

Abstract
The TNF/TNFR pathway is known to influence survival of cancer patients. We hypothesized that single nucleotide polymorphisms (SNPs) in the TNF/TNFR pathway genes related to apoptosis are associated with non-small cell lung cancer (NSCLC) survival. We used 1,185 NSCLC patients in the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial and 984 NSCLC patients in the Harvard Lung Cancer Susceptibility Study as the discovery and validation datasets, respectively. We selected 6788 SNPs in 71 genes in the TNF/TNFR signaling pathway and extracted genotyping data from the PLCO dataset. We performed Cox proportional hazards regression analysis to evaluate associations between the identified SNPs and survival and validated the significant SNPs, which were further analyzed for their functional relevance. IKBKAP rs4978754 CT+TT and TNFRSF1B rs677844 TC+CC genotypes of two validated SNPs (rs4978754 C>T and rs677844 T>C, respectively) as well as their combined genotypes predicted a better overall survival (P=0.004, 0.002 and <0.001, respectively). These two validated SNPs were predicted by the RegulomeDB score to be potentially functional. IKBKAP mRNA expression levels were significantly higher, while TNFRSF1B mRNA expressions were significantly lower in lung cancer tissues than in adjacent normal tissues (P<0.001). The TCGA-based expression quantitative trait loci analysis showed that IKBKAP rs4978754 and TNFRSF1B rs677844 genotypes were significantly associated with their corresponding mRNA expression levels in lung cancer tissues in a recessive model (P=0.035 and 0.045, respectively). We identified two potentially functional SNPs (TNFRSF1B rs677844 T>C and IKBKAP rs4978754 C>T) to be associated with survival in NSCLC patients.
Keywords: non-small cell lung cancer, single nucleotide polymorphism, survival, TNF, TNFR
Introduction
Lung cancer is the leading cause of cancer-related deaths throughout the world, accounting for more than one million deaths annually worldwide. Non-small cell lung cancer (NSCLC) accounts for more than 80% of all lung cancer cases [1]. Since more than 50% of lung cancer cases are diagnosed at a late stage, the survival of most patients remains poor [2], hence, there is an urgent need to better predict which patients are likely to have a poor prognosis. Clinically, the known clinicopathological variables, such as age, sex, performance status, and most importantly tumor stage, are commonly used for predicting prognosis; however, the response of individuals is heterogeneous, suggesting that genetic factors also account for the variability in treatment response.
Several genome-wide association studies (GWASs) of survival identified a number of single nucleotide polymorphisms (SNPs) as susceptibility loci for lung cancer survival [3,4]. GWASs have a great capability of detecting genetic variants for certain complex diseases using the “20–50 most-significant SNPs” strategy; however, the nature of these hypothesis-free studies has obvious limitations in revealing interactions among SNPs from multiple genes. Consequently, a pathway-based approach has recently been proposed not only to jointly consider multiple variants in interacting genes but also to consider their potential biological functions that may be the mechanisms underlying the observed associations. This, hypothesis-based method evaluates the combined effects of genetic variants of genes involved in the same biological pathway, which reduces unnecessary multiple tests and thus greatly strengthens the predictive power [5]. Previous publications that utilized a pathway analysis approach suggest that some SNPs may be related to outcomes of patients with lung cancer [6,7].
Apoptosis, also called programmed cell death, is an essential mechanism of maintaining tissue homeostasis in the organism. Various conditions including cancer result from a deregulation of apoptosis [8]. Genetic variants in apoptosis-related genes have been reported as predictors of prognosis in NSCLC [9,10]. The extrinsic pathway of apoptosis is initiated from the combination of tumor necrosis factor (TNF) and the TNF receptor (TNFR), which induces the cascade of procaspase activation [11]. Some studies found that TNF mediated genes were implicated in lung cancer [12,13].
Nevertheless, the associations between genetic polymorphisms in the TNF/TNFR signaling genes and survival of NSCLC patients remain unclear. The aim of the present study is to identify genetic variants in the TNF/TNFR signaling pathway and determine their effect on survival in patients with lung cancer.
Methods
Discovery dataset
We received approval to use data from the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial, a large population-based randomized trial designed to evaluate the effectiveness of cancer screening and to investigate etiologic factors and early markers of cancers [14]. Participants in the PLCO trial were recruited at 10 centers in the United States between 1993 and 2001. Among this large study population, there were 1,185 Caucasian NSCLC patients with complete follow-up information and GWAS genotyping data, which have been made available for survival analysis. Blood samples were collected at the first screening visit for genomic DNA extraction and used for genotyping with Illumina HumanHap240Sv1.0, HumanHap300v1.1 and HumanHap550v3.0 (dbGaP accession: phs000093.v2.p2 and phs000336.v1.p1) [15,16]. The follow-up time was defined from lung cancer diagnosis to the last follow-up or time of death. Overall survival (OS) was the primary endpoint, and disease-specific survival (DSS) was also recorded. The study protocol was reviewed and approved by the institutional review board of the National Cancer Institute (NCI), and written informed consent was obtained from each participant.
Validation Dataset
After identifying representative significant SNPs in the PLCO database through single locus analysis, we conducted a validation analysis using GWAS data from the Harvard Lung Cancer Susceptibility (HLCS) Study, a population of 984 histology-confirmed Caucasian NSCLC patients from the Massachusetts General Hospital. Blood samples were collected at the time of diagnosis, and DNA was extracted by using the Auto Pure Large Sample Nucleic Acid Purification System (QIAGEN Company, Venlo, Limburg, Netherlands). Genotyping data were obtained using Illumina Humanhap610-Quad arrays, and imputation was performed using MaCH v1.0 based on the 1000 Genomes Project. Details of the participants in the Harvard HLCS study have been described previously [17]. Multivariate Cox regression analysis was also applied to estimate HRs. Only those significant SNPs discovered in the PLCO dataset and validated by the Harvard dataset were subjected to further functional validation.
