

Abstract
Purpose:
The third-generation EGFR inhibitor, osimertinib, is the first mutant selective inhibitor that has received regulatory approval for the treatment of patients with EGFR-mutant lung cancer. Despite the development of highly selective third-generation inhibitors, acquired resistance remains a significant clinical challenge. Recently, we and others have identified a novel osimertinib resistance mutation, G724S, which was not predicted in in vitro screens. Here, we investigate how G724S confers resistance to osimertinib.
Experimental Design:
We combine structure-based predictive modeling of G724S in combination with the two most common EGFR activating mutations, exon 19 deletion (Ex19Del) and L858R, with in vitro drug-response models and patient genomic profiling.
Results:
Our simulations suggest that the G724S mutation selectively reduces osimertinib binding affinity in the context of Ex19Del. Consistent with our simulations, cell lines transduced with Ex19Del/G724S demonstrate resistance to osimertinib, while cells transduced with L858R/G724S are sensitive to osimertinib. Subsequent clinical genomic profiling data further suggests G724S occurs with Ex19Del but not L858R. Furthermore, we demonstrate that Ex19Del/G724S retains sensitivity to afatinib, but not to erlotinib, suggesting a possible therapy for patients at the time of disease relapse.
Conclusions:
Altogether, these data suggest that G724S is an allele-specific resistance mutation emerging in the context of Ex19Del but not L858R. Our results fundamentally reframe the problem of targeted therapy resistance from one focused on the “drug – resistance mutation” pair to one focused on the “activating mutation – drug – resistance mutation” trio. This has broad implications across clinical oncology.
Keywords: Epidermal growth factor receptor (EGFR), non-small cell lung cancer (NSCLC), tyrosine kinase inhibitor (TKI), osimertinib, acquired resistance
Introduction
Oncogenic mutations in the EGFR tyrosine kinase domain are found in ~15–30% of non-small cell lung carcinomas (NSCLC) (1,2). Of these cases, approximately 90% can be attributed to in-frame deletions within exon 19 (Ex19Del) or missense mutations in exon 21 (L858R), which occur with approximately equal prevalence (1,2). Multiple phase III clinical trials have shown that patients with EGFR-mutant tumors experience >70% radiographic response rates (RRs) and a statistically significant improvement in progression-free survival (PFS) when treated with first-generation (erlotinib, gefitinib) or second-generation (afatinib) EGFR tyrosine kinase inhibitors (TKIs) as compared with platinum based chemotherapy (3–6). However, response to these targeted agents is transient, and acquired therapeutic resistance typically develops within 8–10 months. In approximately 60% of cases, resistance is acquired through acquisition of a secondary EGFR mutation, EGFR T790M (7–9). Osimertinib, a mutant-selective third-generation covalent inhibitor, was developed specifically to target T790M. For these reasons, the clinical standard of care for EGFR-mutant NSCLC has been treatment with first or second generation TKIs followed by treatment with osimertinib post-progression on first line therapy (10). Recently, osimertinib became approved as first-line therapy (11).
Unfortunately, resistance mutations may also emerge against osimertinib therapy (12,13). The most well described to date is C797S, which is detected in approximately 10%−19% of patients with first-line and second-line osimertinib resistance (14,15). Mutation of C797 to serine prevents covalent adduct formation between osimertinib and the EGFR kinase domain (16,17). We (18) and others (14,19,20) have also identified G724S as a mutation which is selected for in osimertinib resistant tumors. Unlike C797S, G724S was not predicted based on in vitro studies (21,22), and the precise mechanism whereby G724S mutation confers osimertinib resistance is unknown.
The most fundamental principle of structural biology is that sequence determines structure and structure determines function. To determine the relationship between classical EGFR kinase activating mutations (Ex19Del and L858R), acquired G724S mutation, and osimertinib resistance, we employed an integrated computational / experimental approach. Our results suggest that G724S is a resistance mutation that develops with Ex19Del but not L858R and provide mechanistic insight into this process at the structural level.
Materials and Methods
Inhibitor source and preparation:
EGFR TKIs were purchased from Selleck Chemicals (Houston, TX, USA). All drugs were prepared and stored as a stock solution at 10 mM in DMSO (Sigma-Aldrich, St. Louis, MO, USA).
Cell culture:
293FT cells were purchased from Invitrogen (Carlsbad, CA, USA). NR-6 cells were a gift from Dr. William Pao (23). 293FT and NR-6 cells were cultured in DMEM with 4.5 g/L glucose, L-glutamine & sodium pyruvate (Mediatech, Corning, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA, USA) and penicillin (100 U/mL)/streptomycin (100 μg/mL) (Mediatech). Ba/F3 cells were purchased from DSMZ and were cultured in RPMI 1640 with L-glutamine (Mediatech) supplemented with 10% heat-inactivated FBS, penicillin (100 U/mL)/streptomycin (100 μg/mL), and 1 ng/mL interleukin-3 (IL-3) (Thermo Fisher Scientific, Waltham, MA, USA) until retroviral transduction and subsequent IL-3 withdrawal. Cells were grown in a humidified incubator with 5% CO2 at 37°C and were routinely evaluated for mycoplasma using a Venor GeM Mycoplasma Detection Kit (Sigma-Aldrich).
Immunoblot analysis:
Cells were washed with PBS and lysed in radioimmunoprecipitation analysis buffer (50 mM TrisHCl pH 8.0, 150 mM sodium chloride, 5 mM magnesium chloride, 1% Triton X-100. 0.5% sodium deoxycholate, 0.1% SDS, 40 mM sodium fluoride, 1 mM sodium orthovanadate, and complete Protease Inhibitor Cocktail [Roche Diagnostics, Indianapolis, IN, USA]). Western Lightning ECL reagent (PerkinElmer, Waltham, MA, USA) was used for signal detection. β-actin antibody (A2066) was purchased from Sigma-Aldrich. EGFR (#2232), pEGFR Y1068 (#2234), pEGFR Y1173 (#2244), ERK (#9102), pERK T202/Y204 (#9101), horseradish peroxidase (HRP)-conjugated anti-mouse (#7076) and HRP-conjugated anti-rabbit (#7074) antibodies were purchased from Cell Signaling (Danvers, MA, USA). Each experiment was performed twice.
CellTiter Blue cell viability assay:
Ba/F3 cells were seeded in 96-well plates at a density of 20,000 cells/well and treated with varying concentrations of indicated compounds, with six technical replicates per concentration. After 72 hours, CellTiter Blue Reagent (Promega, Madison, WI, USA) was added to wells according to manufacturer’s instructions, and cells were incubated at 37°C with 5% CO2 for 2 to 4 hours. Absorbance was detected at 590 nm with a Synergy HTX microplate reader (BioTek Instruments, Winooski, VT, USA). Each experiment was performed three times.
Statistical analysis:
All experiments were performed at least three times and the differences were determined by one-way ANOVA. Differences were considered significant when p < 0.05.
