC cell lines (n=59) were incubated with seven different PI3K/mTOR pathway inhibitors. The drugs’ IC70 and area under the curve (AUC) values in wild type cell lines were compared with those in cell lines with mutations in 33 common driver genes using a modified two-sample t-test at a false discovery rate (FDR) of 0.05. Positive and negative correlations between the presence of a mutation and drug sensitivity are indicated by blue and red data points, respectively, with larger points representing significant differences. B. The IC70 values in cell lines with wild type or mutant NOTCH1 and PIK3CA were compared using a Kruskal-Wallis test corrected for multiple comparisons by the Dunn test; p values are indicated in the figure. The dashed black lines indicate each drug’s peak plasma concentration. N.S., not significant. C. The frequency and location of mutations in NOTCH1 in HNSCC cell lines (upper gene map) are compared with those of T-acute lymphoblastic leukemia (T-ALL; lower map, below the gene) and HNSCC patients (upper map, above the gene) based on data from the Catalogue of Somatic Mutations in Cancer and The Cancer Genome Atlas (TCGA). Oncogenic T-ALL NOTCH1 mutations occur in a hotspot of mostly missense mutations within the negative regulatory heterodimerization (NRD) domain or in a second hotspot of mostly truncating mutations near the C-terminus, deleting the proline-glutamic acid-serine-threonine (PEST) domain and increasing the stabilization of activated NOTCH1 in the nucleus. In contrast, the truncating mutations in HNSCC are scattered throughout the gene but not in the PEST domain, and the missense mutations cluster in the extracellular EGF ligand-binding domains. D. Most mutations in the NOTCH1 gene in TCGA HNSCC patient samples are missense mutations outside the C-terminal NRD and PEST domains. E. From our panel of 59 HNSCC cell lines, we identified 24 harboring NOTCH1 and/or PIK3CA mutations. Orange text corresponds to nonsense mutations outside the PEST domain; red text, frameshift mutations outside the PEST domain; blue text, missense mutations outside the NRD and PEST domains; and gray text, in-frame deletions. ¥Non-canonical mutations; †suspected single-nucleotide polymorphisms excluded from NOTCH1 LOF mutants. F. The basal expression levels of NOTCH1 and NOTCH intracellular domain (NICD) proteins in NOTCH1WT and NOTCH1MUT cell lines were compared using a two-sample t-test. The correlation between the expression levels of NICD and NOTCH1 was assessed by Spearman correlation.
Because PIK3CA mutations predict pathway inhibitor response in HNSCC (10–12), we specifically examined their relationship with drug sensitivity. We found no consistent correlation between PIK3CA mutations and drug sensitivity when we included all 12 PIK3CAMUT cell lines (Fig. 1A). Compared with PIK3CAWT cell lines, PIK3CAMUT HNSCC cell lines (excluding three cell lines with uncharacterized mutations) were significantly more sensitive to four drugs and substantially