

Abstract
Purpose:
Human papillomavirus (HPV)-negative head and neck squamous cell carcinomas (HNSCC) commonly bear disruptive mutations in TP53, resulting in treatment resistance. In these patients, direct targeting of p53 has not been successful, but synthetic lethal approaches have promise. Although Aurora A kinase (AURKA) is overexpressed and an oncogenic driver, its inhibition has only modest clinical effects in HPV-negative HNSCC. We explored a novel combination of AURKA and WEE1 inhibition to overcome intrinsic resistance to AURKA inhibition.
Experimental Design:
AURKA protein expression was determined by fluorescence-based automated quantitative analysis of patient specimens and correlated with survival. We evaluated treatment with the AURKA inhibitor alisertib (MLN8237) and the WEE1 inhibitor adavosertib (AZD1775), alone or in combination, using in vitro and in vivo HNSCC models.
Results:
Elevated nuclear AURKA correlated with worse survival among p16(−) HNSCC patients. Alisertib caused spindle defects, G2/M arrest and inhibitory CDK1 phosphorylation, and cytostasis in TP53 mutant HNSCC FaDu and UNC7 cells. Addition of adavosertib to alisertib instead triggered mitotic entry and mitotic catastrophe. Moreover, in FaDu and Detroit 562 xenografts, this combination demonstrated synergistic effects on tumor growth and extended overall survival compared to either vehicle or single agent treatment.
Conclusions:
Combinatorial treatment with adavosertib and alisertib leads to synergistic antitumor effects in in vitro and in vivo HNSCC models. These findings suggest a novel rational combination, providing a promising therapeutic avenue for TP53-mutated cancers.
Keywords: WEE1, Aurora Kinase A, alisertib, adavosertib, HNSCC
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) affects more than a half million people annually worldwide (1). For HNSCC cases not associated with human papillomavirus (HPV), about 50% of patients with locally advanced disease and almost all with distant metastases will succumb to this cancer—despite morbid treatments including cisplatin and irradiation. Targeted agents have shown limited single-agent activity in a cancer whose genomic profile is dominated by mutations in tumor suppressor genes (2) such as TP53-the most commonly mutated gene in HPV-negative (–) HNSCC. Cells with mutations in TP53 evade cisplatin-induced cellular senescence and initiate G2-M mitotic arrest, enabling DNA repair. Although direct restoration of p53 function with clinically translatable methods has not been achieved, synthetic lethal approaches have promise in this subset of HNSCC. High throughput screens point to specific signaling intermediaries as possible candidates for this approach. We have identified Aurora kinase A (AURKA) and WEE1 as two kinases of potential value for co-inhibition in HNSCC (3–5).
Aurora Kinases are a family of three serine-threonine kinases (AURKA, AURKB, and AURKC) important for cell cycle regulation. The centrosomal AURKA has pleotropic roles in centrosome maturation, mitotic entry, spindle assembly, and cytokinesis (6–8). AURKA is negatively regulated by p53 (9). Consequently, AURKA is upregulated in the majority of HPV(−) TP53 mutant HNSCC (4), and correlates with poor prognosis (4, 10) and cisplatin resistance (11). The AURKA inhibitor, alisertib (MLN8237) has a 9% monotherapy response rate in treatment-refractory HNSCC, with responses occurring in HPV(−) disease (12–14). At present, there are no validated biomarkers for alisertib sensitivity, and mechanisms of resistance to AURKA inhibition in HNSCC are poorly understood.
To potentiate AURKA inhibition and optimize synthetic lethal approaches for HNSCC therapy, we considered the role of AURKA in regulating mitotic entry through promotion of CDK1/cyclin B complex activation, an essential step for mitotic entry. CDK1 activation depends on the removal of an inhibitory phosphorylation at tyrosine 15 (Y15), which is mediated by the CDC25 family phosphatases. Activated AURKA levels rise at the end of G2, and are required for CDK1 co-localization to the centrosome (15). AURKA phosphorylation of CDC25b activates its phosphatase activity (16). In parallel, AURKA activates the PLK1 kinase via direct phosphorylation (17); PLK1, in turn, also phosphorylates and activates the CDC25 phosphatases (18), and importantly, phosphorylates and inhibits WEE1, the kinase responsible for introducing the inhibitory CDK1 phosphorylation (19). Together, these events contribute to dephosphorylation of CDK1 and full CDK1/cyclin B activation. Under conditions of AURKA overexpression, cells are characterized by amplified centrosomes and multipolar spindles, genomic instability due to failure to resolve cytokinesis, and activation of multiple pro-oncogenic signaling pathways due to anomalous AURKA phosphorylation of numerous cytoplasmic and nuclear substrates (20). AURKA inhibition or loss also causes characteristic spindle defects, including asymmetric or monopolar spindles, and typically leads to cell cycle arrest at the G2/M transition or in early M phase (20).
WEE1 is upregulated in the setting of DNA damage. It prolongs S phase, phosphorylates Histone H2B to terminate histone synthesis (21), and delays G2/M transition to allow DNA repair (22). For these reasons, WEE1 has been considered as a distinct therapeutic target, with the agent adavosertib now advancing through clinical trials (23–25). Both pre-clinical and clinical data show that WEE1 inhibition leads to DNA damage and accelerated mitotic entry (23, 26–28). Given that AURKA inhibition causes spindle assembly defects but also restricts mitotic entry, we hypothesized that the dual inhibition of AURKA and WEE1 would lead cells to enter mitosis with disordered spindles, generating a more lethal phenotype than results from either inhibitor alone. In this study, we show combination of alisertib with adavosertib causes a striking increase in mitotic catastrophe, and potently limits the growth of HNSCC cells in vitro and xenograft tumors in vivo.
MATERIALS AND METHODS
Fluorescence-based automated quantitative analysis (AQUA) staining.
Eighty-nine HNSCC specimens from the Fox Chase Cancer Center tissue bank were assessed for AURKA expression profile by AQUA staining with IRB approval (29, 30). Tissue microarrays (TMA) were created and 5-µm sections deparaffinized. Heat-induced epitope retrieval was performed in EDTA buffer (pH 9.0). Endogenous peroxidase was blocked with 3% hydrogen peroxidase in methanol. Background Sniper (Biocare Medical, BS966) was used to block nonspecific reactions. Slides were then incubated with primary antibodies anti-AURKA at 1:1000 (Bethyl Laboratories, IHC-00062) and anti-phosphoAURKA at threonine 288 (pAURKA) at 1:1000 (Bethyl Laboratories, IHC-00067) and anti-pan-cytokeratin (DAKO, z0622) diluted in Da Vinci Green (Biocare Medical, PD900) at 4 °C for overnight. Slides were incubated with a secondary antibody to pan-cytokeratin conjugated with Alexa Fluor 555 (Invitrogen). Primary antibodies against AURKA and pAURKA were targeted with Envision reagents (DAKO). Target amplification and visualization were accomplished using a Cy-5-tyramide signal amplification system (TSA; PerkinElmer, AT705A). 4′,6-diamidino-2-phenylindole (DAPI)-containing ProLong Gold mounting medium (Invitrogen) was used to visualize nucleus and preserve signals. For AQUA analysis, high-resolution monochromatic digital images were captured with an automated PM-2000 microscope (Genoptix/Novartis). Images were visually inspected and cropped for unfavorable factors (poor focus, debris). Samples with <5% tumor per field were excluded from analysis. Areas of tumor were distinguished from stromal elements by creating a mask from the cytokeratin signal. The DAPI signal within this mask was then used to identify tumor nuclei. The pAURKA and AURKA signals (AQUA score) were scored on a normalized scale expressed as pixel intensity divided by target area, for the tumor mask or tumor nuclei respectively. Scores for duplicate cores were averaged to obtain a mean score/tumor.
p16 determination.