Molecular Modeling:
Structural models of the EGFR kinase exon 19 deletion mutants (Ex19Del) were generated through complementary use of the structure-prediction software package Rosetta utilizing the REF2015 score function (24–26) and molecular dynamics (MD) simulation with AMBER16 (27). Comparative models of Ex19Del kinase domain were created with RosettaCM (24,25) by modeling the kinase domain sequence sans β3-αC residues E746–A750 for the canonical variant model, or a valine substituted for the range E746–S752 for the rare variant model, and applying PDB IDs 2GS6 and 2GS7 as templates for the active and inactive state models, respectively (28). Active and inactive state Rosetta models of EGFR were minimized and allowed to equilibrate in a rectangular box of TIP4PEW explicit solvent neutralized with monovalent chlorine anions (29,30). Solute was buffered on all sides with 12 Å solvent. Afterward, dual-boost Gaussian accelerated MD (GaMD) simulations were performed to enhance conformational sampling (31–34). Protein-ligand binding free energy calculations were performed with MM/GBSA implemented in the AMBER suite in combination with the quasi-harmonic approximation (QHA) of entropy (35). For a detailed description of model building, molecular dynamics simulations, and binding free energy calculations, please see the Supplementary Methods section.
Genomic profiling of patient samples:
Hybrid capture-based next generation sequencing (NGS) was performed on formalin-fixed paraffin embedded tissue sections or circulating tumor DNA isolated from blood samples in a Clinical Laboratory Improvement Amendments (CLIA)-certified, CAP (College of American Pathologists)-accredited laboratory (Foundation Medicine, Cambridge, MA) as described previously (36,37). Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817).
Results
A G724S-mediated conformational change in the glycine-rich P-loop reduces binding affinity of osimertinib to Ex19Del/G724S but not to L858R/G724S
To determine the structural effects of G724S mutation on osimertinib binding, we performed a series of Gaussian accelerated molecular dynamics (GaMD) simulations (32,34) of wild-type EGFR (WT), Ex19Del (unless otherwise stated, the canonical variant E746_A750del), Ex19Del/G724S, L858R, and L858R/G724S in the drug-unbound (apo) state (Supplementary Table S1). Analysis of our initial simulations suggests G724S may increase P-loop backbone conformation fluctuations (Supplementary Figure S1). These data are intriguing because EGFR has previously been shown to bind osimertinib with a characteristic “bent” P-loop conformation (17), and we hypothesized that G724S could reduce osimertinib binding through disruption of the bent P-loop conformation. Previous literature on protein conformational dynamics has cautioned against inferring functional mechanisms from RMSF statistics alone (38). Therefore, to test our hypothesis, we performed GaMD simulations of Ex19Del, Ex19Del/G724S, L858R, and L858R/G724S reversibly bound with osimertinib. We similarly examined these four mutants with the second-generation, wild-type selective EGFR TKI, afatinib, as a control. Afatinib was selected as a control in our study for multiple reasons. Afatinib has previously been reported to be a potential therapeutic agent in the setting of Ex19Del/G724S-mediated NSCLC based on a patient case report (18). Similar to osimertinib, afatinib is an irreversible EGFR inhibitor that has received regulatory approval for treatment of EGFR-mutant lung cancer.
Osimertinib and afatinib both irreversibly bind EGFR through covalent adduct formation. In order to form an irreversible complex, they must first form a reversible, non-covalent complex (Figure 1A). Disruption of the reversible complex formation is expected to reduce formation of adduct. A previously determined crystallographic structure of EGFR kinase reversibly bound to osimertinib demonstrates that osimertinib binding is accommodated through a well-defined “bent” P-loop conformation (Figure 1B) (17). This bent P-loop conformation allows the F723 phenyl ring to make an energetically favorable contact with the indole ring of osimertinib, contributing to its affinity (17).
Figure 1. Stability of osimertinib in reversible complexes with EGFR mutants.
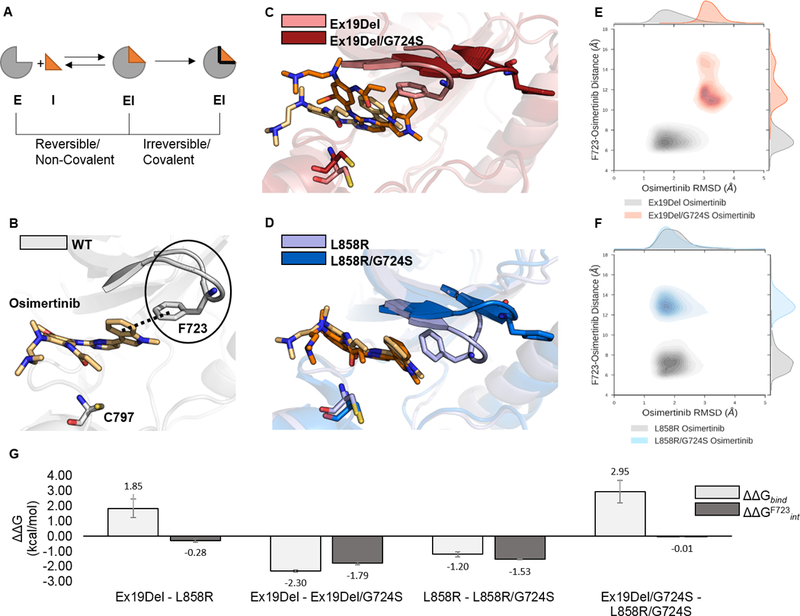
EGFR mutants reversibly bound to osimertinib were simulated with GaMD. A schematic representation of a simplified binding equilibrium for a covalently-binding inhibitor is depicted such that E = Enzyme target, I = Inhibitor, and EI = Enzyme-Inhibitor complex (A). Each simulation was performed in triplicate for a total of 12 independent 250 ns GaMD simulations. Representative images of osimertinib reversibly bound to WT (PDB ID 4ZAU; the solid black line indicates the bent P-loop; the dashed black line indicates the contact between the F723 phenyl and osimertinib indole ring; (B), Ex19Del and Ex19Del/G724S (C), and L858R and L858R/G724S (D) are displayed. Trajectory frames were extracted every 10 ps and plotted as osimertinib RMSD from the equilibrated start structure (x-axis) and distance between the phenyl ring of F723 and the indole ring of osimertinib (y-axis; E – F). RMSD vs. distance plots include data from 3 independent trajectories for each mutant – inhibitor pair (E – F). Select relative osimertinib binding free energies are plotted as averages across 3 independent trajectories; error bars indicate standard error of the mean (G).
ΔGbind = ΔEMM + ΔGsolv – TΔS
ΔGF723int = ΔEMM + ΔGsolv
ΔΔG = ΔG1 – ΔG2
Our GaMD simulations illustrate that G724S rigidifies the tip of the P-loop by stabilizing a β-bend conformation (Figure 1C, D; Supplementary Figure S2). As a result, Ex19Del/G724S and L858R/G724S cannot form a stable bent P-loop conformation when bound to osimertinib. The rigidified P-loop displaces F723 from contact with osimertinib (Figure 1C – F). Interestingly, however, we found evidence of reduced stability of the osimertinib-bound Ex19Del/G724S complex but not the osimertinib-bound L858R/G724S complex. In our simulations, osimertinib maintains an RMSD of 1 – 2 Å from its native binding pose in Ex19Del and L858R. Displacement of F723 from contact with osimertinib is associated with an increase in osimertinib RMSD to 3 – 4 Å in Ex19Del/G724S but not in L858R/G724S (Figure 1E, F). In contrast, afatinib forms a stable reversible complex in all four cases (Ex19del, Ex19del/G724S, L858R, and L858R/G724S) (Supplementary Figure S3). These models suggest that structural perturbations from G724S, which disrupt binding of osimertinib, fail to notably effect binding of afatinib. These data support a potential role for afatinib in treating patients with G724S.