more sensitive to two drugs (Fig. 1B). The median IC70 values of six drugs for the NOTCH1MUT lines were significantly lower than those of the drugs for the NOTCH1WT lines; the seventh drug, BKM120, was almost universally effective in HNSCC cell lines, likely owing to its multiple off-target effects (29). Two cell lines harbored both NOTCH1 and PIK3CA mutations and were sensitive to all the inhibitors tested. There was no mutual exclusivity of these two mutations in the 515 sequenced TCGA HNSCC patient samples, but there was a tendency towards co-occurrence (www.cbioportal.org: accessed on September 15, 2018).
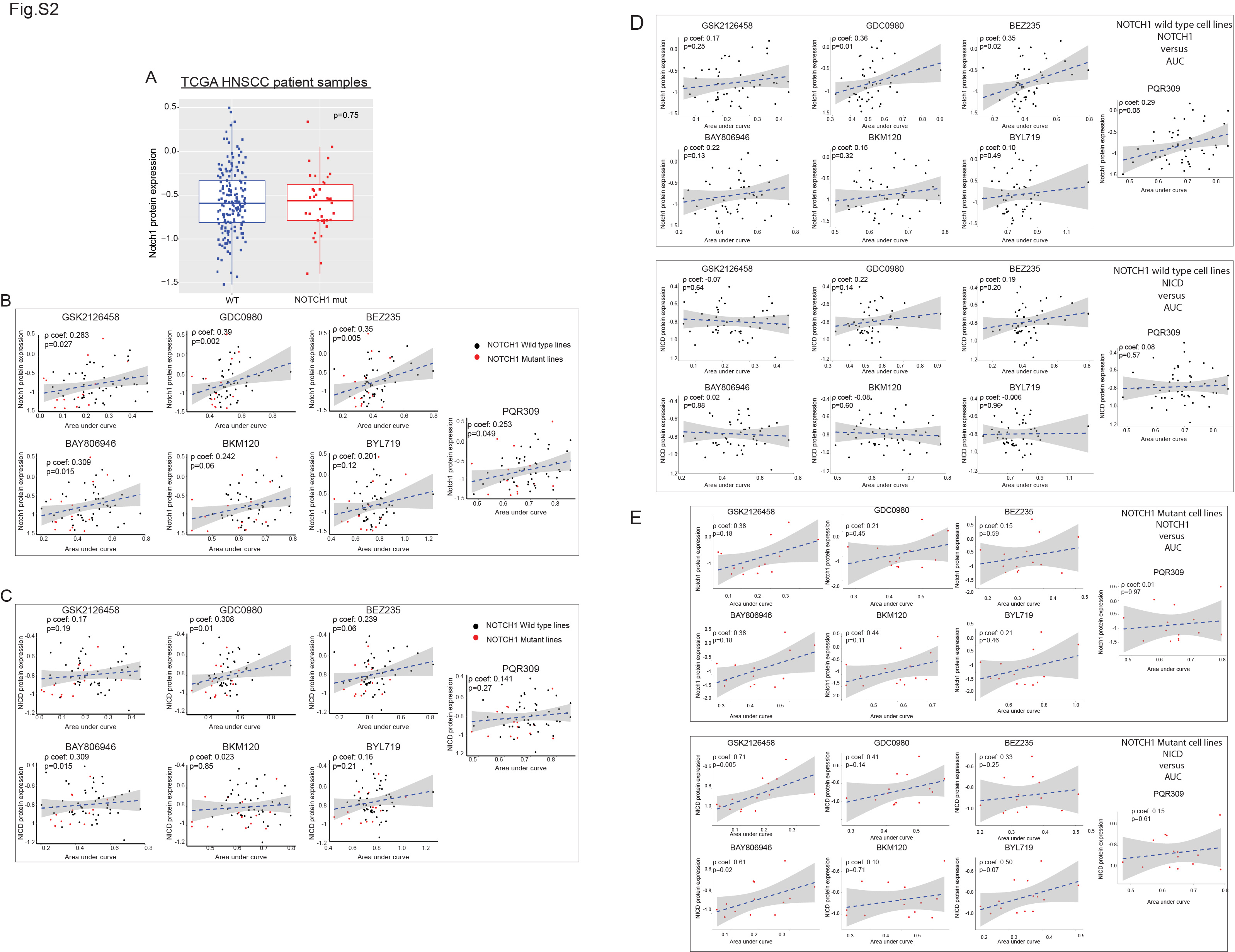
HNSCC patient samples have frequent truncating NOTCH1 mutations (2). We found the distribution of these mutations to be very different from that of the activating mutations found in T-cell acute lymphoblastic leukemia (T-ALL); mutations in HNSCC cell lines were similar to the mutation pattern in HNSCC patient samples (Fig. 1C and D) (30). Of the 17 HNSCC lines we found to harbor NOTCH1 mutations, six had truncating mutations (nonsense mutations and deletions) and 11 had missense mutations. Of those 11, three were excluded from analysis because their NOTCH1 alterations occurred in the nuclear regulatory domain and more likely represented single-nucleotide polymorphisms. Of the 14 HNSCC lines with NOTCH1 LOF mutations (Fig. 1E), two had both bona fide NOTCH1 and PIK3CA mutations. Both NOTCH1 and cleaved NOTCH1 (NOTCH intracellular domain [NICD]) protein levels were lower in NOTCH1MUT lines than in NOTCH1WT lines, although the difference was not significant, likely owing to diverse protein expression levels in NOTCH1WT lines. However, we found a significant positive correlation between NICD and total NOTCH1 protein levels in the cell lines (Fig. 1F). TCGA HNSCC patient samples also showed diverse NOTCH1 protein expression levels regardless of NOTCH1 mutation status (Fig. S2A). NOTCH1 and NICD levels did not consistently correlate with drug sensitivity in NOTCH1MUT or NOTCH1WT lines (Fig. S2B-E).
Basal activation of the PI3K/mTOR pathway and HPV status do not predict sensitivity to PI3K/mTOR pathway inhibitors
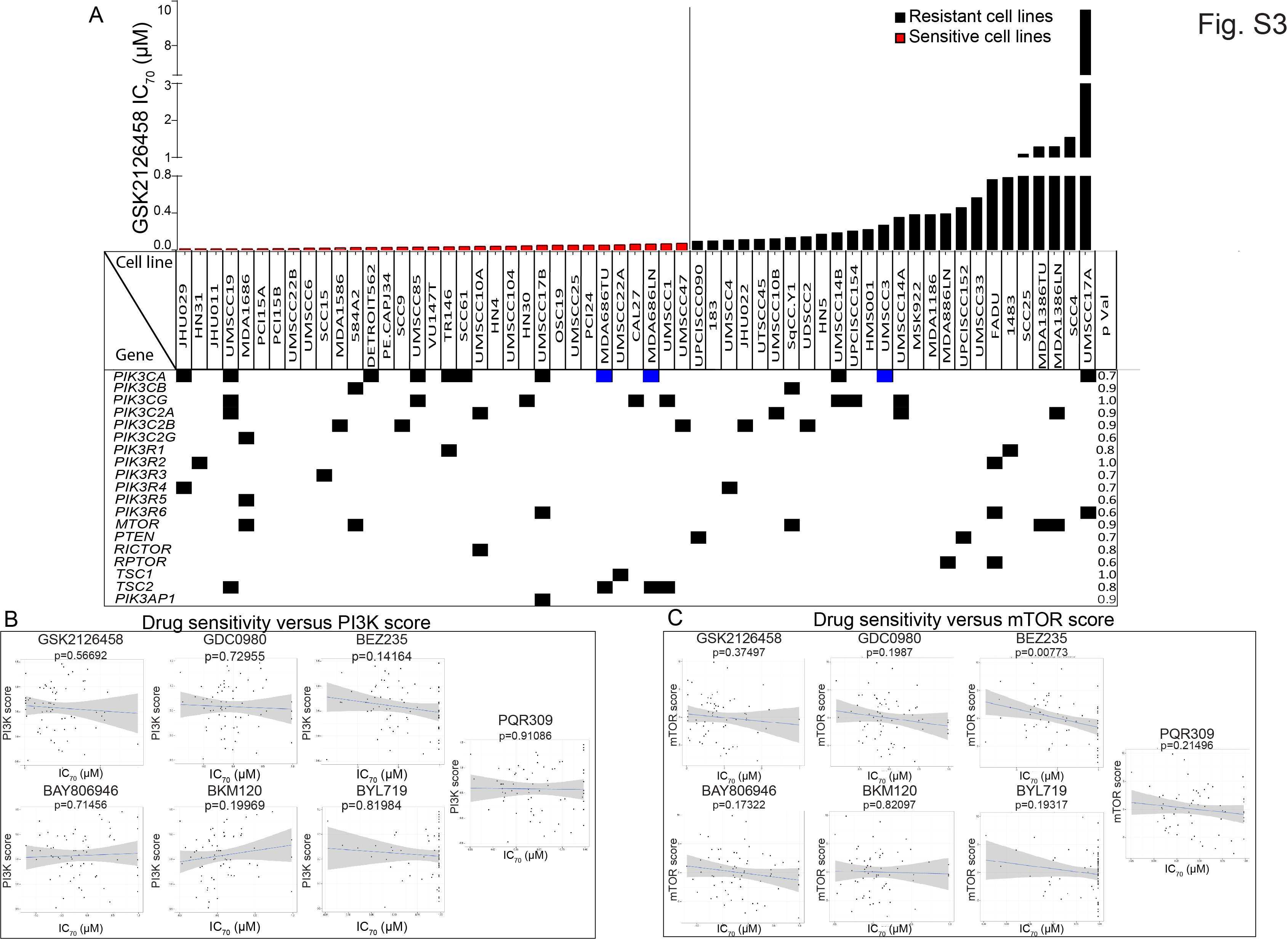
The diverse drug sensitivity suggested that subsets of cell lines have inherent molecular therapeutic vulnerabilities. Although there was a trend for PIK3CAMUT cell lines to be more sensitive to PI3K/mTOR pathway inhibitors, none of the 28 mutations in the PI3K/mTOR pathway described previously (6) consistently correlated with drug sensitivity (Fig. S3A). Likewise, we found no significant correlation between the basal activation of the PI3K/mTOR pathway and drug sensitivity (Fig. S3B and C, Table S3). Of note, drug sensitivity did not correlate with RICTOR amplification or PDK1 protein or gene expression (data not shown). Although HPV-positive HNSCC is molecularly distinct from HPV-negative HNSCC, drug sensitivity did not correlate with HPV status (Fig. S4A and B).
