

Abstract
Tumor angiogenesis and escape of immunosurveillance are two cancer hallmarks that are tightly linked and reciprocally regulated by paracrine signaling cues of cell constituents from both compartments. Formation and remodeling of new blood vessels in tumors is abnormal and facilitates immune evasion. In turn, immune cells in the tumor, specifically in context with an acidic and hypoxic environment, can promote neovascularization. Immunotherapy has emerged as a major therapeutic modality in cancer but is often hampered by the low influx of activated cytotoxic T-cells. On the other hand, anti-angiogenic therapy has been shown to transiently normalize the tumor vasculature and enhance infiltration of T lymphocytes, providing a rationale for a combination of these two therapeutic approaches to sustain and improve therapeutic efficacy in cancer. In this review, we discuss how the tumor vasculature facilitates an immunosuppressive phenotype and vice versa how innate and adaptive immune cells regulate angiogenesis during tumor progression. We further highlight recent results of antiangiogenic immunotherapies in experimental models and the clinic to evaluate the concept that targeting both the tumor vessels and immune cells increases the effectiveness in cancer patients.
Keywords: Angiogenesis, Immunosuppression, antiangiogenic therapy, immunotherapy immune checkpoint inhibitors, innate and adaptive immune cells, tumor hypoxia and acidosis, metabolism
Introduction
The onset of tumor neovascularization is a well-established hallmark of cancer that is initiated at a certain time during tumor progression depending on the tumor type, grade and stage. Blood vessels not only have to provide oxygen and deplete waste products to oblige the demands of a growing tumor but also form niches that enable cancer stem cell maintenance, protection and entail sites of infiltrating immune cells [1-4]. Various mechanisms for the reinstatement of blood vessel growth have been described of which angiogenesis, the sprouting of new vessels from pre-existing capillaries and postcapillary venules is the most commonly used mode [5-7]. Subsequently, tip cells anastomose with tip cells from neighboring sprouts to connect the newly formed vascular structures with the help of bridging macrophages. With the initiation of blood flow, the establishment of a basement membrane, and the recruitment of vessel-stabilizing pericytes, vascular growth is then terminated [5, 6]. Thus, under physiological conditions angiogenesis is tightly regulated by a homeostatic balance of a wealth of proangiogenic and inhibitory factors of which members of the vascular endothelial growth factor (VEGF), and angiopoietin tyrosine kinase receptor families as well as various members of the CXC and CC chemokines are the most prominent molecules to orchestrate this multistep process [1, 8]. The balance of angiogenesis-promoting and inhibiting molecules is, however, lost in tumors. Upon initiating neovascularization, tumors continue to promote angiogenic signaling for several reasons [9]. First, tumors express oncogenes and/or lose tumor suppressor genes that result in the activation of pro-angiogenic signaling pathways [10-12]. Second, expanding tumors exhibit a continually and abnormally growing tumor vasculature with a typical display of irregular-shaped, hyperdilated and poorly pericyte-covered tumor vessels that cause a leaky and sluggish blood flow [13]. These typical signs of poor vessel maturation increase interstitial pressure and generate and maintain a hypoxic and acidic environment that continues to elevate proangiogenic factors thereby further aggravating a pro-angiogenic feedback loop that never ceases.
Effects of low-oxygen tension are mediated by hypoxia-inducible factor (HIF) transcription factors that coordinate a transcriptional program to ensure metabolic and vascular adaptations under hypoxic conditions by various mechanisms [14-16]. Hypoxia upregulates various proangiogenic growth factors and chemokines that directly engage in vessel growth like VEGF, PlGF and Ang2 [17-21]. In addition, numerous experimental models have shown that both hypoxic and acidic conditions in the tumor microenvironment change the availability of metabolites and induce the secretion of molecules in tumors (e.g. CSF1, GM-CSF, G-CSF, CX3CL1, CXCL5, CXCL12, CCL17, CCL22, IL6, Sema3) that attract innate immune cells to the tumor site where they become reprogrammed to serve as an additional source of angiogenic factors [22-28]. It is important to note that innate immune cells elicit a high plasticity in order to quickly adapt and serve the homeostatic repair program. Upon injury, first neutrophils and then macrophages are recruited to the wound that are immunosupportive and angiostatic due to their responsibility to phagocytose and kill bacteria and degrade necrotic tissue. In the subsequent resolution phase, myeloid cells then convert to an angiogenic and immunosuppressive phenotype to support tissue and blood vessel restoration [29]. The ability of myeloid cells to display such opposing properties to either eliminate potential infections or hinder excessive inflammation is indeed pivotal to properly regulate tissue injury and maintain tissue homeostasis [30]. In line with the concept that tumors are wounds that never heal [31], tumors take advantage of the myeloid plasticity by reprogramming these cells to exhibit both immunosuppressive and angiogenic features to enable tumors to escape immunosurveillance and facilitate vascular expansion. These two features have been defined as pivotal hallmarks in cancer propagation and progression [32].