Selection of candidate genes in the pathway
We searched candidate genes in the TNF/TNFR pathway from the Molecular Signatures Database (MsigDB), (http://www.broadinstitute.org/gsea/msigdb/search.jsp), a collection of annotated gene sets for enrichment analysis using ‘TNF or TNFR1 or TNFR2’ as keywords. Duplicate genes in the datasets, pseudogenes and genes in chromosome X were excluded. The selected genes and their ± 2 kb flanking regions were searched to cover the promoter regions for all the candidate genes.
Genotyping data extraction and imputation
We extracted genotyping data for all the common SNPs located in those selected candidate genes using PLINK 1.07. The quality control criteria of SNPs were: minor allele frequency (MAF) ≥ 0.05, genotyping call rate ≥ 95% and P-value for Hardy-Weinberg Equilibrium test ≥1×10-5. Then, we performed imputation with IMPUTE2 [18] according to the 1000 Genomes Project CEU (Northern Europeans from Utah) data (phase 1 release 3 March 2012) [19], and imputed SNPs with info score ≥ 0.8 were used in further analysis. The Manhattan plot of all of the candidate SNPs was generated by using Haploview software (v4.2) [20].
Statistical analysis
Multivariate Cox proportional hazards regression analysis with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy and surgery was carried out in the PLCO dataset to estimate the association between extracted SNPs and survival in an additive genetic model using the GenABEL package in R software [21]. Hazard ratios (HRs) and 95% confidence intervals (95% CIs) for each SNP were calculated. The false discovery rate (FDR) [22] method was first used for multiple testing corrections; however, because the vast majority of the SNPs in the current study were imputed and thus not independent as required by FDR or other correction methods, the false positive report probability (FPRP) method [23] was then applied to perform the multiple testing corrections with a strict cut-off of 0.10. We assigned a prior probability of 0.10 to detect an HR of 2.0 for an association with variant genotypes or minor alleles of the SNPs with P <0.05. The SNPs that were identified with a P-value <0.05 by Cox regression and passed the FPRP threshold were subjected to linkage disequilibrium (LD) analysis. Representative SNPs (r2 >0.8 in LD with other SNPs) identified using PLINK and Haploview software were used for further population validation using the HLCS GWAS dataset and functional validation.
We analyzed the associations of those SNPs identified in the PLCO dataset and validated in the Harvard dataset with OS of NSCLC using co-dominant, dominant and additive models in the PLCO trial. The associations between the combined protective genotypes of those significant SNPs and OS/DSS were also estimated in the PLCO dataset using SAS software.
We also performed stratified analysis to evaluate whether the combined effect of protective genotypes as defined by the genetic score on NSCLC OS/DSS was affected by clinical characteristics, including age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy and surgery. All statistical analyses were performed with SAS software (v9.4; SAS Institute, Cary, NC, USA), if not specified otherwise. HR (95% CI) and P-value were calculated, and P-value <0.05 was considered significant. Kaplan-Meier survival curves and log-ranks test were used to estimate the effects of protective genotypes on the cumulative probability of OS/DSS. Kaplan-Meier curves were plotted using GraphPad Prism 6.0 (GraphPad Software Inc, San Diego, CA, USA).
Functional annotation
We used an in silico approach, in which SNPinfo [24] (http://snpinfo.niehs.nih.gov), RegulomeDB [25] (http://regulomedb.org), and HaploReg v4.1 [26]
(http://archive.broadinstitute.org/mammals/haploreg/haploreg.php) were used to predict SNP-associated potential functions. Survival analyses relevant to the mRNA expression of the validated genes in lung tumor tissue were also performed using available online data (http://kmplot.com/analysis).
We also performed expression quantitative trait loci (eQTL) analysis of the validated SNPs by using expression data of lung cancer tissues from the Cancer Genome Atlas (TCGA) database (dbGaP Study Accession: phs000178.v1.p1) [27], obtained from the Broad TCGA GDAC site (http://gdac.broadinstitute.org). The paired differential expression analyses using expression data of both lung cancer tissues and adjacent normal lung tissues from the TCGA were also compared.
Additional eQTL analysis was performed, in which the correlations between the genotypes of those validated SNPs and their mRNA expression levels were investigated by using RNA sequencing data from lymphoblastoid cells derived from the 373 European descendants in the 1000 Genomes Project [28].
Results
Study population characteristics
Clinical characteristics of NSCLC patients from the PLCO trial have been previously described [29]. In total, the analysis included 1,185 patients, 698 men and 487 women, aged 55 to 74 with an average of 71 years. Smoking status, pack-years, histologic diagnosis, tumor stage and treatment method were recorded. There were 798 (67.3%) deaths during the follow-up with a median survival time of 23.77 months. There were 11 subjects with missing data, including one for pack years, two for tumor stage, and eight for chemotherapy/radiotherapy/surgery. The associations between demographics and clinical characteristics and OS in the PLCO trial are shown in Supplementary Table 1 [29].Overall, characteristics associated with improved survival were age ≤ 71 years, female sex, never smoking, having a lower stage and having had chemotherapy or surgery.
The identified SNPs obtained from the PLCO discovery analysis were further validated by the GWAS dataset from the HLCS study. After applying quality control, 984 histology-confirmed Caucasian patients remained in the HLCS study, who were older than 18 years old, with newly diagnosed, histologically confirmed primary NSCLC, as previously described [17].
Gene and SNPs extraction from the PLCO trial
The overall workflow of the present study is shown in Figure 1. Seventy-one candidate genes in the TNF/TNFR signaling pathway were chosen from MsigDB, and two genes located on chromosome X were removed (Supplementary Table 2). Extraction of genotyping data for these 69 genes and subsequent imputation resulted in 6788 SNPs (656 genotyped and 6132 imputed) in the PLCO dataset for survival analysis. The Manhattan plot shows the locations of all of the candidate SNPs (Supplementary Figure 1).