We also compared basal gene and protein expression levels between resistant and sensitive cell lines (Table S4 and Table S5) and two additional proteomic scores (28). There was no robust correlation between DNA repair and epithelial to mesenchymal transition scores and sensitivity to drugs (r. coefficient<0.4) (Fig. S4C-D). We did not study these differentially expressed genes or proteins or these proteomic scores further owing to both inconsistency of the correlations and a lack of prior validation of the scores in HNSCC.
PI3K/mTOR pathway inhibition induces apoptosis and reduces clonogenic growth in NOTCH1MUT cell lines
We then validated the susceptibility of NOTCH1MUT HNSCC to PI3K/mTOR inhibition with GSK2126458. GSK2126458 induced significant apoptosis in all NOTCH1MUT lines tested; no apoptosis was detected in the two NOTCH1WT/PIK3CAWT lines or three NOTCH1WT/PIK3CAMUT lines (Fig. 2A and B). BAY806946 and PQR309 elicited similar results (Fig. S5A and B). Consistent with these data, GSK2126458, even at concentrations well below the peak plasma concentration value, significantly reduced the number of colonies formed and the total cell number in NOTCH1MUT lines compared with NOTCH1WT lines (Fig. 2C). GSK2126458 induced G1 arrest in all cell lines but increased the sub-G0 fraction in only NOTCH1MUT lines (Fig. S5C). We confirmed that GSK2126458 had dose-dependent target inhibition in representative NOTCH1WT and NOTCH1MUT lines (Fig. 2D).
Figure 2.

PI3K/mTOR signaling inhibition induces cell death in NOTCH1MUT but not NOTCH1WT head and neck squamous cell carcinoma (HNSCC) cell lines in vitro. A. Annexin V/propidium iodide or BrDU-terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) assays were used to measure apoptosis in HNSCC cells with the indicated NOTCH1 and PIK3CA mutations, which were treated with 50 nM GSK2126458 (GSK212) for 24 or 48 hours, respectively. Values are the means ± standard deviations of three independent experiments. *p<0.05, unpaired two-tailed Student t-test. B. Western blotting of cleaved PARP (Cl-PARP) and cleaved caspase 3 (Cl-Caspase 3) levels in mutant and wild type cells after GSK212 treatment for 48 hours. C. NOTCH1MUT and NOTCH1WT HNSCC cells were plated sparsely and treated with GSK212 at the indicated concentrations for 48 hours. At 14-21 days after treatment, the cells were fixed and stained, images were captured using the GelCount Tumour Colony Counter, and colony numbers and the colorimetric intensity of the colonies were determined by measuring the optical density (OD) at 570 nm. Values are the means ± standard deviations of three independent experiments. *p<0.001, unpaired two-tailed Student t-test. D. Three NOTCH1WT lines (black text and bars) and three NOTCH1MUT lines (red text and bars) were treated with increasing concentrations of GSK212 for 4 hours, and levels of PI3K/mTOR pathway proteins were measured by Western blot analysis.
PI3K/mTOR pathway inhibition reduces tumor growth in both orthotopic tongue and subcutaneous xenograft models of NOTCH1MUT HNSCC
To assess the anti-tumor effect of PI3K/mTOR inhibition in NOTCH1MUT HNSCC, we first used two NOTCH1MUT lines to create subcutaneous xenograft models. The GSK2126458-treated tumors regressed, whereas the vehicle-treated control tumors grew significantly. TUNEL staining showed that, consistent with the in vitro data, tumors treated with GSK2126458 had significant apoptosis (Fig. 3A and B).
Figure 3.

PI3K/mTOR pathway inhibitors cause apoptosis and reduce tumor size in NOTCH1MUT head and neck squamous cell carcinoma (HNSCC) in vivo. A and B. The NOTCH1MUT cell lines HN31 and UMSCC22A (UM22A) were injected subcutaneously (SQ) into athymic nude mice. After tumors reached 150 mm3, the mice were randomized to receive either vehicle control (2-hydroxypropyl-β-cyclodextrin) or 1 mg/kg GSK2126458 (GSK212) by oral gavage daily for 28 days. Tumor sizes were measured and tumor volumes were calculated at the indicated times, and the percent change in tumor volume at the end of therapy was calculated using the formula (final tumor volume – initial tumor volume/initial tumor volume) x 100%. The percentages of TUNEL-positive cells were calculated from image quantification at 40X magnification. C-F. HN31 and UM22A were implanted orthotopically (OT) into the anterior tongues of athymic nude mice. After tumors reached 1.5–2.0 mm3, the mice were randomized to receive either vehicle control (16% 2-hydroxypropyl-β-cyclodextrin for GSK212 or 2-hydroxypropyl-β-cyclodextrin for PQR309) or GSK212 (3 mg/kg) or PQR309 (30 mg/kg) by oral gavage daily for 28 days. Tumor sizes were measured, and tumor volumes and percent changes in tumor volumes were calculated as above. For the tumor growth curve analysis, we applied a linear model using generalized least squares with two variables (treatment and day) and an autocorrelation structure of order 1 in residuals. The percentages of TUNEL-positive cells were calculated as described above. Data are means ±standard deviations from 5 mice per group; p values were calculated using an unpaired t-test with the Welch correction.