To further investigate these differences, we applied the molecular mechanics-generalized Born surface area method (MM/GBSA) to compute the binding free energies of osimertinib with Ex19Del, Ex19Del/G724S, L858R, and L858R/G724S. Our calculations predict a 2.3 kcal/mol reduction in osimertinib binding free energy (ΔΔGbind) with Ex19Del/G724S (Figure 1G). Our binding free energy calculations also suggest that osimertinib reversibly binds L858R more tightly than Ex19Del by 1.9 kcal/mol. (Figure 1G). The osimertinib binding free energies computed for Ex19Del and L858R/G724S are indistinguishable, within error, suggesting that the reduction in binding affinity accompanying the addition of the G724S mutation in L858R should not confer osimertinib resistance (Supplementary Table S2).
In addition, energy decomposition analysis supports our qualitative observation that F723 contributes favorably to osimertinib binding in both Ex19Del and L858R (the interaction energy of F723, ΔGF723int, defined ΔGF723int = ΔEMM + ΔGsolv, is approximately −1.8 and −1.5 kcal/mol, respectively), and that addition of G724S prevents this interaction (Figure 1G). As expected based on crystallographic evidence, our simulations show that F723 contributes considerably less to the interaction of EGFR with afatinib (39) (Supplementary Table S2). Consistent with Fassunke et al. (20), our afatinib relative binding free energies are less affected by G724S versus osimertinib (Supplementary Table S2). Altogether, these data suggest G724S may function as a resistance mutation to osimertinib in Ex19Del/G724S, but not in L858R/G724S.
In vitro expression of Ex19Del/G724S, but not L858R/G724S, is associated with osimertinib resistance
To test our simulation predictions, we first examined the ability of osimertinib to inhibit EGFR autophosphorylation of various EGFR single, double, and triple mutants. Of note, to date, G724S has been detected in both the absence and presence of T790M (18,19). Therefore, we modeled all possibilities in our experimental studies. Osimertinib was effective at inhibiting EGFR autophosphorylation in 293FT cells expressing Ex19Del and Ex19Del/T790M, but not in 293FT cells expressing Ex19Del/C797S or Ex19Del/T790M/C797S, as C797S mutation has previously been associated with osimertinib resistance(16,40) (Figure 2A). Likewise, osimertinib was ineffective at blocking autophosphorylation of EGFR Ex19Del/G724S and Ex19Del/T790M/G724S mutants.
Figure 2. EGFR G724S mediates osimertinib resistance in EGFR Ex19Del but not EGFR L858R mutants.
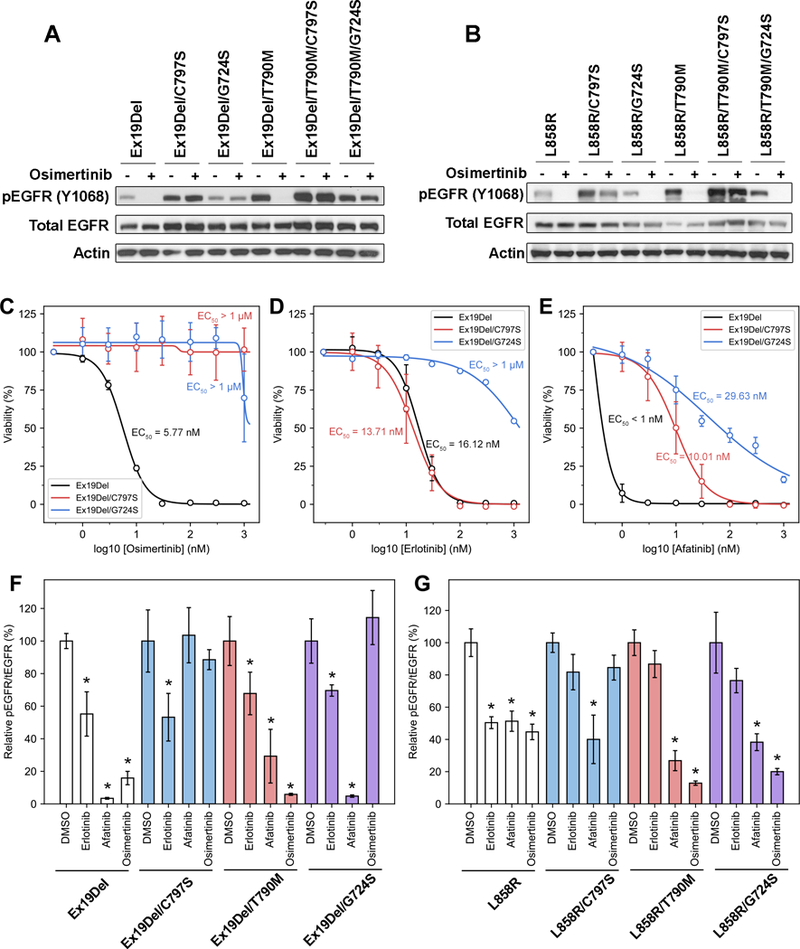
(A) 293FT cell transduced with different EGFR del19 variants were treated with 100 nM osimertinib for 4 hours. Cellular lysates were probed with the indicated antibodies. (B) 293FT cell transduced with different EGFR L858R variants were treated with 100 nM osimertinib for 4 hours. Cellular lysates were probed with the indicated antibodies. Ba/F3 EGFR Ex19Del, Ex19Del19/C979S, Ex19Del/G724S were treated with increasing amount of (C) osimertinib, (D) erlotinib or (E) afatinib for 72 hours. CellTiter Blue assays were performed to assess cell viability. Each point represents three replicates. Data are presented as the mean percentage of viable cells compared to control ± SD. NR6 cells transduced with (F) different EGFR del19 variants or (G) different EGFR L858R variants were treated with either DMSO, 100 nM erlotinib, 100 nM afatinib, or 100 nM osimertinib for 4 hours. Relative pEGFR/tEGFR values are calculated by the density of pEGFR signal divided by the density of tEGFR signal, then normalized by the DMSO-treated group in each cell line. Density of western blots was analyzed by ImageJ. *: p < 0.05 as compared to DMSO-treated group in each cell line. Representative western blots are shown in Supplementary Figure S6.
We also tested the efficacy of osimertinib against L858R variant combinations. Analogous to the Ex19Del data above, phosphorylation of L858R and L858R/T790M were inhibited by osimertinib while C797S containing variants (L858R/C797S and L858R/T790M/C797S) were insensitive to this agent (Figure 2B). In contrast to the Ex19Del variant data, phosphorylation of L858R/G724S and L858R/T790M/G724S were potently inhibited by osimertinib (Figure 2B). These data are consistent with our simulations, which suggested a difference in the drug binding properties between Ex19Del and L858R when combined with G724S mutation. Altogether, these data suggest that G724S functions as a resistance mutation in the context of Ex19Del but not L858R.