Immunohistochemical (IHC) staining for p16 is a widely accepted surrogate for HPV status, as HPV E7 oncoprotein fosters Rb degradation, reducing negative regulation of p16 (31). p16 status was evaluated by IHC analysis using the CINtec Histology Kit (Ventana Medical Systems), according to manufacturer’s instructions. Specimens were deemed p16(+) if strong and diffuse immunostaining was present in at least 70 percent of tumor cells.
Cell culture and chemical compounds.
Human HNSCC TP53 mutation-bearing cell lines were studied. FaDu, Detroit 562 and SCC-9 cell lines were purchased from the American Type Culture Collection (ATCC); the UNC7 is a patient-derived cancer cell line. A normal human tracheobronchial epithelial cell line (NHTBE) was purchased from Lonza. FaDu and Detroit 562 cells were maintained in EMEM media (ATCC) and SCC-9 and UNC7 cells in DMEM/F12 media supplemented with 0.2 µg/mL hydrocortisone (Millipore-Sigma, H0135). All media were supplemented with 10% fetal bovine serum and 1% Antibiotic-Antimycotic (Invitrogen). NHTBE cells were maintained in bronchial epithelial cell growth medium (BEGM) supplemented with BEGM bulletKit (Lonza, CC-3170). Three-dimensional organotypic air-liquid interface was utilized for NHTBE cell culture, as previously described (32). All cell lines were cultured under standard tissue culture conditions (5% CO2 at 37 °C) within less than 8 passages following resuscitation and regularly tested for mycoplasma using a MycoAlert mycoplasma detection kit (Lonza). UNC7 cells were authenticated using STR DNA profiling (Genewiz and the Yale Cell Line Authentication Service). The WEE1 inhibitor (adavosertib) and AURKA inhibitor (alisertib) were purchased from Selleck Chemicals, and dissolved in dimethyl sulfoxide (DMSO) for in vitro experiments.
Whole exome sequencing of UNC7 cells.
UNC7 cells had been previously described as TP53 wild type. We undertook whole exome sequencing (WES) to confirm this; WES was performed by the Yale Center for Genome Analysis as previously described (33). Fastq files from targeted sequence capture were processed in an exome analysis pipeline. Reads were aligned with human genome reference hg19, specifying target regions from a BED file that corresponds to the IDT xGen capture kit. PCR duplicates were removed using Picard tools (v1.118) and then reads were analyzed using GATK (v.3.2–2) for indel calling and realignment, quality recalibration, and variant calling. The resulting VCF file of variants was annotated using vcf2maf (v1.6.13, https://github.com/mskcc/vcf2maf/), filtering common variants based on presence in the ExAC database and 1000Genomes project. Sequences have been submitted to the NCBI Sequence Read Archive (SRA), accession number SRP132099.
Cell viability, soft agar and oncosphere formation assays.
Cell viability and soft agar assays were performed as previously described (32). Briefly, cells were plated in 96-well plates and exposed to the indicated doses of adavosertib (dose range, 50–12,500 nmol/L) and alisertib (dose range, 10–10,000 nmol/L) combination for three days. Cell viability was assessed with the CellTiter-Glo Luminescent Cell Viability Assay (Promega). For soft agar assay, cells were resuspended with 0.4% agarose-containing medium in 6-well plates contained bottom agarose and allowed to grow with exposure to the indicated drug(s). Colonies were stained with p-iodonitrotetrazolium violet (Sigma-Aldrich) to select live colonies, followed by analysis of the stained colonies using Image J software (NIH, USA). For oncosphere formation assay, cells were plated as single cell suspensions in serum-free media supplemented with 50 μg/ml Insulin (Millipore-Sigma), 20 μg/ml EGF (PeproTech), 10 μg/ml basic FGF (PeproTech), 0.4% BSA (Millipore-Sigma), N-2 Plus Media Supplement (Thermo Fisher Scientific), B-27 Supplement (Thermo Fisher Scientific) and 1% Antibiotic-Antimycotic in an ultra-low attachment plate (Fisher Scientific) for 7–9 days. The threshold for defining a colony or oncosphere was 10 cells, as indicated at >50 pixels in Image J or >100 μm on light microscopy.
Flow cytometry assays for cell cycle and apoptosis.
FaDu and UNC7 human HNSCC cells were treated with adavosertib (500 nM) and/or alisertib (250 nM) for 8 or 24 hours and stained with PI/RNase staining buffer (BD Biosciences). Cell cycle distribution was acquired by flow cytometry. For apoptosis assays, FaDu and UNC7 cells were exposed to advaosertib and/or alisertib for 24 hours and followed by staining with Annexin V/PI staining buffer (BD Biosciences). Stained cells were assessed on the BD LSRII Flow cytometer and analyzed with FlowJo software (FlowJo LLC).
In vivo studies.
Six-week-old female athymic nude mice (Jackson laboratory) were subcutaneously injected with 1×106 FaDu (n=8–10 each group) or 3×106 Detroit 562 (n=6 each group) HNSCC cells into the bilateral dorsal flanks. Treatments began at tumor size 200 to 350 mm3 and 150 to 200 mm3 for FaDu and Detroit 562 xenografts, respectively. Tumor-bearing FaDu-xenografted mice received one of the following treatments: 1) vehicle; 2) adavosertib (30 mg/kg, daily, p.o.); 3) adavosertib (90 mg/kg, daily, p.o.); 4) alisertib (30 mg/kg, daily, p.o.); 5) combination adavosertib (30 mg/kg) + alisertib (30 mg/kg, daily, p.o.); or 6) combination adavosertib (90 mg/kg) + alisertib (30 mg/kg, daily, p.o.), for 21 days, whereas, the 90 mg/kg adavosertib dose level was omitted in the Detroit 562 xenografted mice. Tumor volume was calculated as (l × w2) / 2, where l and w refer to the largest and smallest perpendicular dimensions at each measurement. Tumors were monitored daily and mice were euthanized when tumor volume greater than 1,000 mm3 or tumor ulceration was observed, or 26 days after initial dosing, whichever came first. Tumor regression, median tumor volume, and treatment tolerability were also considered. Tumor weight was measured at the endpoint of study. All animals were housed in the Yale University Animal Facility under the guidelines of the Institutional Animal Care and Use Committee (IACUC) and all animal studies were approved by Yale IACUC (#2017–11464).