We also used an orthotopic xenograft model of HNSCC, whose local growth patterns and histology are similar to those of human HNSCC (19). In both NOTCH1MUT models, GSK2126458 and PQR309 significantly reduced tumor size compared with vehicle alone. TUNEL staining showed that the NOTCH1MUT tumors underwent apoptosis at the end of treatment (Fig. 3C-F).
CRISPR-Cas9 NOTCH1 KO sensitizes NOTCH1WT HNSCC to PI3K/mTOR pathway inhibitor–mediated apoptosis
We used four NOTCH1WT lines with NOTCH1 KO to test the hypothesis that abrogating NOTCH1 signaling renders NOTCH1WT cells more sensitive to PI3K/mTOR pathway inhibition. Compared with their parental cell lines, the PJ34 and UMSCC49 lines with NOTCH1 KO showed decreased cell viability after GSK2126458 treatment, with IC70 values that dropped from 0.1 µM to 0.024 µM and from 1.48 µM to 0.36 µM, respectively (Fig. 4A); had a significantly higher rate of apoptosis when treated with PI3K/mTOR inhibitors (Fig. 4B and C); and formed significantly fewer colonies after PI3K/mTOR inhibition (Fig. 4D). However, NOTCH1 KO did not significantly increase the sensitivity of the NOTCH1WT lines FaDU and MDA686LN to PI3K/mTOR pathway inhibition (Fig. S6).
Figure 4.

NOTCH1 knockout (KO) sensitizes two NOTCH1WT head and neck squamous cell carcinoma (HNSCC) cell lines to PI3K/mTOR pathway inhibition in vitro. Two NOTCH1WT HNSCC cell lines (PJ34 and UMSCC49 [UM49]), parental cell lines, and NOTCH1 KO cell lines were treated with PI3K/mTOR inhibitors. A. The CellTiter-Glo assay was used to assess the viability of cells treated for 72 hours with the indicated concentrations of GSK2126458 (GSK212). B. Western blot analysis of apoptosis markers, NOTCH1, and NOTCH intracellular domain (NICD) was performed after treatment with the indicated concentrations of GSK212 for 48 hours. C. Apoptosis in cells treated with 50 nM GSK212 or 200 nM BEZ235 for 48 hours was measured using an Annexin V/propidium iodide assay. Values are the means ± standard deviations of three independent experiments. *p<0.05, unpaired two-tailed Student t-test. D. Cells were treated with 50 nM GSK212 for 48 hours and then incubated in drug-free medium for 14-21 days. Colony areas as percentages of the control were measured using ImageJ. Data are presented as the means ± standard deviations of three independent experiments. *p<0.05, unpaired two-tailed Student t-test.
We also used the γ-secretase inhibitors N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester and dibenzazepine to inhibit the intra-membrane proteolytic cleavage/activation of NOTCH1. The combination of PI3K/mTOR and NOTCH1 signaling inhibitors decreased cell viability in four NOTCH1WT lines (Fig. S7A and B), with most combination indices of less than 1 for a range of fractions affected (Table S6). Consistent with these findings, the combination also increased apoptosis (Fig. S7C-E). As with the NOTCH1-KO lines, combined γ-secretase and PI3K/mTOR inhibition was not synergistic in two NOTCH1WT lines, likely because of the diverse molecular mechanisms of resistance.
PDK1 mediates resistance to PI3K/mTOR pathway inhibition in NOTCH1WT HNSCC
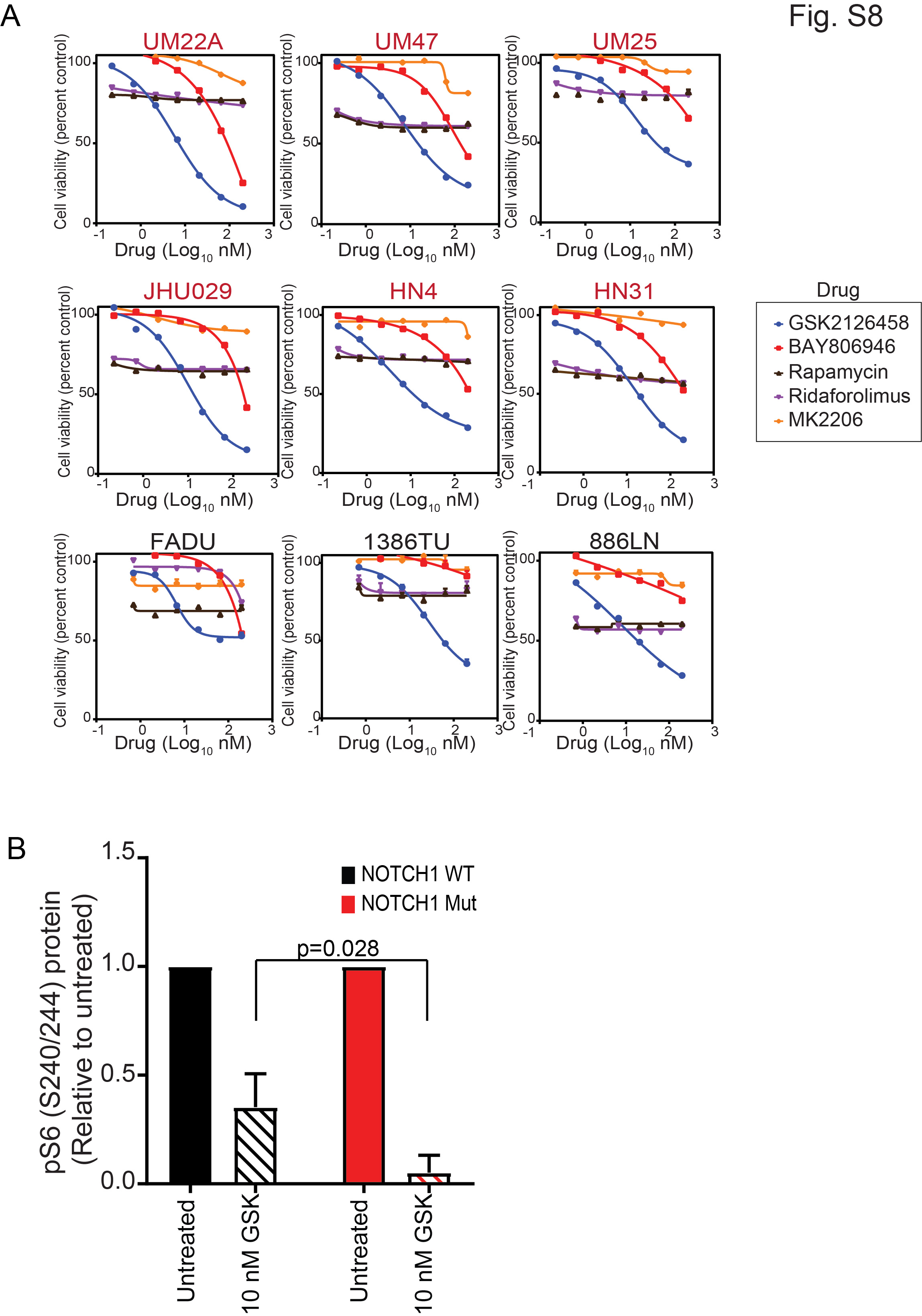
To identify the mechanism of sensitivity in NOTCH1MUT HNSCC, we inhibited components of the PI3K/mTOR pathway in both NOTCH1MUT and NOTCH1WT cell lines. All cell lines were resistant to the mTOR inhibitors (rapamycin and ridoforolimus) and the AKT inhibitor (MK-2206), but both the dual PI3K/mTOR inhibitor (GSK2126458) and pan-PI3K inhibitor (BAY806946) had differential effects on NOTCH1MUT and NOTCH1WT cells (Fig. S8A). These results suggest that an important signaling node that is downstream of PI3K but independent of AKT and mTOR is responsible for the differential sensitivity.