Figure 1.

Study workflow chart. (SNP, single nucleotide polymorphism; PLCO, Prostate, Lung, Colorectal and Ovarian cancer trial; FPRP, false-positive report probability; eQTL,expression quantitative trait loci; ROC, receiver operating characteristic.)
Survival association analyses of the PLCO dataset
In the single locus analysis using the PLCO dataset, 737 SNPs were significantly associated with NSCLC OS (P<0.05). Next, since the majority of the SNPs were imputed and not independent, FPRP were performed and 202 SNPs remained significant after the multiple testing correction. Thirty-three representative SNPs were selected after additional LD analysis (r2>0.8).
Population validation in the Harvard GWAS dataset and the combined analysis
The 33 SNPs identified in the PLCO dataset underwent validation analysis using the genotyping data from the Harvard GWAS dataset. Two validated SNPs, TNFRSF1B rs677844 and IKBKAP rs4978754, remained significantly associated with survival of NSCLC patients in both of the datasets. Specifically, the results showed for TNFRSF1B rs677844, HR=0.83 (95% CI 0.74–0.94, P=0.002) in the PLCO discovery dataset and HR=0.83 (95% CI 0.73–0.96, P=0.010) in the Harvard validation dataset, whereas, for IKBKAP rs4978754, HR=0.77 (95% CI 0.63–0.93, P=0.006) in the PLCO discovery dataset and HR=0.73 (95% CI 0.60–0.89, P=0.001) in the Harvard validation dataset (Supplementary Table 3). The analysis of these two validated SNPs, based on these two combined datasets, showed improved OS of NSCLC patients associated with the rs677844 C and rs4978754 T alleles (P=5.12×10−5 and 4.70×10−5, respectively).
Independent SNPs and further survival analyses with genetic models
We conducted a stepwise Cox regression analysis of selected clinical variables from the PLCO dataset and the two validated SNPs to examine whether these SNPs are independent predictors of survival. This analysis was restricted to the PLCO dataset because it had more detailed clinical variables, including age, sex, smoking status, tumor stage, tumor histology, chemotherapy, radiotherapy, surgery, and top four principal components. There were 10 observations with missing data for clinical variables (two for tumor stage and eight for chemotherapy/radiotherapy/surgery), and 12 observations with missing data for TNFRSF1B rs677844, leading to the removal of 22 observations; therefore, the final analysis included 1163 patients for the stepwise analysis. Both of the SNPs remained significant in the final multivariate model (Table 1).
Table 1.
Predictors of OS obtained from stepwise Cox regression analysis in the PLCO dataset
| Variables 1 | Category | Frequency 2 | HR (95%CI) | P |
|---|---|---|---|---|
| Age | Continuous | 1163 | 1.03 (1.02-1.05) | <0.001 |
| Sex | Male | 691 | 1.00 | |
| Female | 472 | 0.78 (0.67-0.90) | <0.001 | |
| Smoking status | Never | 113 | 1.00 | |
| Current | 416 | 1.65 (1.24-2.21) | 0.001 | |
| Former | 634 | 1.62 (1.23-2.13) | 0.001 | |
| Histology | Adenocarcinoma | 569 | 1.00 | |
| Squamous cell | 280 | 1.15 (0.96-1.39) | 0.138 | |
| others | 314 | 1.26 (1.06-1.50) | 0.008 | |
| Tumor stage | I-IIIA | 646 | 1.00 | |
| IIIB-IV | 517 | 2.83 (2.33-3.44) | <0.001 | |
| Chemotherapy | No | 630 | 1.00 | |
| Yes | 533 | 0.58 (0.49-0.69) | <0.001 | |
| Radiotherapy | No | 754 | 1.00 | |
| Yes | 409 | 0.96 (0.82-1.13) | 0.641 | |
| Surgery | No | 629 | 1.00 | |
| Yes | 534 | 0.21 (0.16-0.27) | <0.001 | |
| TNFRSF1B rs677844 T>C 3 | TT/TC/CC | 640/447/76 | 0.84 (0.75-0.94) | 0.003 |
| IKBKAP rs4978754 C>T 3 | CC/CT/TT | 975/178/10 | 0.78 (0.64-0.94) | 0.010 |
Abbreviations: OS = Overall survival; PLCO = Prostate, Lung, Colorectal and Ovarian cancer trial; HR = Hazard ratio; 95% CI = 95% Confidence interval.
Stepwise analysis included age, sex, smoking status, tumor stage, tumor histology, chemotherapy, radiotherapy, surgery, top four principal components and two SNPs (TNFRSF1B rs677844 and IKBKAP rs4978754 in an additive model).
Ten observations missing of clinical variables (two of tumor stage and eight of chemotherapy/radiotherapy/surgery). Twelve observations missing of TNFRSF1B rs677844; 1163 patients remained for the stepwise analysis.
The leftmost was used as the reference.
The survival analysis with different genetic models for each independent SNP was performed in the PLCO dataset. We found that, under an additive genetic model, TNFRSF1B rs677844 C and IKBKAP rs4978754 T variant alleles were associated with an decreased death risk of NSCLC, with a variant-allele attributed HR of 0.83 (95% CI=0.74–0.94, P=0.002) and 0.77 (95% CI=0.63–0.93, P=0.006), respectively, in the multivariate OS analysis. The univariate and multivariate Cox regression analyses with different genetic models (co-dominant/dominant/additive) of the two SNPs are presented in Table 2. According to these results, TNFRSF1B rs677844 TC+CC and IKBKAP rs4978754 CT+TT genotypes were found to predict a favorable survival in NSCLC patients in a dominant model. Alternatively, as shown in Supplementary Table 4, TNFRSF1B rs677844 TT and IKBKAP rs4978754 CC genotypes were significantly associated with a poor cancer-specific survival in the multivariate analysis (P<0.05 for both).
Table 2.