Immunohistochemistry, immunofluorescence and immunoblotting.
Tumor tissues were fixed with 10% buffered formalin (Fisher Scientific) at 4 °C for overnight. Fixed tissues were embedded with paraffin and sectioned by Yale Pathology Tissue Services. For IHC staining, tissue sections were deparaffinized, rehydrated and subjected to high temperature antigen retrieval in 0.01 M citrate buffer, pH 6.0. Tissues were blocked with Dual Endogenenous Enzyme Block (Agilent-Dako) for 10 min, and followed by incubation with 1% Bovine Serum Albumin (BSA) in PBS/0.1 % Tween-20 for 30 min. Primary antibodies against Ki-67 (Cell Signaling Technology) and cleaved Caspase-3 (Abcam, ab4051) were used for staining following dilution in SignalStain® Antibody Diluent. HRP-conjugated SignalStain® Boost secondary antibodies (Cell Signaling Technology) were used, and followed by DAB+ Chromogen (Agilent-Dako) staining. Immunofluorescence staining and immunoblotting were performed as previously described (32). The following antibodies for immunoblotting and/or immunofluorescent staining were purchased from Cell Signaling Technology: anti-phospho-CDK1 (Y15; #4539), anti-CDK1 (#9116), anti-phospho-AURKA (T288; #3097), anti-AURKA (#14475), anti-phospho-Histone H3 (S10; #3377), anti-phospho-WEE1 (S642; #4910), anti-WEE1 (13084), anti-cleaved PARP (D214; #5625) and anti-γH2A.X (S139; #9718). Anti-β-actin (A2228) antibody was purchased from Millipore-Sigma. For phospho-CDK1 (Y15; pCDK1) staining, tissues were deparaffinized, rehydrated and followed by EDTA-mediated antigen retrieval. Slides were incubated with antibodies against both anti-pCDK1 and anti-cytokeratin (Dako, M3515) simultaneously at 4 °C for overnight. Tissues were then incubated with anti-rabbit Envision system-HRP (Dako, K4003) and anti-mouse secondary antibody conjugated with Alexa Fluor 546 (Invitrogen, A11010) for one hour at room temperature, followed by incubation with Cy5-tyramide signal amplification system (Perkin Elmer, FP1117) and DAPI. Using AQUA method, quantitative immunofluorescence (QIF) score of pCDK1 intensity in tumor was calculated by dividing the target pCDK1 pixel intensities by area in the tumor compartment as defined by cytokeratin expression. Scores were normalized to the exposure time and bit depth at which images were captured, allowing comparison of scores collected at different exposure times. Fields of view (FOV) were visually evaluated and were systematically excluded for staining or imaging artifacts, extensive necrosis, or less than 5% tumor area. Given that the core of the tumors presented extensive necrosis and that pCDK1 expression was enriched in the leading edge, only FOVs in the periphery of each tumor were included in the analysis. AQUA scores of pCDK1 in nuclei in tumor were used for the analysis. A heat-map of pCDK1 in nuclei in tumor was obtained by creating an analysis profile of each whole tissue section using AQUAnalysis software v. 3.1.2.1 (Genoptix Medical Laboratory)
Calculation of drug parameters.
For single-agent analysis, we estimated the IC50 using GraphPad Prism 7 with multiple dose-response models. For drug combinations, we applied the Chou-Talalay method to determine combination index (CI); additive effect (CI = 1), synergism (CI < 1), and antagonism (CI > 1) (34).
Statistical analysis.
Plots and bar graphs depict the mean and standard error of the mean (SEM) as calculated by Student’s t test. Differences between treatment groups were determined by one- or two-way ANOVA followed by Bonferroni’s post-test. A Kaplan-Meier plot was generated to show survival and significance was assessed by log-rank (Mantel-Cox) test. For pCDK1 QIF analysis, differences between treatment groups were determined by one-way ANOVA followed by Tukey’s multiple comparisons test. Analyses were conducted using GraphPad Prism 7 and differences were considered to be significant at P < 0.05.
RESULTS
Elevated nuclear AURKA expression is associated with poor survival in HNSCC.
To investigate if expression of AURKA and/or pAURKA (T288) conferred higher clinical aggressiveness in HNSCC patients, we conducted AQUA™ automated immunofluorescence staining on a total of 89 clinically annotated surgical HNSCC tumor specimens for which p16 status was available; of these, 20 were p16-positive and 69 were p16-negative (Table 1). Given the oncogenic role of nuclear AURKA (35), we further determined both nuclear and cytoplasmic AURKA and pAURKA expression, dichotomized at the median. Analyses were conducted in the total population of 89 patients, as well as in the 23 patients treated with surgery only, and adjusted for T stage in the 75 patients for whom T stage was known. Notably, pAURKA expression was predominantly nuclear (cytoplasmic median score 3021.82, range 636.19–10,480.58; nuclear median score 5909.15, range 1031.1–12953.89). AURKA was detected in the nuclear compartment and at centrosomes, along mitotic spindle microtubules and in the cytoplasm of mitotically proliferating cells (cytoplasmic median score 1375.03, range 415.09–4,122.53; nuclear median score 1816.77, range 416.72–4,776.05). Among 75 patients with available T stage information, higher pathologic T stage is associated with lower pAURKA AQUA score in the nuclear (P = 0.0008), and cytoplasmic compartments (P = 0.0005), but did not correlate with N stage. Cytoplasmic AURKA and pAURKA expression did not correlate with overall survival (OS). For the total population, higher AURKA expression correlated with worse OS (P = 0.0488) and among the 23 patients treated with surgery only, who did not receive post-operative therapy, higher nuclear pAURKA expression was also significantly associated with worse OS (P = 0.0133; Fig. 1A–C). The median OS of 36 months in HNSCC patients with high nuclear AURKA expression tended to be lower compared to 92 months among low expressers (Fig. 1D). This effect was largely driven by the correlation of nuclear AURKA expression with outcome among p16(−) patients. Median OS in 69 p16(−) patients was 93.6 vs. 35.9 months (P = 0.08) for those with low and high nuclear AURKA expression, respectively, while there was no difference for high compared with low nuclear AURKA expression among p16(+) patients (53.3 months vs. 57.2 months, P = 0.679; Fig. 1D).
Table 1.