1. Angiogenic factors promote immunosuppression
There is increasing evidence that tumor-supporting inflammation and angiogenesis, although being distinct and separable processes, are closely related events that are in part regulated by common chemokines [32, 33]. VEGF and Ang2 are proangiogenic factors that directly and indirectly convey immunosuppression in endothelial cells as well as innate and adaptive immune cells [34-37]. VEGF induces an immunosuppressive vasculature by on different levels (Fig.1). It downregulates the expression of the vascular adhesion molecules I-CAM1 and V-CAM1 that are necessary for T-cells to cross the endothelial layer and transit into the tumor. VEGF and Ang2 have been shown to enhance the expression of the negative checkpoint regulator programmed cell death protein 1 (PD-1)– programmed cell death 1 ligand 1 (PD-L1) in endothelial cells thus disabling the cytotoxic function of PD1+-T cells [38-41], In addition, elevated FasL levels on tumor ECs is known to trigger apoptosis of Fas-expressing CD8+T cells [42]. Tumor-associated ECs also preferentially attract immunosuppressive Tregs by upregulating the multifunctional endothelial receptor CLEVER-1/stabilin-1 [43]. All these mechanisms consequently lead to a potent barrier that disables cytotoxic T cell infiltration into the tumor. VEGF, however, not only facilitates VEGFR-2 signaling in endothelial cells but also in VEGFR2+ immune cells. Thereby, it stimulates the expression of PD-L1 in dendritic cells and blocks the maturation and thus, functionality of dendritic cells due to their impaired ability to present tumor antigens [44]. VEGF/VEGFR2 signaling further suppresses proliferation of effector T cells but increases and promotes tumor homing of Tregs [32, 44]. This effect is dependent on neuropilin-1 (nrp1) because in an experimental tumor model, nrp1 depletion resulted in reduction of Tregs with a concomitant increase in cytotoxic T cells [45]. VEGF also upregulates several negative immune checkpoint receptors besides PD-1 (e.g. CTLA-4, Lag-3, TIM-3) on T cells contributing to T-cell anergy and exhaustion [46]. Finally, elevated levels of VEGF and/or Ang-2 as well as other tumor-secreted factors promote the recruitment and expansion of innate immune cell populations including TAMs, TEMs, neutrophils, MDSC and immature dendritic cells that secrete molecules fostering an immunosuppressive and angiogenic tumor microenvironment.
Figure 1. VEGF/VEGFR signaling promotes angiogenesis and suppresses antitumor immunity.