Next, we attempted to define strategies to overcome osimertinib resistance mediated by G724S mutation. In particular, we focused on the efficacy of earlier generations of wild-type selective EGFR TKIs (Supplementary Table S3). Previous studies have demonstrated that C797S-containing EGFR variants, which are resistant to osimertinib, retain sensitivity to the first generation EGFR TKIs (erlotinib, gefitinib) (16). We sought similar strategies for G724S-containing EGFR variants. We quantitatively evaluated several TKIs on Ex19Del-series mutants by stably transducing Ex19Del EGFR variants into Ba/F3 cells and measuring IL-3-independent growth at multiple inhibitor concentrations (Figure 2 C-E). As expected, growth of cells expressing EGFR Ex19del/C797S and EGFR Ex19del/G724S was insensitive to osimertinib. Cell lines expressing Ex19del/C797S and Ex19del/G724S were also cross-resistant to another mutant-selective EGFR-TKI, rociletinib (Supplementary Figure S4). In accord with previous data (16), cells expressing Ex19Del/C797S were sensitive to the effects of the first generation EGFR TKI, erlotinib (Supplementary Table S3), with an EC50 paralleling that of the original Ex19Del single mutant (16.12 nM vs. 13.71 nM, respectively, Figure 2D). However, the Ex19Del/G724S mutant was insensitive to the effects of erlotinib (EC50 > 1 μM). Our structural data suggested that afatinib may retain efficacy against the Ex19Del/G724S double mutant (Supplementary Figure S3). In accord with these data, the growth of cells expressing this double mutant was inhibited with an EC50 of 29.63 nM afatinib (Figure 2E). Likewise, autophosphorylation of the Ex19Del/G724S in stably transduced NR6 cells was potently inhibited by afatinib, but not erlotinib or osimertinib (Figure 2F, Supplementary Figure S5), while the autophosphorylation of the L858R/G724S was potently inhibited by both afatinib and osimertinib (Figure 2G, Supplementary Figure S5).
Importantly, previous in vitro screens failed to identify G724S as a resistance mutation (21,22). Our data suggest that this may be because these screens generated missense mutants beginning with WT, L858R, or L858R/T790M. Our data suggest that G724S functions as a resistance mutation in the context of Ex19Del but not L858R. Moreover, our results provide additional evidence that afatinib, but not osimertinib or erlotinib, can function effectively as an inhibitor of Ex19Del/G724S.
G724S emerges as a resistance mutation in Ex19Del but not L858R-mediated NSCLC
To date, four independent reports (14,18–20) have identified G724S as an emergent mutation in patients who have developed acquired resistance to osimertinib, with the frequency of G724S being 13% (higher than the frequency of C797S) in a recent paper by Fassunke and colleagues (20). Interestingly, all of these patients harbored Ex19Del as the original activating mutation (14,18–20). Our computational and experimental data suggest that G724S confers resistance to osimertinib in Ex19Del but not L858R; nevertheless, it is possible that L858R/G724S exists in a subset of EGFR-mutant NSCLC patients. To investigate the prevalence of EGFR G724S mutation, we analyzed data from tissue and plasma DNA samples within the Foundation Medicine database (Supplementary Table S4). Consistent with our computational and experimental evidence, G724S co-occurred with an Ex19Del variant in 15/19 cases, and L858R/G724S was not identified (Figure 3A). Given that the likelihood of observing Ex19Del versus L858R in EGFR-mutant NSCLC is approximately equal (41), it is exceedingly unlikely that L858R activating mutation would not be found in any of our patient samples without an additional bias.
Figure 3. Prevalence of oncogenic EGFR mutations in NSCLC patient samples with G724S.
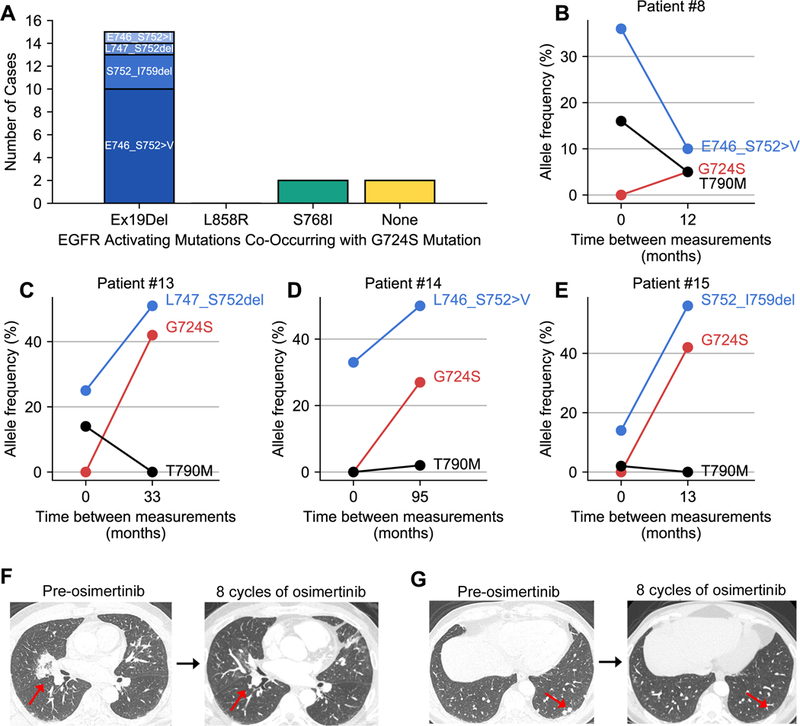
(A) Bar chart depicting the number of cases of each oncogenic EGFR mutation associated with G724S in NSCLC patient samples with genomic profiling obtained through Foundation Medicine (total n=19). (B-E) Allelic frequencies for the specific Ex19Del variant, T790M, and G724S are plotted versus time between measurements for four cases for which tissue genomic profiling results were available at two independent time points. (F-G) Radiographic images for Patient #15 taken prior to osimertinib therapy (left) and after 8 cycles of osimertinib (right). The red arrows in the CT scan images show sites of disease that responded to osimertinib.
In four cases (all Ex19Del variants), we were able to obtain tissue genomic profiling data at two unique time points. In three of these cases (Figure 3C-E), G724S allelic frequency is positively correlated with Ex19Del allelic frequency over time and decline of the T790M allele. Moreover, G724S is not present in the tumor biopsy from any of these four patients prior to Ex19Del; that is, the mutant allele frequency (MAF) of G724S starts at zero in all of these matched cases (Figure 3B – E). These data suggest that G724S emerges in a fraction of Ex19Del patients to promote disease progression.
To highlight one particular case (patient #15, Figure 3E), a 54 year old Caucasian gentleman never smoker was diagnosed with stage IV lung adenocarcinoma after presenting with abdominal pain. Tumor mutational testing was positive for an EGFR Ex19Del mutation. He was treated with first line erlotinib plus bevacizumab with partial response. Fifteen months after starting this combination therapy, he experienced progression of disease with enlargement of bilateral pulmonary nodules and a ground glass opacity in the left upper lobe. Repeat biopsy confirmed metastatic lung adenocarcinoma and tumor genetic testing at that time revealed the presence of EGFR Ex19Del and T790M mutations. He was thereafter treated with osimertinib and had a partial response lasting thirty months (Figure 3F). He experienced progression of disease with new metastases to the skull, liver, and bone. Tumor genetic testing of a repeat biopsy revealed the presence of EGFR Ex19Del, loss of T790M mutation, and gain of EGFR G724S mutation. He was treated with radiation therapy to the skull followed by systemic therapy with carboplatin and pemetrexed. Approximately four months after starting cytotoxic chemotherapy, he developed symptomatic pleural and pericardial effusions, which ultimately resulted in his demise.