Characteristics of study population
| Mean | SD | Freq. | % | ||
|---|---|---|---|---|---|
| Age | 62.0696 | 12.9403 | |||
| Gender | Female | 30 | 33.71 | ||
| Male | 59 | 66.29 | |||
| Race | White | 79 | 88.76 | ||
| Black | 7 | 7.87 | |||
| Other | 3 | 3.37 | |||
| Registry general stage | Distant Metastasis | 13 | 14.61 | ||
| Localized | 19 | 21.35 | |||
| Regional both direct extension and lymph | 19 | 21.35 | |||
| Regional by direct extension | 20 | 22.47 | |||
| Regional to lymph nodes | 18 | 20.22 | |||
| Pathologic T stage | 1 | 11 | 12.36 | ||
| 2 | 20 | 22.47 | |||
| 3 | 13 | 14.61 | |||
| 4 | 31 | 34.83 | |||
| N/A | 14 | 15.73 | |||
| Pathologic Nodal stage | 0 | 32 | 35.96 | ||
| 1 | 11 | 12.36 | |||
| 2 | 32 | 35.96 | |||
| N/A | 14 | 15.73 | |||
| Registry grade | Moderately differentiated | 48 | 53.93 | ||
| Poorly differentiated | 28 | 31.46 | |||
| Undifferentiated | 5 | 5.62 | |||
| Well differentiated | 8 | 8.99 |
N/A: Not available; SD: Standard deviation; Freq.: Frequency
Figure 1. Overexpressed nuclear AURKA is associated with a poor overall survival of HPV-negative HNSCC patients.

Nuclear expression of pAURKA/AURKA was assessed in 89 HNSCC surgical specimens using AQUA analysis. AURKA expression was classified into high or low AURKA expression based on relation to median AURKA levels. (A) Kaplan Meier curves were generated for all patients (n = 89) regarding AURKA level in nucleus regardless HPV positivity (HR 1.9, 95% CI 1.05–3.46). (B) Kaplan Meier curves were generated for 23 patients who got surgery only regarding pAURKA status after adjustment for T stage. (C) Representative AQUA images of pAURKA (top) and AURKA (bottom) in HNSCC surgical specimens. Scale bars: white: 100 µm, yellow: 10 µm. (D) Kaplan Meier curve for all patients (n = 89) who were grouped into 4 categories: p16-/Low AURKA; p16+/Low AURKA; p16−/High AURKA; p16+/High AURKA- classified by HPV positivity (p16) and AURKA status (HR 1.84, 95% CI 0.89–3.79).
Whole exome sequencing of UNC7 cells.
UNC7 cells had previously been described as TP53 wild type, but were not genomically well-characterized. We thus undertook whole exome sequencing to determine variants in both TP53 alleles in UNC7 cells. Interestingly, we identified two mutations of TP53 including a frameshift mutation by deletion in one allele and a missense mutation in the other (Chr17:7572991. TP53. c.1118delA (p.K373Rfs*49) and Chr17:7578403.TP53.c.527G>A (p.C176Y); Supplementary Fig. S1); of note, each is predicted as a deleterious mutation (36).
Concomitant inhibition of AURKA and WEE1 synergistically suppresses cell growth and survival of HNSCC cells.
We first evaluated the effect of the AURKA inhibitor, alisertib (MLN8237) used alone or in combination with the WEE1 inhibitor, adavosertib (AZD1775) in the TP53 mutant FaDu, Detroit 562, SCC-9 and UNC7 human HNSCC cell lines. Of note, all cell lines have previously been identified as HPV(−) and p16-inactive (37–39). IC50 values for alisertib and adavosertib were established by assessing cell viability after 3 days treatment. In Fig. 2A, FaDu, Detroit 562 and SCC-9 cells were slightly more sensitive to both adavosertib (FaDu: IC50 = 533.9 nM; Detroit 562: IC50 = 407 nM; SCC-9: IC50 = 487.7 nM) and alisertib (FaDu: IC50 = 1,274 nM; Detroit 562: IC50 = 92.38 nM; SCC-9: IC50 = 1,052 nM) compared to UNC7 cells (adavosertib: IC50 = 728.4 nM; alisertib IC50 = 2,942 nM). Synergism was determined by the CI calculated using the Chou-Talalay method (34). Strikingly, we observed that combination treatment with adavosertib and alisertib synergistically suppressed cell viability of all four FaDu (CI = 0.492), Detroit 562 (CI = 0.705), SCC-9 (CI = 0.683) and UNC7 cell lines (CI = 0.765; Fig. 2A).
Figure 2. Concomitant inhibition of AURKA and WEE1 synergistically suppresses cell growth and survival of HNSCC cells.

(A) Dose-response curve of human HNSCC cell lines, FaDu, Detroit 562, SCC-9 and UNC7 treated with adavosertib (AZD1775, orange), alisertib (MLN8237, blue), and adavosertib + alisertib (red), from a 3-day CellTiter-Glo assay. Cell viability was normalized to vehicle-treated control cells. (B) FaDu cells were plated with low density of agarose (0.4 %) and treated with the indicated drugs in anchorage-independent growth condition. A colony was defined as anything containing more than 10 cells, as indicated >50 pixels in Image J. Representative image of colony formation in soft agar. Scale bar: 100 µm. (C and D) FaDu and UNC7 cells were cultured with the conditioned media for oncosphere formation assay for 7–9 days and then formed spheres were transferred to standard tissue culture condition for additional 2 days. (C) Representative image of oncospheres (top) and re-differentiated spheres (bottom). Scale bar: 100 µm. (D) The number of oncospheres were counted using light microscopy. (E) Representative image of NHTBE cells morphology captured at 3 days after exposure to vehicle, adavosertib (500 nM), alisertib (250 nM), or adavosertib + alisertib in organotypic air-liquid interface culture condition. Scale bar: 100 µm. (F) Dose-response curve of NHTBE cells treated with adavosertib (orange), alisertib (blue), and adavosertib + alisertib (red), from a 3-day CellTiter-Glo assay. Cell viability was normalized to vehicle-treated control cells. Graphs depict Mean ± SEM from triplicated and/or three times repeated independent experiments. Statistical significance was assessed by Student’s t-test (ns: not significant; *: P < 0.05; **: P < 0.005).
Using maximally synergistic doses of adavosertib and alisertib, we assessed the inhibitory effect of the combination treatment for anchorage-independent growth of the FaDu and UNC7 cells in soft agar assay. We found that UNC7 cells unable to form colonies in semi-solid agarose conditions. Combined administration of adavosertib and alisertib inhibited colony formation of FaDu cells much more effectively than vehicle or either of the single agents, yielding fewer and smaller soft agar colonies (Fig. 2B). Furthermore, the combination therapy was more effective than each single agent at reducing oncosphere formation of both FaDu and UNC7 cells in conditioned three-dimensional (3D) cell culture (Fig. 2C and D). To test whether drug effects were permanent or reversible, oncospheres were transferred to standard tissue culture dishes after 8 days in 3D culture, then allowed to further grow for an additional 2 days. Vehicle-treated cells rapidly reattached and proliferated as monolayers, and 2 days after removal of drugs and replating, cells exposed to single agent treatment reattached much less effectively and exhibited reduced proliferation. In contrast, cells exposed to the combination did not proliferate, indicating that these very small oncospheres were likely non-viable (Fig. 2C).