Associations of the two validated SNPs in the TNF/TNFR pathway with OS of NSCLC in the PLCO dataset
| OS Univariate analysis | OS Multivariate analysis 1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Frequency | Frequency | |||||||
| Genotype | All | Death (%) | HR (95% CI) | P | All | Death (%) | HR (95% CI) | P |
| TNFRSF1B rs677844 T > C | ||||||||
| n=1173 | n=1163 | |||||||
| TT | 647 | 449 (69.4) | 1.00 | 640 | 443 (69.2) | 1.00 | ||
| TC | 450 | 295 (65.6) | 0.86 (0.74-0.99) | 0.040 | 447 | 292 (65.3) | 0.81 (0.69-0.94) | 0.006 |
| CC | 76 | 48 (63.2) | 0.84 (0.62-1.13) | 0.236 | 76 | 48 (63.2) | 0.74 (0.54-0.99) | 0.046 |
| Trend | 0.037 | 0.002 | ||||||
| TC+CC | 526 | 343 (65.2) | 0.85 (0.74-0.98) | 0.028 | 523 | 340 (65.0) | 0.80 (0.69-0.92) | 0.002 |
| Genotype missing 2 | 12 | 12 | ||||||
| Phenotype missing 3 | N/A | 10 | ||||||
| IKBKAP rs4978754 C > T | ||||||||
| n=1185 | n=1175 | |||||||
| CC | 991 | 676 (68.2) | 1.00 | 984 | 669 (68.0) | 1.00 | ||
| CT | 184 | 117 (63.6) | 0.91 (0.75-1.10) | 0.330 | 181 | 115 (63.6) | 0.74 (0.60-0.90) | 0.003 |
| TT | 10 | 5 (50.0) | 0.58 (0.24-1.41) | 0.230 | 10 | 5 (50.0) | 1.03 (0.43-2.50) | 0.943 |
| Trend | 0.159 | 0.006 | ||||||
| CT+TT | 194 | 122 (62.9) | 0.89 (0.73-1.08) | 0.222 | 191 | 120 (62.8) | 0.74 (0.61-0.91) | 0.004 |
| Genotype missing 2 | 0 | 0 | ||||||
| Phenotype missing 3 | N/A | 10 | ||||||
Abbreviations: SNPs = Single nucleotide polymorphisms; OS = Overall survival; NSCLC = Non-small cell lung cancer; PLCO = Prostate, Lung, Colorectal and Ovarian cancer trial; HR = Hazard ratio; 95% CI = 95% Confidence interval; N/A = Not applicable.
Adjusted for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery, and top four principal components.
Twelve observations missing of TNFRSF1B rs677844.
Two observations missing of tumor stage and eight observations missing of chemotherapy/radiotherapy/surgery in the PLCO dataset.
Combined analyses of the two SNPs
As TNFRSF1B rs677844 TC+CC and IKBKAP rs4978754 CT+TT genotypes were associated with a favorable prognosis in NSCLC patients, we combined rs677844 TC+CC and rs4978754 CT+TT genotypes into a genetic score to assess their combined protective effect. The NSCLC patients were divided into three groups with a genetic score of zero, one and two. As shown in Table 3, the increase in per score unit was correlated with an increased OS after adjustment for age, sex, smoking, tumor stage, histology, chemotherapy, radiotherapy, surgery and the top four principal components (Ptrend<0.001). Next, all the patients were dichotomized into a low-protective genotypes group (score 0–1) and a high-protective genotype group (score 2), and the multivariate OS analysis showed that the latter group had a 0.53-fold decrease in risk of death (HR=0.53, 0.40–0.72; P<0.001). The results of univariate and multivariate DSS analysis of the combined protective genotypes were similar (Supplementary Table 5). These results are depicted graphically with Kaplan-Meier survival curves in Supplementary Figure 2.
Table 3.
Combined protective genotypes of the two validated SNPs and OS of NSCLC in PLCO Trial
| OS Univariate analysis |
OS Multivariate analysis 2 |
|||||||
|---|---|---|---|---|---|---|---|---|
| Frequency | Frequency | |||||||
| Genotype | All | Death (%) | HR (95% CI) | P | All | Death (%) | HR (95% CI) | P |
| NPG 1 | ||||||||
| n=1173 | n=1163 | |||||||
| 0 | 546 | 379 (69.4) | 1.00 | 541 | 374 (69.1) | 1.00 | ||
| 1 | 537 | 362 (67.4) | 0.93 (0.80-1.07) | 0.294 | 533 | 359 (67.4) | 0.88 (0.76-1.02) | 0.100 |
| 2 | 90 | 51 (56.7) | 0.68 (0.51-0.91) | 0.009 | 89 | 50 (56.2) | 0.50 (0.37-0.68) | <0.001 |
| Trend | 0.016 | <0.001 | ||||||
| 0-1 | 1083 | 741 (68.4) | 1.00 | 1074 | 733 (68.3) | 1.00 | ||
| 2 | 90 | 51 (56.7) | 0.71 (0.53-0.94) | 0.016 | 89 | 50 (56.2) | 0.53 (0.40-0.72) | <0.001 |
| Genotype missing 3 | 12 | 12 | ||||||
| Phenotype missing 4 | N/A | 10 | ||||||
Abbreviations: SNPs = Single nucleotide polymorphisms; OS = Overall survival; NSCLC = Non-small cell lung cancer; PLCO = Prostate, Lung, Colorectal and Ovarian cancer trial; NPG = Number of protective genotypes; HR, Hazard ratio; 95% CI = 95% Confidence interval; N/A = Not applicable.
Protective genotypes were IKBKAP rs4978754 CT+TT and TNFRSF1B rs677844 TC+CC.
Multivariate Cox hazards regression analyses were adjusted for age, sex, smoking, stage, histology, chemotherapy, radiotherapy, surgery, and top four principal components.
Twelve observations missing of TNFRSF1B rs677844.