Of note, G724S was also detected with the oncogenic missense mutant S768I in 2/19 cases, Shan and colleagues previously demonstrated that S768I stabilizes the active conformation by improving hydrophobic packing between the αC-helix and the ß9-strand. G724S also occurred as an individual missense mutation in 2/19 cases (Figure 3A). The latter suggests that G724S could potentially be independently oncogenic. Indeed, G724S could support oncogenic growth of Ba/F3 cells (Supplementary Figure S6A). Of note, the G724S single mutant exhibits a TKI sensitivity profile very similar to Ex19Del in that this mutant can be effectively inhibited by erlotinib, afatinib, and osimertinib (Figure 4, Supplementary Figure S6B). In addition, we identified nine cases of EGFR G724S as an isolated mutation in patients with small-cell lung carcinoma, bladder urothelial carcinoma, glioblastoma, breast cancer, and colorectal cancer (Supplementary Table S5). These data are consistent with recent evidence implicating G724S as an oncogenic driver in colorectal cancer (42) and suggest that patients with tumors harboring an isolated G724S mutation could be treated with FDA-approved EGFR TKIs, such as afatinib.
Figure 4. The EGFR G724S single mutant can be effectively inhibited by EGFR TKIs.
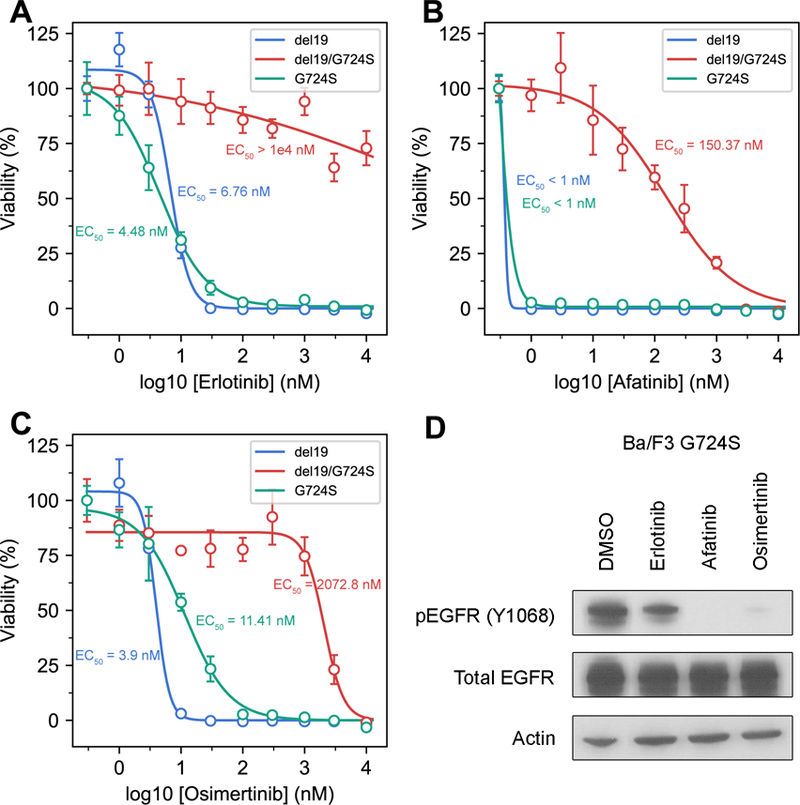
Ba/F3 cells stably expressing EGFR Ex19Del, G724S, and Ex19Del/G724S were treated with increasing amounts of (A) erlotinib, (B) afatinib or (C) osimertinib for 72 hours. CellTiter Blue assays were performed to assess cell viability. Each point represents four replicates. Data are presented as the mean percentage of viable cells compared to control ± SD. (D) Ba/F3 cells transduced with EGFR G724S were treated with either DMSO, 100 nM erlotinib, 100 nM afatinib, or 100 nM osimertinib for 4 hours. Cellular lysates were probed with the indicated antibodies.
The catalytically active conformation of EGFR is better stabilized by E746_S752>V/G724S than by E746_A750del/G724S
Unexpectedly, all of the Ex19Del alterations co-occurring with G724S in patient tumor samples were rare variants (Supplementary Table S4). The Ex19Del variant occurring most frequently with G724S in this cohort was E746_S752>V (10/19), followed by S752_I759del (3/19), E746_S752>I (1/19), and L747_S752del (1/19). For context, approximately 67% of Ex19Del cases are attributed to the canonical variant, E746_A750del, while less than 2% are attributed E746_S752>V (43). To better understand this enrichment in Ex19Del rare variants, we performed GaMD simulations for E746_S752>V and E746_S752>V/G724S in the apo-state and in reversible complex with osimertinib.
We utilized MM/GBSA to compute the relative binding free energies between the two sets of Ex19Del variants. The results displayed large statistical uncertainty in the calculation of the binding free energies (Supplementary Table S2), that we attribute to increased P-loop fluctuations in E746_S752>V and E746_S752>V/G724S relative to WT and the other variants (Supplementary Figure S1, Supplementary Movies 1 – 2). The majority of this difference is attributable to increased fluctuations in E746_S752>V, and just as in the cases of WT and E746_A750del the additional fluctuations associated with G724S in E746_S752>V occur primarily at the tip of the P-loop (Supplementary Figure S1). Nevertheless, E746_S752>V, but not E746_S752>V/G724S, is able to stabilize a favorable contact between F723 and the indole ring of osimertinib, consistent with results obtained in the previous E746_A750del and E746_A750del/G724S osimertinib-binding simulations (Supplementary Table S2).
EGFR kinase activation is achieved through asymmetric dimerization of an acceptor EGFR kinase αC-helix with a donor kinase αH-helix. The acceptor kinase is the catalytically active dimer subunit (28). In a seminal paper on EGFR dynamics, Shan and colleagues demonstrated that common oncogenic mutations increase activity by stabilizing the αC-helix inward conformation to promote asymmetric dimerization (44). We hypothesized that the unexpected enrichment of the E746_S752>V/G724S double mutant in clinical samples may result from increased stabilization of the αC-helix inward conformation in E746_S752>V/G724S relative to E746_S752>V. To test this hypothesis, we performed a detailed analysis of the conformational free energy landscape profiles of each EGFR variant in the apo-state.
Consistent with Shan and colleagues, results from our GaMD simulations of WT, E746_A750del, and L858R demonstrate increased stabilization of the αC-helix inward conformation compared to WT (44). Additionally, our simulations show that E746_S752>V stabilizes the αC-helix inward conformation relative to WT. Critically, our computational analyses suggest that E746_S752>V/G724S stabilizes the active αC-helix inward conformation even more than E746_S752>V (Figure 5E). In contrast, E746_A750del/G724S visits αC-in conformations less frequently than E746_A750del (Figure 5D). These results suggest that E746_S752>V/G724S could lead to enhanced dimerization-dependent activation compared to E746_S752>V, while E746_A750del/G724S could lead to reduced dimerization-dependent activation compared to E746_A750del.