Next, we evaluated the off-target cytotoxicity of the combination in normal human tracheobronchial epithelial (NHTBE) cells under an optimized 3D-organotypic air-liquid interface culture condition, as previously performed (32). Cells were treated with adavosertib, alisertib, or the combination by introduction of the drugs into the bottom chamber of the plate after full differentiation of the 3D culture. Notably, neither single agent nor combination treatment was cytotoxic toward NHTBE cells (Fig. 2E and F). Taken together, the combinatorial inhibition of AURKA and WEE1 by alisertib and adavosertib was synergistically superior to either single drug in inhibition of cell viability, anchorage-independent growth and oncosphere formation without severe toxicity.
Combined inhibition of AURKA and WEE1 affects mitotic machinery in HNSCC cells.
WEE1 and AURKA are opposing regulators of mitotic entry, through actions on the mitotic spindle and the phosphorylation of CDK1 (40, 41). Thus, we next examined mitotic morphology of FaDu and UNC7 cells treated with single agents or the combination. Control- and adavosertib-treated cells displayed a typical mitotic morphology with condensed chromosomes aligned at the metaphase plate, whereas loosened chromatin and abnormal tripolar spindles were observed in alisertib-treated cells (Fig. 3A). Interestingly, the combination treatment of adavosertib with alisertib resulted in highly abnormal mitoses with unaligned, dispersed chromosomes and disorganized multipolar spindles, as well as distorted and ballooned cellular morphology, indicating mitotic catastrophe-mediated cell death (Fig. 3A) (42). Additionally, this combination resulted in a dramatic >95% increase in the proportion of cells which were pHH3-positive with abnormal mitotic morphology (Fig. 3B and C). A slight increase in pHH3-positive cells was observed with alisertib single-agent treatment compared to control, but without the marked degree of abnormal mitotic morphology observed in combination-treated cells. Consistent with these markers of accelerated mitotic entry and mitotic catastrophe, cell cycle distribution following combination treatment showed an increase in the G2/M population (Fig. 3D and Supplementary Fig. S2).
Figure 3. Combined inhibition of AURKA and WEE1 affects mitotic morphology in HNSCC cells.

(A–C) FaDu and UNC7 cells were treated with adavosertib (500 nM), alisertib (250 nM), or adavosertib + alisertib for 24 hours and followed by immunofluorescent staining with anti-tubulin (Green) and anti-pHH3 (S10; Red). Nucleus was stained with DAPI. (A) Representative images of mitotic cells were captured by confocal microscopy. Scale bar: 10 µm. (B) Percentage of pHH3-positive cells in all DAPI-positive FaDu (left) and UNC7 (right) cells. Fields were randomly captured and counted DAPI-positive cells at least over 400 cells. (C) Percentage of normal or abnormal mitotic cells in pHH3-positive FaDu (left) and UNC7 (right) cells. (D) Cells were treated with the indicated drug singly or in combination for 8 and 24 hours and followed by assessing cell cycle distribution of FaDu (top) and UNC7 (bottom) cells by FACS analysis. (E) Cells were treated with the indicated drug singly or in combination for 8 and 24 hours. Cell lysates were subjected to SDS-PAGE and immunoblotting with the indicated antibodies.
Active WEE1 phosphorylates CDK1 at Tyr15 (pCDK1), resulting in blockade of mitotic entry, whereas CDK1 dephosphorylation by AURKA-PLK1-mediated CDC25B activation promotes mitotic entry (16). We analyzed activation of CDK1 and associated proteins after 8 or 24 hours of drug treatment by western blotting. Although inhibitory phosphorylation of CDK1 was increased after 24 hours of alisertib exposure, this effect was abrogated by the addition of adavosertib in both FaDu and UNC7 cells. Together, these results indicate that the addition of WEE1 inhibition to AURKA inhibition may abrogate the pCDK1-mediated mechanism of alisertib resistance in HNSCC cells (Fig. 4E).
Figure 4. Combined treatment of adavosertib and alisertib synergistically triggers apoptosis induction in HNSCC cells.

(A) FaDu (left) or UNC7 (right) cells were treated with adavosertib (500 nM), alisertib (250 nM), or adavosertib + alisertib for 24 hours and followed by staining with Annexin V and PI for apoptosis by FACS analysis. Representative image of apoptosis induction responding to the treatments determined by FACS analysis (top) and percentage of apoptotic cells (bottom). Graphs depict Mean ± SEM from three times repeated independent experiments. Statistical significance was assessed by Student’s t-test (**: P < 0.005). (B) The indicated cell lines were exposed to the drugs singly or in combination for 24 hours. Cell lysates were subjected to SDS-PAGE and immunoblotting with anti-cleaved PARP antibody for apoptosis induction.
Combined treatment of alisertib and adavosertib synergistically triggers enhanced apoptosis in HNSCC cells.
Given that the combinatorial inhibition of AURKA with WEE1 resulted in much greater mitotic derangement compared to either control or single agent in HNSCC cells, we next employed flow cytometry to examine whether the combination synergistically triggers cell death. Adavosertib and alisertib each resulted in modest apoptotic death in FaDu (adavosertib: 19.52%, alisertib: 12.2%) and UNC7 (adavosertib: 13.22%, alisertib: 13.18%) HNSCC cells compared to control cells (FaDu: 7.28%, UNC7: 6.94%), while the combination significantly enhanced induction of apoptosis (FaDu: 38.4%, UNC7: 16.41%, P < 0.002; Fig. 4A). Indeed, the induction of apoptotic cell death was also confirmed by an increase of PARP cleavage following combination treatment (Fig. 4B). Taken with the morphologic evidence for mitotic catastrophe presented above, these data suggest that the accumulated mitotic abnormalities produced by alisertib prime cells for mitotic catastrophe and cell death, but that because AURKA inhibition induces CDK1 phosphorylation and arrests the cell cycle prior to mitotic entry, actual cytotoxic effects are minimized.
In vivo efficacy of the combination treatment of alisertib and adavosertib.