Two observations missing of tumor stage and eight observations missing of chemotherapy/radiotherapy/surgery in the PLCO dataset.
eQTL analyses
We compared mRNA expression levels of TNFRSF1B and IKBKAP in lung cancer tissues with that in adjacent normal lung tissues, which were obtained in the TCGA dataset, and we found that TNFRSF1B mRNA expression levels in lung cancer tissues were significantly lower than that in normal tissues (P<0.001), while IKBKAP mRNA expression levels in lung cancer tissues were significantly higher than that in normal tissues (P<0.001) (Figure 2).
Figure 2.
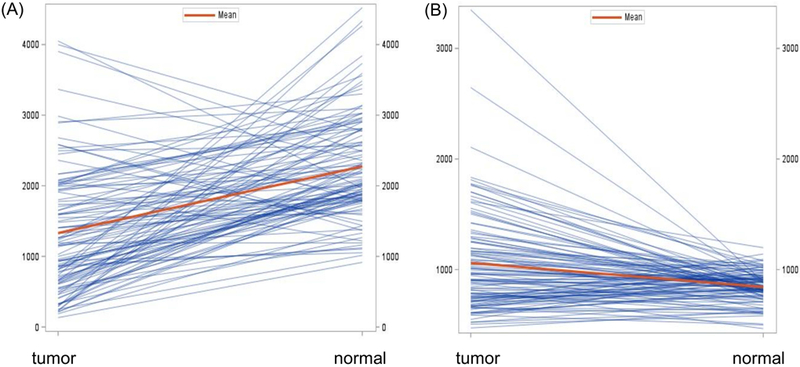
Comparison of mRNA expression levels of TNFRSF1B and IKBKAP between adjacent normal lung and lung cancer tissues in TCGA. (A) The TNFRSF1B mRNA expression levels in lung cancer tissues were significantly lower than that in normal tissues (P<0.001); (B) The IKBKAP mRNA expression levels in lung cancer tissues were significantly higher than that in normal tissues (P<0.001).
In addition, the eQTL analysis of rs677844 and rs4978754 in the TCGA database showed that the TNFRSF1B rs677844 genotypes were significantly associated with the corresponding mRNA expression levels in lung cancer tissues in a recessive model (Precessive=0.045), which was not found in other genotype models (Padditive=0.702). Similarly, IKBKAP rs4978754 genotypes were significantly associated with the corresponding mRNA expression levels in lung cancer tissues in a recessive model (Precessive=0.035), which was not found in other genotype models (Padditive=0.423) (Figure 3).
Figure 3.
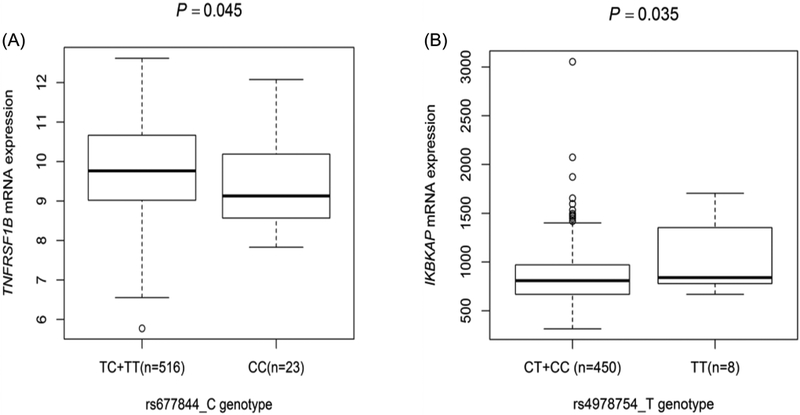
Correlation between IKBKAP rs4978754, TNFRSF1B rs677844 and their corresponding mRNA expression levels in the TCGA dataset. (A) TNFRSF1B rs677844 genotypes were significantly associated with the corresponding mRNA expression levels in lung cancer tissues in a recessive model (P=0.045); (B) IKBKAP rs4978754 genotypes were significantly associated with the corresponding mRNA expression levels in lung cancer tissues in a recessive model (P=0.035).
The eQTL analysis in the 1000 Genomes Project showed that TNFRSF1B rs677844 and IKBKAP rs4978754 genotypes were non-significantly associated with their levels of the corresponding mRNA expression in the lymphoblastoid cells in an additive model (Padditive = 0.155 and 0.693, respectively).
Functional validation analyses in silico
In a functional prediction analysis of the two validated SNPs, the RegulomeDB score was 4 for TNFRSF1B rs677844 and 5 for IKBKAP rs4978754. The results of SNPinfo, RegulomeDB and HaploReg v4.1 are shown in Supplementary Table 6.
Based on the online survival analysis software, high expression levels of TNFRSF1B and IKBKAP in lung cancer tissue were associated with a favorable survival (Supplementary Figure 3) in the TCGA dataset. According to the National Center for Biotechnology Information (NCBI) online data (www.ncbi.nlm.nih.gov/pubmed), these two SNPs are located in potential functional area of their corresponding genes Supplementary Figure 4 and Supplementary Figure 5.
Stratified analyses for the effect of combined genotypes on OS
Stratified analysis in the PLCO dataset was further performed to evaluate whether the combined effect of protective genotypes as defined by the genetic score on NSCLC OS was affected by other clinical covariates, including age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery, and principal components. Patients with a high-score risk protective genotypes exhibited significantly favorable survival in subgroups of current smoker, adenocarcinoma, IIIB-IV tumor stage, received radiotherapy and without surgery (all P<0.05). There was an evidence for heterogeneity among the three subgroups in tumor histology (P=0.012) with a multiplicative interaction (P=0.026). No heterogeneity was found in the other subgroups. The results are shown in Supplementary Table 7. The detail associations of subgroup analysis of tumor histology is shown in the Supplementary Table 8.