Figure 5. Conformational free energy landscape of EGFR kinase domain mutants.
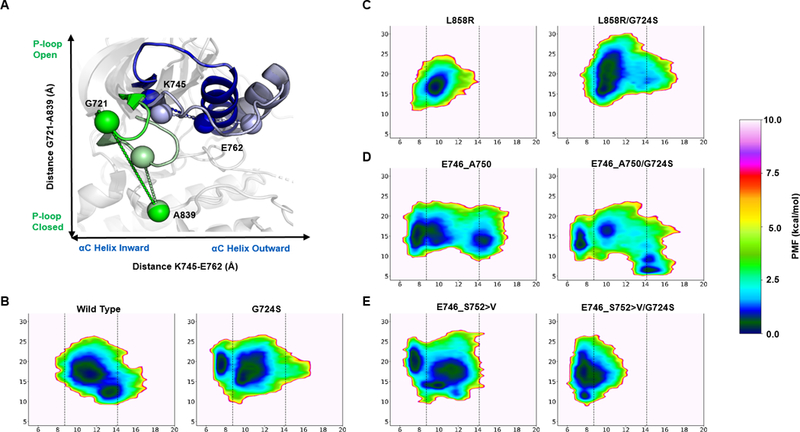
The reaction coordinate reference for the conformational free energy landscape of EGFR kinase mutants is indicated on a model of WT in the active (PDB ID 2ITX; bold colors) and inactive (PDB ID 3GT8; faded colors) conformations (A). Green spheres represent the distance (Å) between Hα1 of G721 and Cß of A839. Blue spheres represent the distance between Cß of K745 and Cß of E762. The potential of mean force (PMF) with respect to the positions of the αC helix (x-axis) and P-loop (y-axis) are plotted for WT and G724S, L858R and L858R/G724S, E746_A750 and E746_A750/G724S, and E746_S752>V and E746_S752>V/G724S (B). The left and right vertical dashed lines on the free energy plots (C-E) indicate center-of-mass distances between K745 and E762 in active (PDB ID 2GS6) and inactive (PDB ID 2GS7) EGFR kinase, respectively. The left vertical dashed lined therefore represents the canonical EGFR kinase αC-helix inward conformation, while the right vertical dashed line represents the canonical EGFR kinase αC-helix outward conformation. All depicted simulations start from the active (αC-helix inward, activation loop outward) conformation. The energetic reweighting factor was approximated with cumulant expansion to the 2nd order. Free energy landscapes from the 500 ns GaMD simulations are depicted here.
Collectively, these data support G724S as a resistance mutation in Ex19Del over L858R, and that specific Ex19Del mutants may preferentially co-occur with G724S, potentially driven by differences in active conformation stability in the presence of G724S. In addition, our results suggest that G724S (as a single point mutation) also stabilizes the αC-helix inward conformation, consistent with reports that G724S may function as an oncogenic variant in colorectal cancer (42) (Figure 5A – D; Supplementary Figure S7). Our data more broadly suggest that the underlying activating mutation profile of EGFR influences the development of drug resistance mutations. This has important implications for clinical management of patients with EGFR-mutant NSCLC.
Discussion
Notable advancements have been observed through the development of increasingly selective inhibitors of mutant oncoproteins (11). The design and clinical implementation of mutant-selective third generation EGFR TKIs, such as osimertinib, are an excellent example. Unfortunately, despite these advances, the development of resistance mutations to TKI therapy remains a significant barrier in attaining the best outcomes for patients with EGFR-mutant NSCLC. In addition to the previously identified C797S resistance mutation, our results demonstrate osimertinib resistance may emerge in the form of G724S mutations within the P-loop of the EGFR kinase domain. However, unlike C797S, our results also suggest that G724S-mediated resistance preferentially occurs in Ex19Del but not L858R. Indeed, extensive atomic-detail simulations at the structural level, multiple independent in vitro models, and patient genomic profiling all demonstrate G724S to be an Ex19Del-specific resistance mechanism to osimertinib. Retrospectively, we identified multiple patient cases now observed in the literature where patients with EGFR Ex19Del–mutant NSCLC displayed tumor progression post-osimertinib treatment in the presence of G724S (14,18–20). Together with the data we have presented here, these case studies suggest G724S functions as a resistance mutation in an allele-specific manner. To our knowledge, ours is the first evidence directly demonstrating that the underlying activating mutation (e.g. Ex19Del vs. L858R) influences the emergence of resistance mutations under selective pressure from a specific TKI.
Enhanced αC-helix stabilization in L858R results from polar interactions between the substituted arginine and neighboring negatively charged amino acids. In contrast, enhanced αC-helix stabilization in Ex19Del mutations likely results from alterations at the ß3-αC interface. Structural superimposition of our active state deletion models onto EGFR WT shows that the position of L747 in WT is occupied by S752 (WT numbering) in E746_A750del and by the inserted valine in E746_S752>V (Supplementary Figure S8). Our data suggest that the P-loop conformational changes induced by G724S lead to destabilization of the αC-helix inward conformation in the presence of polar ß3-αC interface substitutions.
Interestingly, Ex19Del/G724S displays phospho-EGFR levels similar to Ex19Del, but reduced phospho-EGFR compared to Ex19Del/C797S (Figure 2A). Our modeling suggests that stabilization of the αC-helix can vary between mutants upon introduction of G724S (Figure 5). Similarly, C797S may preferentially stabilize the αC-helix inward conformation of specific Ex19Del variants. C797 is a critical member of the structurally distinct catalytic spine (C-spine). The C-spine does not contribute to the interface formed by the glycine rich loop and ß3-αC linker region. Nevertheless, previous network analysis by McClendon et al. (45) suggests that the dynamics of the glycine rich loop and the C-spine may be highly correlated. We therefore suspect C797S may influence inter-domain correlations.
Fundamentally, our observations are similar to a concept familiar to clinical oncologists – that sequence variations in mutant proteins can impact drug binding. Osimertinib was developed to bind T790M with higher affinity than non-T790M EGFR mutants (46). Here, we show that sequence variations corresponding to the original activating mutation should also be taken into account when considering mechanisms of TKI resistance. Our findings have several important and immediate clinical implications. First, we further knowledge on a novel osimertinib resistance mutation that was not predicted by in vitro studies (21,22). Recent studies have shown that G724S may be as prevalent as C797S in osimertinib resistant tumors (20). However, there are critical differences. While C797S containing EGFR mutants (e.g., Ex19Del/C797S) regain sensitivity to first-generation EGFR TKIs, erlotinib and gefitinib, the same G724S containing EGFR variants are cross-resistant to these inhibitors. In fact, there is an ongoing phase I clinical trial (NCT03122717) of osimertinib plus gefitinib combination therapy in patients with treatment naïve advanced EGFR-mutant NSCLC. This trial aims to test the hypothesis that circumventing C797S-mediated osimertinib resistance with gefitinib will prolong response. This concept will clearly not apply for patients with G724S mediated osimertinib resistance. However, our results support a role for afatinib therapy in treating Ex19Del patients with disease progression on osimertinib via C797S or G724S in the absence of T790M (Figure 2). Furthermore, in cases where G724S is potentially an independent oncogenic driver of other cancers (Supplementary Table S5), our results suggest possible treatment strategies with existing FDA-approved inhibitors. This level of evidence is critical to nominate variants of uncertain clinical significance, such as isolated G724S mutation, for eligibility into clinical trials such as NCI MATCH (NCT02465060).