We then investigated PDK1’s role in the differential sensitivity, because PDK1 is activated by PI3K, mediates resistance to PI3K inhibition in breast cancer (31), and can regulate p70S6K, which was inhibited more robustly in NOTCH1MUT than in NOTCH1WT HNSCC (Fig. S8B). PI3K inhibition using three different drugs reduced PDK1 expression in only NOTCH1MUT lines (Fig. 5A, 5B S9A). To assess PDK1’s role in PI3K/mTOR inhibition–induced apoptosis in NOTCH1MUT HNSCC, we induced PDK1’s overexpression in NOTCH1MUT HNSCC cells. Compared with parental lines, NOTCH1MUT cells overexpressing PDK1 had higher expression levels of p-AKT, p-S6, and p-4EBP1 (Fig. 5C), and they had decreased apoptosis after GSK2126458 treatment (Fig. 5D and E). As expected, PDK1 overexpression did not inhibit the effect of GSK2126458 on phosphatidylinositol (3,4,5)-trisphosphate (PIP3)-dependent molecules such as p-AKT (T308) (Fig. 5E). In contrast, PDK1 overexpression did inhibit the effect of GSK2126458 on PIP3-independent molecules such as c-Myc and p-TSC2 (Fig. S9B).
Figure 5.

PDK1 mediates resistance to PI3K/mTOR pathway inhibition in NOTCH1WT head and neck squamous cell carcinoma (HNSCC) lines. A. Two NOTCH1WT lines and four NOTCH1MUT lines were treated with 50 nM GSK2126458 (GSK212) for 24 hours and then subjected to Western blot analysis with the indicated antibodies to assess the expression of 3-phosphoinositide dependent kinase 1 (PDK1) and phosphorylated PDK1. B. Protein expression from Western blotting was quantified using ImageJ and normalized using β-actin as a control, and the relative fold control from untreated was calculated. Bars indicate means ± standard errors of the means from two NOTCH1WT cell lines and four NOTCH1MUT cell lines. *p<0.05, one-tailed Student t-test. C. We established NOTCH1MUT lines (HN31, PCI15B, and UMSCC22A) with PDK1 overexpression, which was confirmed by Western blot analysis. D and E. Parental and PDK1-overexpressing NOTCH1MUT cells were treated with 50 nM GSK212 for 24 or 48 hours, and apoptosis was assessed by measuring levels of cleaved PARP (Cl-PARP) and cleaved caspase 3 (Cl-Caspase 3) by Annexin V/propidium iodide assay (D) and Western blot analysis (E). The percentage of apoptotic cells are expressed as the means ± standard deviations of three independent experiments. *p<0.001, two-way ANOVA corrected for multiple comparisons by the Tukey test.
PDK1 inhibition with GSK2334470 alone had little effect on cell viability or apoptosis regardless of NOTCH1 status (Fig. 6). Therefore, we hypothesized that both AKT and PDK1 inhibition are necessary for apoptosis following PI3K inhibition. In all cell lines tested, AKT inhibition plus PDK1 inhibition decreased cell viability with combination indices consistent with a greater-than-additive effect (Fig. 6A, Fig. S9C and D, and Table S7). The combination also led to apoptosis in all cell lines tested (Fig. 6B and C). In NOTCH1MUT lines, the effect of the combination was similar to that achieved with PI3K/mTOR inhibition alone (Fig. 6B).
Figure 6.

AKT inhibition and PDK1 inhibition synergistically induce apoptosis in NOTCH1WT head and neck squamous cell carcinoma (HNSCC) lines A. NOTCH1WT HNSCC cell lines were treated with increasing concentrations of MK2206, GSK2334470 (GSK233), or the MK2206-GSK233 combination at a fixed 1:1 ratio for 72 hours, and cell viability was measured with the CellTiter-Glo assay. The combination index (CI) values were calculated with CalcuSyn, and the fraction affected (Fa) and CI are plotted as heatmaps for all four cell lines. B. NOTCH1WT (black text) and NOTCH1MUT (red text) HNSCC cells were treated with 50 nM GSK2126458 (GSK212), 10 µM GSK233, 10 µM MK2206, or the GSK233-MK2206 combination for 48 hours. Western blot analysis showed that the combination increased cleaved PARP (Cl-PARP) and cleaved caspase 3 (Cl-Casp3) in the NOTCH1WT lines. C. Four NOTCH1WT HNSCC cell lines were treated with 50 nM GSK212, 10 µM GSK233, 10 µM MK2206, or the GSK233-MK2206 combination for 48 hours. Apoptosis was measured by Annexin V/propidium iodide assay, and the percentages of apoptotic cells are expressed as the means ± standard deviations of three independent experiments. *p<0.001, **p<0.05, two-way analysis of variance corrected for multiple comparisons with the Tukey test.