Discussion
Most of the earlier candidate-gene studies had investigated only a few variants at a time, and even the later “pathway-based” studies typically interrogated a relatively small number of variants in a limited number of genes due to the prohibitive cost of genotyping. The published GWAS datasets, however, provide a great opportunity for investigators to look into the massive genotyping data that may harbor information on significant variants that may have been missed by the original GWAS due to the stringent criteria imposed on multiple testing correction.
The TNF/TNFR signaling responds to cellular stress and inflammatory signals to activate pro-apoptotic pathways and cytokine cascades, which includes two receptors: TNFR1 and TNFR2. TNFR1 is well known to mediate the extrinsic apoptosis pathways through the activation of caspase-8 or caspase-10 and then the downstream caspases, which results in cellular apoptosis [30]. TNFR2 lacks a death domain and is incapable of mediating apoptosis directly, but can regulate cellular functions, such as extracellular matrix remodeling and growth via cooperation with other receptors [31]. Some studies have showed that TNF/TNFR also acts as a tumor promoter, contributing to tumor growth and metastasis in different kinds of cancer [32].
Considering the complicated role of the TNF/TNFR signaling in cancer, the goal of the present pathway-based study was to determine whether genetic variants in the pathway genes could predict survival of NSCLC patients. To achieve this, we investigated the associations between thousands of SNPs in the TNF/TNFR pathway genes with survival of NSCLC patients in the PLCO GWAS dataset and validated the findings in the Harvard GWAS dataset. As a result, we found that two SNPs in two genes, TNFRSF1B rs677844 T>C and IKBKAP rs4978754 C>T, were predictors of NSCLC survival. Those carrying rs677844 C allele or CC+TC genotypes and those carrying rs4978754 T allele or TT+CT genotypes had a significantly longer survival time.
In addition, the eQTL analysis also showed that TNFRSF1B rs677844 and IKBKAP rs4978754 genotypes were significantly associated with their corresponding mRNA expression levels in lung cancer tissues in a recessive model in the TCGA database. However, in the eQTL analysis of data obtained from the 1000 Genomes Project, we failed to find a correlation between these two SNPs and their mRNA expression levels in lymphoblastoid cells to support the observed associations as a possible underlying molecular mechanism. Therefore, the molecular mechanisms under the observed associations may be due to some biologic processes other than affecting the gene expression at the transcription level and need additional investigations.
rs677844 is located in TNFRSF1B named as TNF receptor superfamily member 1B on chromosome 1, encoding a member of the TNF-receptor superfamily, found on circulating T lymphocytes, which plays an important role in the extrinsic pathway of apoptosis [33]. In an early North American study of 225 NSCLC patients treated with chemoradiotherapy or radiotherapy alone, the authors investigated only five potentially functional polymorphisms of TNF-α and TNFRSF1B genes and found that the TNFRSF1B +676 GG genotype was associated with a significantly better OS of NSCLC [34]. In another Korean study of 382 NSCLC patients, among 32 SNPs in 21 apoptotic pathway genes genotyped, four SNPs in four apoptotic genes (TNFRSF1B rs1061624, BCL2 rs2279115, BIRC5 rs9904341, and CASP8 rs3769818) were found to be significantly associated with OS but not with response to chemotherapy [35]. These studies either had a small sample size, included an Asian patient population, or had no validation.
IKBKAP rs4978754 is located on chromosome 9, and the gene encodes the I kappa B kinase complex-associated protein, also known as elongator complex protein 1 (ELP1), an inhibitor of kappa light polypeptide enhancer in B cells [36]. IKBKAP has been suggested to be responsible for multiple cellular processes, including DNA demethylation, exocytosis, tRNA modification, actin organization, cell migration and survival [37]. It has been reported that this IKBKAP protein decreased proliferation of other cancers, such as prostate cancer [38], but few studies have focused on associations between IKBKAP SNPs and lung cancer survival. Therefore, the role of IKBKAP in lung cancer survival remains to be further investigated.
In the current study, we also found that TNFRSF1B mRNA expression levels in lung cancer tissues were significantly lower than that in normal lung tissues and that IKBKAP mRNA expression levels in lung cancer tissues were significantly higher than that in normal tissues. In addition, the mRNA expression levels of TNFRSF1B in lung carcinoma tissue were reported to be lower than that in normal lung tissues [39], which is consistent with the results of the present study. The Kaplan-Meier survival curve drawn based on the available online data demonstrated that low mRNA expression, compared with high expression, of TNFRSF1B in lung tumor tissue was associated with a poor outcome. However, the effect of TNFRSF1B on lung cancer survival remains unclear. As to IKBKAP, few published reports had explored its role in tumorigenesis and development of lung cancer. Taken together, the exact biological mechanism underlying the association between IKBKAP and survival remains to be investigated.
The present study has some limitations. First, since the genetic variant analysis can be influenced by different ethnic backgrounds, our findings may not be generalizable to other ethnic populations, because the only available GWAS datasets were from Caucasian populations. Second, the PLCO and Harvard GWAS datasets had different distributions of baseline characteristics, which partially explains why some significant top SNPs were not validated in the Harvard GWAS dataset. Third, we failed to find a correlation between the two significant SNPs and their mRNA expression levels in the eQTL analysis using the data derived from lymphoblastoid cells in the 1000 Genomes Project. Although the analysis in the TCGA database showed that TNFRSF1B rs677844 and IKBKAP rs4978754 genotypes were significantly associated with their corresponding mRNA expression levels in lung cancer tissues, the positive results were demonstrated only in a recessive model, and the number of minor homozygote carriers was rather small. Hence, the associations between the SNPs and their transcript levels need further investigations.
conclusion
In conclusion, we conducted a pathway-based genetic variants analysis in the PLCO and Harvard GWAS datasets. We identified two potential functional SNPs in two genes, rs677844 T>C (TNFRSF1B) and rs4978754 C>T (IKBKAP), that were associated with survival in NSCLC patients. Those carrying rs677844 CC+TC or rs4978754 TT+CT genotypes had a significantly longer survival time. Further experiments will be needed to explore functions of their encoded proteins that may provide the mechanisms underlying the observed associations.