These clinical consequences are rooted in structural perturbations to EGFR kinase. Detailed mechanistic understanding of these perturbations can provide critical insight to guide therapeutic intervention. Just prior to submission of the present manuscript, Fassunke et al. published investigations into the structural basis of EGFR G724S-mediated osimertinib (20). The authors coupled structure-based alignment of EGFR WT to EGFR D770_N771insNPG (exon 20 mutation) with P-loop RMSF calculations derived from short, single-trajectory cMD simulations. Specifically, Fassunke et al. demonstrated an elevated RMSF in both WT and E746_A750del when G724S is introduced. From that result, the authors postulated two potential, opposing mechanisms of G724S-mediated third-generation TKI resistance: (1) steric repulsion of the inhibitor, or (2) loss of important interactions with the inhibitor. However, RMSF calculations alone are rarely sufficient to provide detailed mechanistic insights (38). Moreover, osimertinib resistance occurs in Ex19Del/G724S variants (Figure 2) but not G724S single mutants (Figure 4). The broad mechanisms previously posited do not provide adequate detail to address these data.
Here, we performed multiple independent GaMD enhanced sampling simulations in the presence and absence of osimertinib or afatinib totaling over 23 µs. For each EGFR mutant, we computed the relative binding free energies of osimertinib and afatinib as well as the conformational free energy landscape profiles of the apo-state structures. While our RMSF calculations are consistent with Fassunke et al., our results further suggest that G724S hyper-stabilizes a β-bend conformation of the glycine-rich P-loop. This prevents contact of the F723 phenyl ring with the osimertinib indole ring. Our calculations suggest that L858R reversibly binds osimertinib with higher affinity than Ex19Del, and consequently loss of the F723 – osimertinib contact fails to disrupt binding in L858R. In Ex19Del, the addition of G724S destabilizes the reversible complex necessary for covalent adduct formation (Figure 1, Supplementary Table S2).
Moreover, we identified differences in P-loop conformational preferences between Ex19Del/G724S and L858R/G724S. (Figure 4A – D; Supplementary Figure S7). In addition to our findings in Figure 1, it is possible that L858R/G724S is less poised to accommodate substrate binding vs. Ex19Del/G724S, resulting in L858R/G724S functioning as a catalytically inefficient receiver kinase in an asymmetric dimer (Supplementary Figure S7G – H); however, additional experiments would be required to test this hypothesis. It is also possible that L858R/G724S conformations may be less primed to support dimerization compared with L858R. The αC-helix of L858R/G724S bows outward over the course of the simulation, suggesting increased local instability. Despite still favoring the active state relative to WT, it is possible that with longer simulation times the αC-helix of L858R/G724S would more rapidly transition to a state incapable of supporting asymmetric dimerization than L858R (Figure 4C; Supplementary Figure S7G – H).
Importantly, our simulations also suggest that G724S increases the stability of the EGFR active conformation in the E746_S752 variant of Ex19Del, but reduces stability of the E746_A750del variant. Greater stability of the active αC-inward conformation in E746_S752>V/G724S offers a possible explanation for the enrichment of the rare variant Ex19Del in the Foundation Medicine cohort of NSCLC patients with G724S. Interestingly, of the four patients with genomic profiling data presented in Fassunke et al., all of them saw an increase in molecular fraction of G724S post-osimertinib therapy, and all of them had uncommon variants of Ex19Del (20).
These findings have implications in other, non-EGFR-mutant cancers as well. For example, ALK (anaplastic lymphoma kinase) rearrangements can be found in approximately 5% of NSCLC cancers (47). Over a dozen fusion partners have been identified across ALK+ cancers (47). Even the most frequently occurring fusion partner in ALK+ NSCLC, echinoderm microtubule-associated protein-like 4 (EML4), has > 10 identified unique fusion variants (48). In addition, on-target acquired resistance to first-and second-generation ALK TKIs occurs in the form of approximately a dozen unique missense mutations (49). Recent data suggests that a particularly recalcitrant ALK solvent front mutation, G1202R, is more likely to cause resistance in the context of EML4-ALK E6;A20 (V3) fusion rather than the more common EML4-ALK E13;A20 (V1) fusion (50). A structural basis for this observation was not presented; however, analogous to our current study, it could be that the unique structural and biochemical properties of the original activating mutation foreshadowed the development of a specific resistance mutation.
In summary, we have employed an interdisciplinary computational and experimental approach which provides evidence that on-target osimertinib resistance in EGFR-mutant NSCLC occurs in an allele-specific manner dependent on the underlying activating mutation. Our data support a potential structural mechanism for Ex19Del/G724S osimertinib resistance, and open the door for further studies on TKI-EGFR interactions. We hope these mechanistic studies will be exploited to develop novel EGFR TKIs that circumvent multiple drug resistance mutations. Finally, we hope that insights from our investigations will be applied to develop increasingly effective targeted therapies for additional genetically-defined cancers.
Supplementary Material
Statement of Translational Significance.
Osimertinib is the first mutant-selective EGFR tyrosine kinase inhibitor (TKI) to receive regulatory approval. Despite impressive clinical benefit, acquired resistance to osimertinib presents a significant challenge. Herein, we describe the emergence of G724S as a new mutation within the EGFR kinase domain that is detected at the time of osimertinib resistance. Notably, our in silico, in vitro, and patient level data show that G724S is an allele-specific resistance mutation emerging in the context of EGFR exon 19 deletion (Ex19Del) but not EGFR L858R. The findings are immediately translatable for: (1) Surveillance – patients with Ex19Del mutations are susceptible to G724S-mediated resistance; (2) Treatment – our results provide structural and in vitro evidence for the efficacy of afatinib in treating patients with osimertinib resistance mediated by the emergence of G724S mutation.
Acknowledgements
We gratefully acknowledge guidance from Dr. Yinglong Miao in performing GaMD simulations. We would also like to thank Jeffrey Mendenhall, Merrida Childress, and Darren Tyson for their valuable feedback and helpful discussions.
Financial support: Work in the Lovly laboratory is supported through the National Institutes of Health (NIH) and National Cancer Institute (NCI) R01CA121210, P01CA129243, and P30CA68485. CML was also supported by a Damon Runyon Clinical Investigator Award, a LUNGevity Career Development Award, a V Foundation Scholar-in-Training Award, a Lung Cancer Foundation of America / International Association for the Study of Lung Cancer Lori Monroe Scholarship. Work in the Meiler laboratory is supported through the NIH (R01GM080403, R01GM099842, R01GM073151). BPB is supported through the NIH by a Ruth L. Kirschstein NRSA fellowship (F30DK118774). DW was supported by a Ruth L. Kirschstein NRSA Fellowship (T32HL094296).