The above data suggested that combined AURKA/WEE1 inhibition could prove to be a useful combination for anticancer therapy in TP53-mutated cancer. We next explored combinatorial inhibition of AURKA with WEE1 in FaDu and Detroit 562 cells xenografted in vivo mouse models. Tumors were established at 200 to 350 mm3 for FaDu xenograft and 150 to 200 mm3 for Detroit 562 xenograft prior to initiation of alisertib, adavosertib or combination treatment. In FaDu xenografts, the humane endpoint was reached at 22 days after implantation in vehicle-treated mice. In addition, tumor growth was not reduced by single agent therapy, with tumors treated with 30 mg/kg alisertib or 30 or 90 mg/kg adavosertib growing as rapidly as those in vehicle-treated mice. In contrast, combination treatment dramatically decreased tumor growth relative to vehicle or single agent treatment, with essentially no increase in tumor size (Fig. 5A). Consistent with the observation of tumor size and weight at endpoint of study, tumor volume change was significantly decreased by the combination treatment of alisertib with adavosertib compared to vehicle and single agent (Fig. 5B and supplementary Fig. S3A). Moreover, only combination treatment prolonged survival of mice (log-rank P < 0.0001; Fig. 5C). Consistent with the antitumor effect of the combination in the FaDu xenograft model, we observed similar effects on tumor size, tumor weight, tumor growth and survival in Detroit 562 xenografts as well (Fig. 5D–F and supplementary Fig. S4A). Given the sensitivity of Detroit 562 cells to alisertib relative to other HNSCC cells, these tumors responded to alisertib alone, as shown by the reduced tumor growth compared to vehicle (Fig. 5D–F). None of the treatments or combinations significantly reduced murine weight or induced observable toxicity (Supplementary Fig. S3B and S4B).
Figure 5. Co-inhibition of AURKA and WEE1 enhances the antitumor efficacy and extends survival of HNSCC in vivo model.

(A) Representative image and weight (g) of FaDu-xenografted tumors (n = 8–9) exposed to vehicle, adavosertib (ADV; 30 mg/kg or 90 mg/kg, daily, p.o.), alisertib (ALI; 30 mg/kg, daily, p.o.), or the combinations of alisertib with either adavosertib (30 mg/kg) or adavosertib (90 mg/kg). Scale bar: 1 cm. (B) Tumor volume was monitored twice a week. Humane endpoint was considered when tumor volume reached 1,000 mm3 following IACUC policy. (C) Overall survival of FaDu-xenografted mice treated with the drugs as single or combination (n = 8–9). (D) Representative image and weight (g) of Detroit 562-xenografted tumors (n = 6) exposed to vehicle, adavosertib (ADV; 30 mg/kg, daily, p.o.), alisertib (ALI; 30 mg/kg, daily, p.o.), or the combinations of alisertib with adavosertib (30 mg/kg). Scale bar: 1 cm. Graphs depict Mean ± SEM. Significant differences between the combinations and drug alone are exhibited. Statistical significance was assessed by Student’s t-test (**: P < 0.005). (E) Tumor volume was monitored every other day. Graphs depict Mean ± SEM. Significant differences between the combinations and drug alone are shown. Statistical significance was assessed by Student’s t-test (*: P < 0.05; **: P < 0.005) (F) Overall survival of Detroit 562-xenografted mice treated with the drugs as single or combination (n = 6). Log-rank (Mantel-Cox) testing was calculated by Prism 7. Significant differences between the combinations and drug alone are exhibited. (H) Representative IHC staining images of Ki-67 and cleaved-caspase 3 in FaDu-xenografted mice treated with the indicated drugs. Scale bar: 100 µm. (G) Representative immunofluorescent staining images of pCDK1 (Y15; Red), cytokeratin (Green) and DAPI (blue) in the FaDu-xenografted mice treated with the indicated drugs. Images in small box were enlarged with high power. Scale bars: 100 µm (large images) and 50 µm (small images).
Histologic evaluation of the xenograft tumors indicated that expression of the cell proliferation maker Ki-67 was decreased by combination therapy, whereas apoptotic cells, indicated by cleaved-Caspase 3, were dramatically increased by the combination compared to vehicle or to either single agent (Fig 5G). pCDK1expression in tumor leading edge was almost eliminated by the adavosertib and combination treatments, consistent with our in vitro results (Fig. 5H and supplementary Fig. S5). Further, the combination of alisertib with adavosertib resulted in aberrant nuclear and cellular morphologies, including balloon-like enlargement, characteristic of mitotic catastrophe. Together, these results suggest that the drug combination of alisertib with adavosertib synergistically impairs CDK1-mediated mitotic arrest, precipitating mitotic catastrophe and enhancing apoptosis in HNSCC in vitro and in vivo.
DISCUSSION
In this study, we investigated the combined inhibition of WEE1 and AURKA using adavosertib and alisertib in HNSCC. Although AURKA has been found to be a therapeutic target in kinomic screens (3), clinical trial data in HNSCC show only a modest response rate of 9%, with no complete responses. Here, our studies describe an intrinsic effect of AURKA inhibition, phosphorylation of CDK1, which results in cell cycle arrest and may protect from cell death despite the profound effects AURKA inhibition exerts on centrosome maturation. This intrinsic source of resistance to AURKA inhibition may itself be susceptible to pharmacologic manipulation. Our data imply that the combined treatment could be an effective therapeutic approach for HNSCC patients, particularly those harboring TP53 mutation.
AURKA not only regulates spindle formation and centrosome maturation, but also fosters the G2/M transition. CDK1 is essential to this transition, and its kinase activity is governed by WEE1 and CDC25. (Fig. 6). WEE1 is a serine/threonine kinase that phosphorylates CDK1 at tyrosine15 and threonine 14, thereby inactivating it. CDC25 is a phosphatase that dephosphorylates the above residues, leading to CDK1 activation (43). In G2 phase, AURKA complexes with BORA to activate PLK1, which can in turn activate CDC25 (44) and target WEE1 stability for degradation (44, 45). AURKA also directly phosphorylates and activates CDC25 (16). With the resulting increased ratio of CDC25 to WEE1 activity, the active form of CDK1 predominates. Moreover, AURKA has been shown to phosphorylate p53 at serine 215 and serine 315, targeting p53 to MDM2-dependent degradation (46, 47). Degradation of p53 may reduce p21 transcription and consequently reduce inhibition of the active CDK1 complex. AURKA is expected to activate CDK1 and lead to mitotic entry via interactions with PLK1, CDC25, and p53.
Figure 6. Schematic model of the effect by the combined inhibition of AURKA and WEE1 in this study.