Supplementary Material
Supplementary Figure 1. Manhattan plot of 6788 SNPs of the TNF/TNFR pathway genes in the PLCO trial. The statistical values across the autosomes for associations between 6788 SNPs and overall survival are plotted as -log10 P values. The blue horizontal line indicates P = 0.05 and the red line indicates FPRP = 0.10.
Supplementary Figure 2. Kaplan-Meier survival curves for NSCLC patients of two validated SNPs and combined protective genotypes in the PLCO trial.(A) Three groups of combined protective genotypes for overall survival; (B) Dichotomized groups of combined protective genotypes for overall survival; (C) Three groups of combined protective genotypes for disease-specific survival; (D) Dichotomized groups of combined protective genotypes for disease-specific survival.
Supplementary Figure 3. Kaplan-Meier analysis for patients with NSCLC by the two genes. Based on online survival analysis software (www.kmplot.com/analysis) (A) High TNFRSF1B expression (B) High IKBKAP expression were associated with better survival of NSCLC.
Supplementary Figure 4. rs677844 (the vertical yellow line indicated by the red arrow) is located in potential functional area of TNFRSF1B based NCBI online data.(Chr1:12,239,973–12,248,662).
Supplementary Figure 5. rs4978754 (the vertical yellow line indicated by the red arrow) is located in potential functional area of IKBKAP based on NCBI online data. (Chr9:111,643,321–111,650,840).
Acknowledgments
The authors thank the National Cancer Institute for access to data collected by the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by NCI. The author would also like to acknowledge dbGaP repository for providing the cancer genotyping datasets. The accession numbers for the datasets of lung cancer are phs000336.v1.p1 and phs000093.v2.p2. Funding support for the GWAS of Lung Cancer and Smoking was provided through the NIH Genes, Environment and Health Initiative [GEI] (Z01 CP 010200). The human subjects participating in the GWAS derive from The Environment and Genetics in Lung Cancer Etiology (EAGLE) case-control study and the Prostate, Lung Colon and Ovary Screening Trial and these studies are supported by intramural resources of the National Cancer Institute. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the Gene Environment Association Studies, GENEVA Coordinating Center (U01HG004446). Assistance with data cleaning was provided by the National Center for Biotechnology Information. Funding support for genotyping, which was performed at the Johns Hopkins University Center for Inherited Disease Research, was provided by the NIH GEI (U01HG004438). PLCO was also supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. The authors thank PLCO screening center investigators and staff, and the staff of Information Management Services Inc. and Westat Inc. Most importantly, we acknowledge trial participants for their contributions that made this study possible. The results published here are in whole or part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute The Cancer Genome Atlas (TCGA) Research Network can be found at “http://cancergenome.nih.gov“. The TCGA SNP data analyzed here are requested through dbGAP (accession#: phs000178.v1.p1).
Funding Sources: Qingyi Wei was supported by the V Foundation for Cancer Research grant (D2017–19), a start-up funds from Duke Cancer Institute, Duke University Medical Center and support from the Duke Cancer Institute as part of the P30 Cancer Center Support Grant (Grant ID: NIH CA014236). The Harvard Lung Cancer Susceptibility Study was supported by NIH grants 5U01CA209414, CA092824, CA074386, and CA090578 to David C. Christiani.
Abbreviations
- NSCLC
non-small cell lung cancer
- SNP
single nucleotide polymorphism
- PLCO
Prostate, Lung, Colorectal and Ovarian cancer trial
- GWASs
genome-wide association studies
- TNFR
tumor necrosis factor receptor
- TNF
tumor necrosis factor
- MSigDB
Molecular signatures database
- OS
overall survival
- DSS
disease specific survival
- FPRP
false positive report probability
- LD
linkage disequilibrium
- eQTL
expression quantitative trait loci
- TCGA
the Cancer Genome Atlas
Footnotes
Conflict of interest statement: None declared.
Supplementary material
Supplementary material can be found at online.
Ethics approval and consent to participates: The study protocol was reviewed and approved by the institutional review board of the National Cancer Institute (NCI), and written informed consent was obtained from each participant. The study was performed in accordance with the Declaration of Helsinki.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- 2.Basumallik N, Agarwal M. Cancer, Lung, Small Cell (Oat Cell) StatPearlsed. Treasure Island (FL), 2018. [Google Scholar]
- 3.Wang JZ, Xiang JJ, Wu LG, et al. A genetic variant in long non-coding RNA MALAT1 associated with survival outcome among patients with advanced lung adenocarcinoma: a survival cohort analysis. BMC Cancer 2017;17(1):167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boland JM, Maleszewski JJ, Wampfler JA, et al. Pulmonary Invasive Mucinous Adenocarcinoma and Mixed Invasive Mucinous/Non-Mucinous Adenocarcinoma- A Clinicopathological and Molecular Genetic Study with Survival Analysis. Hum Pathol 2018;71:8–19. [DOI] [PubMed] [Google Scholar]
- 5.Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet 2007;81:1278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song X, Wang S, Hong X, et al. Single nucleotide polymorphisms of nucleotide excision repair pathway are significantly associated with outcomes of platinum-based chemotherapy in lung cancer. Sci Rep 2017;7(1):11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sausville LN, Jones CC, Aldrich MC, Blot WJ, Pozzi A, Williams SM. Genetic variation in the eicosanoid pathway is associated with non-small-cell lung cancer (NSCLC) survival. PLoS One 2017;12(7):e0180471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elmore S Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim M, Kang HG, Lee SY, et al. Comprehensive analysis of DNA repair gene polymorphisms and survival in patients with early stage non-small-cell lung cancer. Cancer Sci 2010;101(11):2436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee EB, Jeon HS, Yoo SS, et al. Polymorphisms in apoptosis-related genes and survival of patients with early-stage non-smallcell lung cancer. Ann Surg Oncol 2010;17(10):2608–18. [DOI] [PubMed] [Google Scholar]