Discussion
With this study, we are the first to establish NOTCH1 LOF mutations in HNSCC as a significant therapeutic vulnerability to PI3K/mTOR pathway inhibition. We found that, unlike PIK3CAMUT HNSCC cell lines, which underwent only cell cycle arrest following PI3K/mTOR inhibition, NOTCH1MUT HNSCC cell lines also underwent apoptosis. Likewise, PI3K/mTOR inhibitors decreased NOTCH1MUT HNSCC tumor size in vivo. Selective mTOR or AKT inhibitors did not have differential effects on NOTCH1WT or NOTCH1MUT HNSCC. This led us to examine PDK1, whose expression decreased following PI3K/mTOR inhibition in only NOTCH1MUT HNSCC, supporting the hypothesis that persistent PDK1 activation leads to PI3K/mTOR inhibition resistance in NOTCH1WT HNSCC despite robust AKT inhibition. Consistent with this hypothesis, a combination of drugs that inhibit both PDK1 and AKT led to apoptosis.
Two other lines of evidence support the conclusion our findings. First, the PI3K inhibitor PX-886 significantly reduced tumor growth in two NOTCH1MUT HNSCC PDX models (11). Second, NOTCH pathway activation confers resistance to PI3K/mTOR inhibitors via c-Myc activation in breast cancer (32). Although we found that basal c-Myc gene or protein expression did not predict response, and c-Myc expression did not differ according to NOTCH1 mutation status (data not shown), c-Myc may be differentially regulated downstream of PDK1 in a PIP3-independent manner.
Although activating mutations in PIK3CA also occur in HNSCC and other solid tumors, PI3K/mTOR inhibitors have had limited clinical success. Compared with PIK3CAWT HNSCC, PIK3CAMUT HNSCC cell lines and PDX models are more sensitive to PI3K/mTOR pathway inhibitors (4,10–13,33). However, PI3K/mTOR inhibition does not cause significant apoptosis in PIK3CAMUT HNSCC cell lines (10). These published findings are consistent with those of the present study and with clinical findings demonstrating that PIK3CAMUT tumors tend to be more sensitive than PIK3CAWT tumors to PI3K/mTOR pathway inhibitors but still have modest clinical responses (4,7,8,34). Because HNSCC tumors with NOTCH1 mutations undergo cell death (rather than simple growth arrest) in response to PI3K/mTOR inhibitors, we hypothesize that drugs targeting the PI3K/mTOR pathway have greater clinical efficacy in this genomic subtype that in PIK3CAMUT or NOTCH1WT HNSCC.
Resistance to PI3K/mTOR pathway inhibitors manifests as a lack of sustained pathway inhibition despite the use of potent agents. For example, mTORC1 inhibition blocks S6 kinase–dependent inhibition of insulin receptor substrate 1, leading to insulin-like growth factor 1 receptor and then PI3K/AKT activation (35). A second feedback loop occurs because AKT activation inhibits the nuclear localization of forkhead box class O, a transcriptional driver of receptor tyrosine kinase (RTK) expression. AKT inhibition relieves this feedback suppression, leading to RTK expression and subsequent extracellular signal-regulated kinase (ERK) activation (36). Similarly, in HNSCC, the addition of mitogen-activated protein kinase kinase (MEK)/ERK or RTK pathway inhibition enhances anti-tumor effects of PI3K/mTOR inhibition (10,37–39). However, we did not find differential effects on p-ERK after PI3K/mTOR inhibition based on NOTCH1 status (data not shown). A third mechanism is the sustained PDK1 signaling that mediates residual mTORC1 activity despite potent PI3Kα inhibition in PI3Kα inhibitor–resistant PIK3CAMUT breast cancers (31). Our findings suggest that sustained PDK1 activity is a mechanism of resistance to PI3K/mTOR inhibition in NOTCH1WT HNSCC. Additionally, other factors may influence response of HNSCC to PI3K/mTOR pathway inhibitors such as TP53 and TGFβ (40).
The mechanism underlying this differential effect on PDK1 in HNSCC is unknown. Although crosstalk between the PI3K/mTOR and NOTCH1 pathways has been extensively studied in T-ALL, in which NOTCH1 acts as an oncogene (30), the way in which these pathways interact in solid tumors is unknown. In T-ALL, the activation of hes family bHLH transcription factor 1 (HES1), a downstream target of NOTCH1, suppresses PTEN expression, leading to PI3K activation and resistance to NOTCH1 inhibition (30). In the present study, NOTCH1 inhibition did not affect AKT activation in HNSCC. We speculate that in NOTCH1WT HNSCC, PDK1 is activated independently of PI3K. In T-ALL (41) and thymocytes (42), PDK1 activation downstream of NOTCH1 signaling mediates survival and proliferation.
One candidate through which PDK1 becomes activated is collagen 9 type alpha 1 (COL11A1). In ovarian cancer cells, COL11A1 binds to PDK1 and attenuates its ubiquitination and degradation, leading to chemoresistance (43); and in mouse embryonic fibroblasts, COL11A1 expression is downregulated after NOTCH1 inhibition (44). A second candidate is peroxisome proliferator-activated receptors, which are transcription factors induced by NOTCH1 inhibition in fatty liver (45) and can activate PDK1, leading to increased keratinocyte survival (46). In addition, the NOTCH1 pathway can confer drug resistance through a variety of other mechanisms, including epithelial-to-mesenchymal transition (47), and by promoting the cancer stem cell phenotype (48), which leads to tumor proliferation despite PI3K pathway inhibition.
In our study, NOTCH1 inhibition did not sensitize some NOTCH1WT HNSCC cell lines to PI3K/mTOR inhibitors. We speculate that other diverse mechanisms mediate resistance to PI3K/mTOR pathway inhibitors, including PTEN loss (49) and AXL activation (13). However, the consistent sensitivity of NOTCH1MUT HNSCC to multiple PI3K/mTOR pathway inhibitors supports the vulnerability of this molecular subtype despite the varied effects of NOTCH1 manipulations in NOTCH1WT HNSCC.