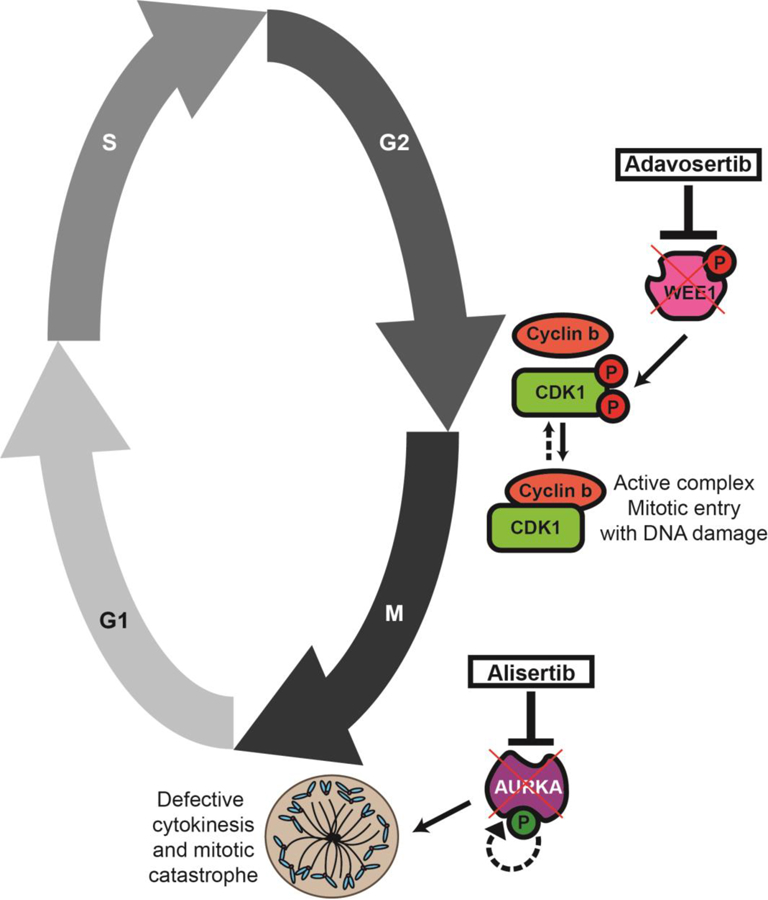
The addition of the WEE1 inhibitor adavosertib would prevent the checkpoint kinase WEE1 with dephosphorylating CDK1 therefore leading to mitotic entry in the presence of DNA damage. When the AURKA inhibitor alisertib is concomitantly added, a failure in cytokinesis including spindle formation and centrosome maturation would in turn lead to mitotic catastrophe and cell death.
Here, we have demonstrated that AURKA inhibition with the kinase inhibitor alisertib leads not only to abnormal spindle assembly, but also to a block in mitotic entry. We report that G2 arrest—driven by CDK1 inhibition—compromises cytotoxicity of AURKA inhibition. We have further demonstrated that this effect can be overcome by addition of the WEE1 inhibitor adavosertib, which induces mitotic entry through its inhibition of CDK1 phosphorylation. In vitro cell viability, soft agar, and oncosphere formation assays all demonstrated clear synergy of the combination of adavosertib with alisertib in targeting TP53 mutant HNSCC cells, but not normal cells (Fig. 2) and the synergistic suppression on tumor growth was further confirmed in two HNSCC-mediated xenograft in vivo models (Fig. 5)
Mechanistic studies show that combined AURKA and WEE1 inhibition induces cells to transition through the G2/M-checkpoint in the setting of DNA damage and defective spindle assembly, resulting in mitotic catastrophe. Western Blot analysis conducted after treatment of both FaDu and UNC7 cells with alisertib demonstrated increased pCDK1 (Y15; inactive form) and reduced pHH3 (S10) compared to untreated cells, suggestive of reduced mitotic entry, especially at eight hours treatment. Addition of the WEE1 inhibitor adavosertib to alisertib resulted in decreased pCDK1, increased pHH3, and increased gamma-H2AX (a DNA damage marker), to levels resembling cells treated with single-agent adavosertib (Fig. 3E). Combination-treated cells also exhibited an increased level of cleaved-PARP compared to cells treated with either drug alone, suggesting induced apoptosis and synergy (Fig. 4). It is worth noting the significant increase in proportion of mitotic cells in combination-treated cells, as determined by pHH3-positivity in immunofluorescence staining (Fig. 3B). A rise in pHH3 at 24 hours was not detected on western blotting (Fig. 3D); this is likely due to the high rate of cell death for mitotic cells exposed to the combination treatment, so that the absolute quantity of pHH3-positive cells is not increased despite the dramatic rise in the proportion of mitotic cells (Fig. 4). Therefore, the small population of pHH3-positive cells was visualized in single cell microscopy. Structurally, combination-treated cells displayed complete spindle disarray and lack of chromatin organization. While cells treated singly with the WEE1 inhibitor adavosertib did not display spindle defects, cells treated singly with alisertib displayed mono- or tri-polar spindles, but lacked the spindle disarray seen with combination therapy. Together these results provide further evidence of synergy and support the suggestion that cells with impaired spindle assembly have increased sensitivity to mitotic kinase inhibitor-mediated mitotic catastrophe.
The WEE1 inhibitor adavosertib employed here has recently been reported to have broader effects, including S phase effects and activity as an inhibitor of PLK1 (48, 49). Thus, alternative explanations for the effects detected here could relate to off-target effects of adavosertib. Most importantly, though, in considering the possibility of translating our observations to clinical advances for patients with TP53-mutated cancers, the combination of adavosertib and alisertib appeared not only highly synergistic, but combination-treated xenograft mice showed this dramatic response to the combination therapy without notable toxicity and with stable weight.
In conclusion, we have demonstrated that the combination of adavosertib and alisertib synergistically enhances cancer cell death in TP53-mutated HNSCC cell lines, and inhibits tumor growth in xenograft in vivo models. Treatment of malignant cells with alisertib leads to defects in spindle assembly; however, due to increased deactivating phosphorylation of CDK1, mitotic entry may be impaired, preventing the expected cytotoxicity of AURKA inhibition. Addition of the WEE1 inhibitor adavosertib overcame resistance to alisertib possibly by abrogating CDK1 phosphorylation and inducing mitotic catastrophe as cells with DNA damage and defective spindles/centrosomes progressed though the G2/M checkpoint. Based on the data presented, we recommend further clinical investigation combining adavosertib and alisertib in HNSCC patients harboring TP53 mutation, and exploration of this drug combination in other TP53-mutated malignancies known to have high AURKA expression.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Aurora kinase A (AURKA) has been extensively investigated as a target for cancer therapeutics due to its roles in regulating mitotic entry and in the function of the centrosome and mitotic spindle. AURKA expression and protein activity are upregulated in many human cancers. However, the clinical activity of AURKA inhibition has been disappointing. Here, we confirmed a relationship of elevated nuclear AURKA expression to poor prognosis in HPV-unrelated head and neck squamous cell carcinoma (HNSCC), an observation that is compatible with observed AURKA upregulation in cancers with loss of p53 function. We investigated approaches to augment the efficacy of AURKA inhibition. AURKA inhibition with alisertib (MLN8237) results in expected abnormalities in spindle polarity of cancer cells, but is associated with inhibitory phosphorylation of CDK1 at tyrosine 15, resulting in G2 arrest and cytostasis rather than cell death. Addition of the WEE1 inhibitor adavosertib (AZD1775) abrogates CDK1 phosphorylation and results in mitotic catastrophe, synergistically eradicating HNSCC both in vitro and in vivo. This result is the first demonstration of adavosertib sensitizing to the anticancer effects of alisertib in any tumor. Our study suggests that AURKA should be reexamined as an important therapeutic target in HNSCC, and that combinatorial treatment with AURKA and WEE1 inhibition offers promise as synthetic lethal therapy in cancers dependent on AURKA.