Abbreviations:
- EGFR
epidermal growth factor receptor
- TKI
tyrosine kinase inhibitor
- NSCLC
non-small cell lung cancer
- MD
molecular dynamics
- GaMD
Gaussian accelerated molecular dynamics
- cMD
conventional molecular dynamics
- WT
wild-type
- MAF
mutant allele frequency
- MM/GBSA
molecular mechanics-generalized Born surface area method
- PME
Particle Mesh Ewald
Footnotes
Author disclosures: JSR, VAM, SA, and ABS are employees of Foundation Medicine, Inc., a wholly-owned subsidiary of Roche. LB has served in Takeda, Astra Zeneca, Genentech, Pfizer, LOXO, Eli Lilly. CML has served as a consultant for Pfizer, Novartis, Astra Zeneca, Genoptix, Sequenom, ARIAD, Takeda, Foundation Medicine, Blueprints Medicine, and Cepheid and has received research funding (to her university) from Novartis and Xcovery. BPB, YZ, DW, YY, VH, HQ, ZD, JAS, and JM report no conflicts of interest.
References
- 1.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101(36):13306–11 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350(21):2129–39 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362(25):2380–8 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 4.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11(2):121–8 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 5.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13(3):239–46 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 6.Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31(27):3327–34 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 7.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19(8):2240–7 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart EL, Tan SZ, Liu G, Tsao MS. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res 2015;4(1):67–81 10.3978/j.issn.2218-6751.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol 2018;4(11):1527–34 10.1001/jamaoncol.2018.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang JC, Ahn MJ, Kim DW, Ramalingam SS, Sequist LV, Su WC, et al. Osimertinib in Pretreated T790M-Positive Advanced Non-Small-Cell Lung Cancer: AURA Study Phase II Extension Component. J Clin Oncol 2017;35(12):1288–96 10.1200/JCO.2016.70.3223. [DOI] [PubMed] [Google Scholar]
- 11.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(2):113–25 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 12.Papadimitrakopoulou VAWY, Han J, Ahn M, Ramalingam SS, John T, Okamoto I, Yang JC, Bulusu KC, Laus G, Collins B, Barrett JC, Chmielecki J, Mok TS. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol 2018;29(Suppl_8). [Google Scholar]
- 13.Ramalingam SSCY, Zhou C, Ohe Y, Imamura F, Cho BC, Lin M, Majem M, Shah R, Rukazenkov Y, Todd A, Markovets A, Barrett FC, Chmielecki J, Gray J. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol 2018;29(Suppl_8). [Google Scholar]
- 14.Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov 2018. 10.1158/2159-8290.CD-18-1022. [DOI] [PMC free article] [PubMed]
- 15.Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J Clin Oncol 2018;36(9):841–9 10.1200/JCO.2017.74.7576. [DOI] [PubMed] [Google Scholar]
- 16.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nature medicine 2015;21(6):560–2 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yosaatmadja Y, Silva S, Dickson JM, Patterson AV, Smaill JB, Flanagan JU, et al. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J Struct Biol 2015;192(3):539–44 10.1016/j.jsb.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Oztan A, Fischer S, Schrock AB, Erlich RL, Lovly CM, Stephens PJ, et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung cancer 2017;111:84–7 10.1016/j.lungcan.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Peled N, Roisman LC, Miron B, Pfeffer R, Lanman RB, Ilouze M, et al. Subclonal Therapy by Two EGFR TKIs Guided by Sequential Plasma Cell-free DNA in EGFR-Mutated Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2017;12(7):e81–e4 10.1016/j.jtho.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 20.Fassunke J, Muller F, Keul M, Michels S, Dammert MA, Schmitt A, et al. Overcoming EGFR(G724S)-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat Commun 2018;9(1):4655 10.1038/s41467-018-07078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Z, Boggon TJ, Kobayashi S, Jin C, Ma PC, Dowlati A, et al. Resistance to an irreversible epidermal growth factor receptor (EGFR) inhibitor in EGFR-mutant lung cancer reveals novel treatment strategies. Cancer Res 2007;67(21):10417–27 10.1158/0008-5472.CAN-07-1248. [DOI] [PubMed] [Google Scholar]
- 22.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res 2015;21(17):3913–23 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 2009;119(10):3000–10 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bender BJ, Cisneros A 3rd, Duran AM, Finn JA, Fu D, Lokits AD, et al. Protocols for Molecular Modeling with Rosetta3 and RosettaScripts. Biochemistry 2016;55(34):4748–63 10.1021/acs.biochem.6b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, et al. High-resolution comparative modeling with RosettaCM. Structure 2013;21(10):1735–42 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput 2017;13(6):3031–48 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D.A. Case DSCTECIIITADREDTJGHGA, Kollman PA. AMBER2017 2017.
- 28.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006;125(6):1137–49 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983;79(2):926–35 10.1063/1.445869. [DOI] [Google Scholar]
- 30.Joung IS, Cheatham TE 3rd,. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 2008;112(30):9020–41 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys 2004;120(24):11919–29 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 32.Miao Y, McCammon JA. Gaussian Accelerated Molecular Dynamics: Theory, Implementation, and Applications. Annu Rep Comput Chem 2017;13:231–78 10.1016/bs.arcc.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miao Y, McCammon JA. Graded activation and free energy landscapes of a muscarinic G-protein-coupled receptor. Proc Natl Acad Sci U S A 2016;113(43):12162–7 10.1073/pnas.1614538113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miao Y, Feher VA, McCammon JA. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J Chem Theory Comput 2015;11(8):3584–95 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller BR 3rd, McGee TD Jr., Swails JM, Homeyer N, Gohlke H, Roitberg AE MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput 2012;8(9):3314–21 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 36.Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, et al. Analytical Validation of a Hybrid Capture-Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA. J Mol Diagn 2018;20(5):686–702 10.1016/j.jmoldx.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31(11):1023–31 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farmer J, Kanwal F, Nikulsin N, Tsilimigras MCB, Jacobs DJ. Statistical Measures to Quantify Similarity between Molecular Dynamics Simulation Trajectories. Entropy 2017;19(12) 10.3390/e19120646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther 2012;343(2):342–50 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 40.Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol 2016;9(1):59 10.1186/s13045-016-0290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang YL, Yuan JQ, Wang KF, Fu XH, Han XR, Threapleton D, et al. The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget 2016;7(48):78985–93 10.18632/oncotarget.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho J, Bass AJ, Lawrence MS, Cibulskis K, Cho A, Lee SN, et al. Colon cancer-derived oncogenic EGFR G724S mutant identified by whole genome sequence analysis is dependent on asymmetric dimerization and sensitive to cetuximab. Molecular cancer 2014;13:141 10.1186/1476-4598-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi Y, Mitsudomi T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci 2016;107(9):1179–86 10.1111/cas.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shan Y, Eastwood MP, Zhang X, Kim ET, Arkhipov A, Dror RO, et al. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012;149(4):860–70 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 45.McClendon CL, Kornev AP, Gilson MK, Taylor SS. Dynamic architecture of a protein kinase. Proc Natl Acad Sci U S A 2014;111(43):E4623–31 10.1073/pnas.1418402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4(9):1046–61 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin JJ, Riely GJ, Shaw AT. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov 2017;7(2):137–55 10.1158/2159-8290.CD-16-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol 2013;31(8):1105–11 10.1200/JCO.2012.44.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular Mechanisms of Resistance to First-and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 2016;6(10):1118–33 10.1158/2159-8290.CD-16-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo-Jack I, et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J Clin Oncol 2018;36(12):1199–206 10.1200/JCO.2017.76.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.