This is the first study to establish a therapeutic vulnerability of NOTCH1MUT HNSCC to any class of drugs. Because NOTCH1 LOF mutations are common in other squamous cell carcinomas, including lung (8%) and esophageal (21%) carcinomas (50), our findings have wide-ranging implications. Our finding that NOTCH1 LOF mutations predict response to PI3K inhibitors may lead to the first biomarker-driven targeted therapy for HNSCC. In addition, targeting PDK1 may sensitize NOTCH1WT HNSCC to PI3K/mTOR pathway inhibitors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of translational relevance.
The genomic landscape of head and neck squamous cell carcinoma (HNSCC) is dominated by non-targetable tumor suppressor genes such as NOTCH1, which is mutated in 18% of HNSCC. Molecularly targeted therapies directed towards activated oncogenes are common for cancers generally but rare for cancers driven by the loss of tumor suppressor function. Our in vitro and in vivo studies revealed that PI3K inhibition causes apoptosis in only HNSCC with NOTCH1 loss-of-function mutations, independent of PIK3CA mutation status, via differential regulation of PDK1. This study, the first to establish a therapeutic vulnerability of HNSCC with NOTCH1 mutations to any class of drugs, may inform the development of the first biomarker-driven targeted therapy for HNSCC. Because NOTCH1 loss-of-function mutations are common in other squamous cell carcinomas, including lung (8%) and esophageal (21%) carcinomas, our findings likely have implications beyond HNSCC.
Acknowledgments:
We thank Doriano Fabbro, PhD, of PIQUR Therapeutics AG for guidance regarding PQR309 use in experiments and Joe Munch in MD Anderson’s Department of Scientific Publications for editing the manuscript.
Financial support: This work was supported by philanthropic contributions to The University of Texas MD Anderson Cancer Center’s Oropharynx Discovery Program (JNM, FMJ); by grants from the National Institutes of Health/National Institute of Dental and Craniofacial Research (U01DE025181, to JNM and MJF; R01 DE024179–01A1, to MJF); and by PIQUR Therapeutics AG. This work used the services of MD Anderson’s Flow Cytometry and Cellular Imaging Core and Bioinformatics Shared Resource, which are supported by the National Institutes of Health through MD Anderson’s Cancer Center Support Grant (P30CA016672).
Footnotes
Conflict of interest disclosure: Faye M. Johnson has received research funding from PIQUR Therapeutics and Trovagene. Other authors have no conflicts of interest to declare.
References
- 1.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517(7536):576–82 doi 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011;333(6046):1154–7 doi 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iglesias-Bartolome R, Martin D, Gutkind JS. Exploiting the head and neck cancer oncogenome: widespread PI3K-mTOR pathway alterations and novel molecular targets. Cancer Discov 2013;3(7):722–5 doi 10.1158/2159-8290.CD-13-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov 2013;3(7):761–9 doi 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodon J, Tabernero J. Improving the Armamentarium of PI3K Inhibitors with Isoform-Selective Agents: A New Light in the Darkness. Cancer Discov 2017;7(7):666–9 doi 10.1158/2159-8290.CD-17-0500. [DOI] [PubMed] [Google Scholar]
- 6.Lui VWY, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer discovery 2013;3(7):761–9 doi 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol 2012;30(8):777–82 doi 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jimeno A, Shirai K, Choi M, Laskin J, Kochenderfer M, Spira A, et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann Oncol 2015;26(3):556–61 doi 10.1093/annonc/mdu574. [DOI] [PubMed] [Google Scholar]
- 9.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(2):113–25 doi 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 10.Mazumdar T, Byers LA, Ng PK, Mills GB, Peng S, Diao L, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther 2014;13(11):2738–50 doi 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keysar SB, Astling DP, Anderson RT, Vogler BW, Bowles DW, Morton JJ, et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol Oncol 2013;7(4):776–90 doi 10.1016/j.molonc.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Wawrose JS, Gooding WE, Garraway LA, Lui VW, Peyser ND, et al. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: a rational approach to preclinical model selection. Mol Cancer Res 2014;12(4):571–82 doi 10.1158/1541-7786.MCR-13-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015;27(4):533–46 doi 10.1016/j.ccell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010;376(9737):235–44 doi 10.1016/s0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 15.Kalu NN, Mazumdar T, Peng S, Shen L, Sambandam V, Rao X, et al. Genomic characterization of human papillomavirus-positive and -negative human squamous cell cancer cell lines. Oncotarget 2017;8(49):86369–83 doi 10.18632/oncotarget.21174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin Cancer Res 2011;17(23):7248–64 doi 10.1158/1078-0432.CCR-11-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalu NN, Mazumdar T, Peng S, Tong P, Shen L, Wang J, et al. Comprehensive pharmacogenomic profiling of human papillomavirus-positive and -negative squamous cell carcinoma identifies sensitivity to aurora kinase inhibition in KMT2D mutants. Cancer Lett 2018;431:64–72 doi 10.1016/j.canlet.2018.05.029. [DOI] [PubMed] [Google Scholar]
- 18.Zhang M, Singh R, Peng S, Mazumdar T, Sambandam V, Shen L, et al. Mutations of the LIM protein AJUBA mediate sensitivity of head and neck squamous cell carcinoma to treatment with cell-cycle inhibitors. Cancer Lett 2017. doi 10.1016/j.canlet.2017.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myers JN, Holsinger FC, Jasser SA, Bekele BN, Fidler IJ. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clin Cancer Res 2002;8(1):293–8. [PubMed] [Google Scholar]
- 20.Munster P, Aggarwal R, Hong D, Schellens JH, van der Noll R, Specht J, et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin Cancer Res 2016;22(8):1932–9 doi 10.1158/1078-0432.ccr-15-1665. [DOI] [PubMed] [Google Scholar]
- 21.Bendell JC, Kurkjian C, Infante JR, Bauer TM, Burris HA 3rd, Greco FA, et al. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Invest New Drugs 2015;33(2):463–71 doi 10.1007/s10637-015-0218-6. [DOI] [PubMed] [Google Scholar]
- 22.Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, et al. BAY 80–6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Mol Cancer Ther 2013;12(11):2319–30 doi 10.1158/1535-7163.mct-12-0993-t. [DOI] [PubMed] [Google Scholar]