Footnotes
Disclosure of Potential Conflicts of Interest: Barbara Burtness reports consulting fees from Astra-Zeneca.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moser R, Xu C, Kao M, Annis J, Lerma LA, Schaupp CM, et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20:4274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehra R, Serebriiskii IG, Burtness B, Astsaturov I, Golemis EA. Aurora kinases in head and neck cancer. Lancet Oncol 2013;14:e425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Science signaling 2010;3:ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 2007;5:1–10. [DOI] [PubMed] [Google Scholar]
- 7.Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer 2005;5:42–50. [DOI] [PubMed] [Google Scholar]
- 8.Kollareddy M, Dzubak P, Zheleva D, Hajduch M. Aurora kinases: structure, functions and their association with cancer. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2008;152:27–33. [DOI] [PubMed] [Google Scholar]
- 9.Marxer M, Ma HT, Man WY, Poon RY. p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of Aurora kinases. Oncogene 2014;33:3550–60. [DOI] [PubMed] [Google Scholar]
- 10.Reiter R, Gais P, Jutting U, Steuer-Vogt MK, Pickhard A, Bink K, et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res 2006;12:5136–41. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Zhang J. AURKA is a predictor of chemotherapy response and prognosis for patients with advanced oral squamous cell carcinoma. Tumour Biol 2015;36:3557–64. [DOI] [PubMed] [Google Scholar]
- 12.Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol 2015;16:395–405. [DOI] [PubMed] [Google Scholar]
- 13.Barr PM, Li H, Spier C, Mahadevan D, LeBlanc M, Ul Haq M, et al. Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J Clin Oncol 2015;33:2399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedberg JW, Mahadevan D, Cebula E, Persky D, Lossos I, Agarwal AB, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol 2014;32:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 2003;114:585–98. [DOI] [PubMed] [Google Scholar]
- 16.Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci 2004;117:2523–31. [DOI] [PubMed] [Google Scholar]
- 17.Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008;455:119–23. [DOI] [PubMed] [Google Scholar]
- 18.Lobjois V, Froment C, Braud E, Grimal F, Burlet-Schiltz O, Ducommun B, et al. Study of the docking-dependent PLK1 phosphorylation of the CDC25B phosphatase. Biochem Biophys Res Commun 2011;410:87–90. [DOI] [PubMed] [Google Scholar]
- 19.Saini P, Li Y, Dobbelstein M. Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget 2015;6:13072–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr., Golemis EA Aurora A kinase (AURKA) in normal and pathological cell division. Cellular and molecular life sciences : CMLS 2013;70:661–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahajan K, Fang B, Koomen JM, Mahajan NP. H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat Struct Mol Biol 2012;19:930–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahajan K, Mahajan NP. WEE1 tyrosine kinase, a novel epigenetic modifier. Trends Genet 2013;29:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol 2015;33:3409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboklatin in Patients With Advanced Solid Tumors. J Clin Oncol 2016;34:4371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J Clin Oncol 2016;34:4354–61. [DOI] [PubMed] [Google Scholar]
- 26.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther 2010;9:514–22. [DOI] [PubMed] [Google Scholar]
- 27.Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther 2015;14:608–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov 2012;2:524–39. [DOI] [PubMed] [Google Scholar]
- 29.Mehra R, Zhu F, Yang DH, Cai KQ, Weaver J, Singh MK, et al. Quantification of excision repair cross-complementing group 1 and survival in p16-negative squamous cell head and neck cancers. Clin Cancer Res 2013;19:6633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med 2002;8:1323–7. [DOI] [PubMed] [Google Scholar]
- 31.Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Brandsma J, et al. Molecular classification identifies a subset of human papillomavirus--associated oropharyngeal cancers with favorable prognosis. J Clin Oncol 2006;24:736–47. [DOI] [PubMed] [Google Scholar]
- 32.Lee JW, Park HS, Park SA, Ryu SH, Meng W, Jurgensmeier JM, et al. A Novel Small-Molecule Inhibitor Targeting CREB-CBP Complex Possesses Anti-Cancer Effects along with Cell Cycle Regulation, Autophagy Suppression and Endoplasmic Reticulum Stress. PLoS One 2015;10:e0122628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Z-M, Zhao B, Bai Y, Iamarino A, Gaffney SG, Schlessinger J, et al. Early and multiple origins of metastatic lineages within primary tumors. Proceedings of the National Academy of Sciences 2016;113:2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 35.Tatsuka M, Sato S, Kanda A, Miki T, Kamata N, Kitajima S, et al. Oncogenic role of nuclear accumulated Aurora-A. Mol Carcinog 2009;48:810–20. [DOI] [PubMed] [Google Scholar]
- 36.Masica DL, Li S, Douville C, Manola J, Ferris RL, Burtness B, et al. Predicting survival in head and neck squamous cell carcinoma from TP53 mutation. Hum Genet 2015;134:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper T, Biron VL, Fast D, Tam R, Carey T, Shmulevitz M, et al. Oncolytic activity of reovirus in HPV positive and negative head and neck squamous cell carcinoma. J Otolaryngol Head Neck Surg 2015;44:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sewell A, Brown B, Biktasova A, Mills GB, Lu Y, Tyson DR, et al. Reverse-Phase Protein Array Profiling of Oropharyngeal Cancer and Significance of PIK3CA Mutations in HPV-Associated Head and Neck Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20:2300–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin D, Abba MC, Molinolo AA, Vitale-Cross L, Wang Z, Zaida M, et al. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget 2014;5:8906–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGowan CH, Russell P. Cell cycle regulation of human WEE1. The EMBO journal 1995;14:2166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. The EMBO journal 1995;14:1878–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004;23:2825–37. [DOI] [PubMed] [Google Scholar]
- 43.Dunphy WG, Kumagai A. The cdc25 protein contains an intrinsic phosphatase activity. Cell 1991;67:189–96. [DOI] [PubMed] [Google Scholar]
- 44.Toyoshima-Morimoto F, Taniguchi E, Nishida E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep 2002;3:341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Vugt MA, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell 2004;15:799–811. [DOI] [PubMed] [Google Scholar]
- 46.Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004;36:55–62. [DOI] [PubMed] [Google Scholar]
- 47.Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem 2004;279:52175–82. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka N, Patel AA, Tang L, Silver NL, Lindemann A, Takahashi H, et al. Replication Stress Leading to Apoptosis within the S-phase Contributes to Synergism between Vorinostat and AZD1775 in HNSCC Harboring High-Risk TP53 Mutation. Clin Cancer Res 2017;23:6541–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright G, Golubeva V, Remsing Rix LL, Berndt N, Luo Y, Ward GA, et al. Dual Targeting of WEE1 and PLK1 by AZD1775 Elicits Single Agent Cellular Anticancer Activity. ACS Chem Biol 2017;12:1883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.