- 11.Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 2009;9(7):501–7. [DOI] [PubMed] [Google Scholar]
- 12.Wang B, Song N, Yu T, et al. Expression of tumor necrosis factor-alpha-mediated genes predicts recurrence-free survival in lung cancer. PLoS One 2014;9(12):e115945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei S, Niu J, Zhao H, et al. Association of a novel functional promoter variant (rs2075533 C>T) in the apoptosis gene TNFSF8 with risk of lung cancer-a finding from Texas lung cancer genome-wide association study. Carcinogenesis 2011;32(4):507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gohagan JK, Prorok PC, Hayes RB, Kramer BS. The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial of the National Cancer Institute: history, organization, and status. Control Clin Trials 2000;21(6 Suppl):251S–72S. [DOI] [PubMed] [Google Scholar]
- 15.Tryka KA, Hao L, Sturcke A, et al. NCBI’s database of genotypes and phenotypes: dbGaP. Nucleic acids research 2014;42:D975–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mailman MD, Feolo M, Jin Y, et al. The NCBI dbGaP database of genotypes and phenotypes. Nature genetics 2007;39(10):1181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhai R, Yu X, Wei Y, Su L, Christiani DC. Smoking and smoking cessation in relation to the development of co-existing non-small cell lung cancer with chronic obstructive pulmonary disease. Int J Cancer 2014;134:961–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarthy MI, Hirschhorn JN. Genome-wide association studies:potential next steps on a genetic journey. Hum Mol Genet 2008;17:R156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet 2013;93:779–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21(2):263–5. [DOI] [PubMed] [Google Scholar]
- 21.Aulchenko YS, Ripke S, Isaacs A, et al. GenABEL:an R library for genome-wide association analysis. Bioinformatics 2007;23:1294–6. [DOI] [PubMed] [Google Scholar]
- 22.Benjiamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 1995;57:289–300. [Google Scholar]
- 23.Wacholder S, Chanock S, Garcia-Closas M, EI Ghormli L, Rothmanet N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst 2004;96(6):434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu ZL, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res 2009;37:W600–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward LD, kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 2012;40:D930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lappalainen T, Sammeth M, Friedländer MR, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013;501(7468):506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Liu H, Ready NE, et al. Genetic variants in ABCG1 are associated with survival of nonsmall-cell lung cancer patients. Int J Cancer 2016;138(11):2592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kischkel FC, Lawrence DA, Tinel A, et al. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem 2001;276(49):46639–46. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Subleski J, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol 2008;180(10):6467–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka T, Imamura T, Yoneda M, et al. Enhancement of active MMP release and invasive activity of lymph node metastatic tongue cancer cells by elevated signaling via the TNF-α-TNFR1-NF-κB pathway and a possible involvement of angiopoietin-like 4 in lung metastasis. Int J Oncol 2016;49(4):1377–84. [DOI] [PubMed] [Google Scholar]
- 33.Waters JP, Pober JS, Bradley JR, et al. Tumor necrosis factor and cancer. J Pathol 2013;230(3):241–8. [DOI] [PubMed] [Google Scholar]
- 34.Guan X, Liao Z, Ma H, et al. TNFRSF1B +676 T>G polymorphism predicts survival of non-small cell lung cancer patients treated with chemoradiotherapy. BMC Cancer 2011;11:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SY, Kang HG, Yoo SS, et al. Polymorphisms in DNA repair and apoptosis-related genes and clinical outcomes of patients with non-small cell lung cancer treated with first-line paclitaxel-cisplatin chemotherapy. Lung Cancer 2013;82(2):330–9. [DOI] [PubMed] [Google Scholar]
- 36.Cheng WW, Tang CS, Gui HS, et al. Depletion of the IKBKAP ortholog in zebrafish leads to hirschsprung disease-like phenotype. World J Gastroenterol 2015;21(7):2040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheishvili D, Maayan C, Cohen-Kupiec R, et al. IKAP/Elp1 involvement in cytoskeleton regulation and implication for familial dysautonomia. Hum Mol Genet 2011;20:1585–94. [DOI] [PubMed] [Google Scholar]
- 38.Martinez HD, Hsiao JJ, Jasavala RJ, Hinkson IV, Eng JK, Wright ME. Androgen-sensitive microsomal signaling networks coupled to the proliferation and differentiation of human prostate cancer cells. Genes Cancer 2011;2(10):956–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharjee A, Richards WG, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 2001;98(24):13790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Manhattan plot of 6788 SNPs of the TNF/TNFR pathway genes in the PLCO trial. The statistical values across the autosomes for associations between 6788 SNPs and overall survival are plotted as -log10 P values. The blue horizontal line indicates P = 0.05 and the red line indicates FPRP = 0.10.
Supplementary Figure 2. Kaplan-Meier survival curves for NSCLC patients of two validated SNPs and combined protective genotypes in the PLCO trial.(A) Three groups of combined protective genotypes for overall survival; (B) Dichotomized groups of combined protective genotypes for overall survival; (C) Three groups of combined protective genotypes for disease-specific survival; (D) Dichotomized groups of combined protective genotypes for disease-specific survival.
Supplementary Figure 3. Kaplan-Meier analysis for patients with NSCLC by the two genes. Based on online survival analysis software (www.kmplot.com/analysis) (A) High TNFRSF1B expression (B) High IKBKAP expression were associated with better survival of NSCLC.
Supplementary Figure 4. rs677844 (the vertical yellow line indicated by the red arrow) is located in potential functional area of TNFRSF1B based NCBI online data.(Chr1:12,239,973–12,248,662).
Supplementary Figure 5. rs4978754 (the vertical yellow line indicated by the red arrow) is located in potential functional area of IKBKAP based on NCBI online data. (Chr9:111,643,321–111,650,840).