- 23.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30(3):282–90 doi 10.1200/jco.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 24.De Buck SS, Jakab A, Boehm M, Bootle D, Juric D, Quadt C, et al. Population pharmacokinetics and pharmacodynamics of BYL719, a phosphoinositide 3-kinase antagonist, in adult patients with advanced solid malignancies. British journal of clinical pharmacology 2014;78(3):543–55 doi 10.1111/bcp.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salphati L, Pang J, Plise EG, Lee LB, Olivero AG, Prior WW, et al. Preclinical assessment of the absorption and disposition of the phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor GDC-0980 and prediction of its pharmacokinetics and efficacy in human. Drug Metab Dispos 2012;40(9):1785–96 doi 10.1124/dmd.112.046052. [DOI] [PubMed] [Google Scholar]
- 26.Wicki A, Brown N, Xyrafas A, Bize V, Hawle H, Berardi S, et al. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13). Eur J Cancer 2018;96:6–16 doi 10.1016/j.ejca.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 27.Beaufils F, Cmiljanovic N, Cmiljanovic V, Bohnacker T, Melone A, Marone R, et al. 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class I PI3K/mTOR Inhibitor as Clinical Candidate in Oncology. J Med Chem 2017;60(17):7524–38 doi 10.1021/acs.jmedchem.7b00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrarotto R, Goonatilake R, Yoo SY, Tong P, Giri U, Peng S, et al. Epithelial-Mesenchymal Transition Predicts Polo-Like Kinase 1 Inhibitor-Mediated Apoptosis in Non-Small Cell Lung Cancer. Clin Cancer Res 2016;22(7):1674–86 doi 10.1158/1078-0432.CCR-14-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohnacker T, Prota AE, Beaufils F, Burke JE, Melone A, Inglis AJ, et al. Deconvolution of Buparlisib’s mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention. Nature communications 2017;8:14683 doi 10.1038/ncomms14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of gamma-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal 2014;26(1):149–61 doi 10.1016/j.cellsig.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer Cell 2016;30(2):229–42 doi 10.1016/j.ccell.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol 2011;7(11):787–93 doi 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zumsteg ZS, Morse N, Krigsfeld G, Gupta G, Higginson DS, Lee NY, et al. Taselisib (GDC-0032), a Potent beta-Sparing Small Molecule Inhibitor of PI3K, Radiosensitizes Head and Neck Squamous Carcinomas Containing Activating PIK3CA Alterations. Clin Cancer Res 2016;22(8):2009–19 doi 10.1158/1078-0432.ccr-15-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai Y, Dodhia S, Su GH. Dysregulations in the PI3K pathway and targeted therapies for head and neck squamous cell carcinoma. Oncotarget 2017;8(13):22203–17 doi 10.18632/oncotarget.14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, Valera JC, Zhao X, Chen Q, Silvio Gutkind J. mTOR co-targeting strategies for head and neck cancer therapy. Cancer Metastasis Rev 2017;36(3):491–502 doi 10.1007/s10555-017-9688-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011;19(1):58–71 doi 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Martin D, Molinolo AA, Patel V, Iglesias-Bartolome R, Degese MS, et al. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst 2014;106(9) doi 10.1093/jnci/dju215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamaguchi K, Iglesias-Bartolomé R, Wang Z, Callejas-Valera JL, Amornphimoltham P, Molinolo AA, et al. A synthetic-lethality RNAi screen reveals an ERK-mTOR co-targeting pro-apoptotic switch in PIK3CA(+) oral cancers. Oncotarget 2016;7(10):10696–709 doi 10.18632/oncotarget.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohan S, Vander Broek R, Shah S, Eytan DF, Pierce ML, Carlson SG, et al. MEK Inhibitor PD-0325901 Overcomes Resistance to PI3K/mTOR Inhibitor PF-5212384 and Potentiates Antitumor Effects in Human Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 2015;21(17):3946–56 doi 10.1158/1078-0432.CCR-14-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herzog A, Bian Y, Vander Broek R, Hall B, Coupar J, Cheng H, et al. PI3K/mTOR inhibitor PF-04691502 antitumor activity is enhanced with induction of wild-type TP53 in human xenograft and murine knockout models of head and neck cancer. Clin Cancer Res 2013;19(14):3808–19 doi 10.1158/1078-0432.CCR-12-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K, Nam KT, Cho SH, Gudapati P, Hwang Y, Park DS, et al. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J Exp Med 2012;209(4):713–28 doi 10.1084/jem.20111470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly AP, Finlay DK, Hinton HJ, Clarke RG, Fiorini E, Radtke F, et al. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J 2007;26(14):3441–50 doi 10.1038/sj.emboj.7601761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu YH, Chang TH, Huang YF, Chen CC, Chou CY. COL11A1 confers chemoresistance on ovarian cancer cells through the activation of Akt/c/EBPbeta pathway and PDK1 stabilization. Oncotarget 2015;6(27):23748–63 doi 10.18632/oncotarget.4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abad M, Hashimoto H, Zhou H, Morales MG, Chen B, Bassel-Duby R, et al. Notch Inhibition Enhances Cardiac Reprogramming by Increasing MEF2C Transcriptional Activity. Stem cell reports 2017;8(3):548–60 doi 10.1016/j.stemcr.2017.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song NJ, Yun UJ, Yang S, Wu C, Seo CR, Gwon AR, et al. Notch1 deficiency decreases hepatic lipid accumulation by induction of fatty acid oxidation. Scientific reports 2016;6:19377 doi 10.1038/srep19377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell 2002;10(4):721–33. [DOI] [PubMed] [Google Scholar]
- 47.Liu W, Zhang J, Gan X, Shen F, Yang X, Du N, et al. LGR5 promotes epithelial ovarian cancer proliferation, metastasis, and epithelial-mesenchymal transition through the Notch1 signaling pathway. Cancer medicine 2018. doi 10.1002/cam4.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB, et al. Targeting Notch signalling pathway of cancer stem cells. Stem cell investigation 2018;5:5 doi 10.21037/sci.2018.02.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 2015;518(7538):240–4 doi 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang M, Biswas S, Qin X, Gong W, Deng W, Yu H. Does Notch play a tumor suppressor role across diverse squamous cell carcinomas? Cancer medicine 2016;5(8):2048–60 doi 10.1002/cam4.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.