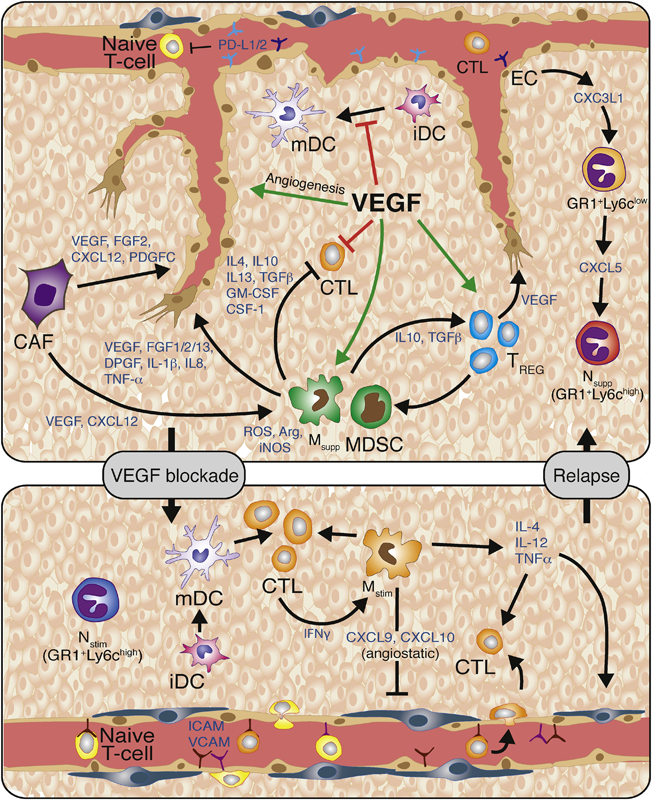
VEGF promotes endothelial cell proliferation, migration and survival. VEGF also promotes immunosuppression by inducing endothelial cell expression of the PD1 ligands PD-L1 and PD-L2 that interact with PD-1 on T-cells resulting in reduced T-cell proliferation and effector function. VEGF also inhibits T-cell-adhesion to the luminal surface of the vasculature and subsequent extravasation into the tumor by blocking TNFα-induced expression of VCAM and ICAM. VEGF can also directly block dendritic cell function by inhibiting dendritic cell maturation and inducing PD-L1 expression on mature dendritic cells. It inhibits the proliferation and effector function of cytotoxic T-cells, while inducing regulatory T-cell (Treg) proliferation. Tregs secrete various cytokines and growth factors, including IL-10, IL-4, IL-13, TGFβ1, GM-CSF, and CSF-1, which recruit angiogenic and immune-suppressive MDSC and TAMs. MDSC and TAMs then produce reactive oxygen species, nitric oxide, and arginase to suppress T-cell proliferation, viability, and activity. Thus, inhibition of VEGF should restore many of these phenotypes. Congruently, VEGF-blockade enables the endothelium to facilitate T-cell infiltration due to vessel normalization accompanied with elevated levels of I-CAM and V-CAM lymphocyte adhesion molecules. Furthermore, VEGF inhibition enables dendritic cell maturation and function, and thus increases levels of intratumoral effector T-cells. Anti-VEGF therapy also increases the presence of Th1/M1-immunosupportive/angiostatic myeloid cells (e.g; macrophages, neutrophils). Subsequently, VEGF-blockade should unleash the anti-tumor immune response and lead to increased tumor cell apoptosis. Further vessel pruning, however, creates hypoxic areas that drive the recruitment and polarization of immunosuppressive and angiogenic innate immune cells. Therefore, therapeutic approaches that promote immunosurveillance, can enhance and sustain the efficacy of antiangiogenic therapy. Abbreviations: VEGF, vascular endothelial growth factor; PDL1/2, programmed death ligand 1/2; VCAM, vascular cell adhesion molecule; ICAM, intercellular adhesion molecule; iDC, immature dendritic cell; DC, dendritic cell; CTL, cytotoxic T-cell; Treg, regulatory T-cell; ROS, reactive oxygen species; NO, nitric oxide; Arg1, arginase-1; IL-10, -4, -13, -12, - 23, -1b, -8, interleukin-10, -4, -13, -12, -23, -1b, -8; TGFβ1, transforming growth factor-β1; GM-CSF, granulocyte/monocyte-colony stimulating factor; CSF-1, colony stimulating factor-1; G- or M- MDSC, granulocytic or monocytic-myeloid derived suppressor cell; M2- TAM, M2-polarized macrophage; FGF-1, -2, -13, fibroblast growth factor-1, -2, -13; PDGF, platelet-derived growth factor; TNFα, tumor necrosis factor-α NK-cell, natural killer cell; Th1, T-helper 1.
2. Immune cells promote angiogenesis
2.1. Innate immune cells promote angiogenesis
As discussed above, tumor-associated macrophages (TAM) and tumor-associated neutrophils (TAN) can either convey angiostatic and immunosupportive or proangiogenic and immunosuppressive features, but in tumors are commonly found to polarize to an immunosuppressive as well as angiogenic phenotype [47-49]. In addition, immature Gr1+ cells with either a mononuclear or granular morphology have been identified in tumors that promote immunosuppressive functions and are therefore also termed myeloid-derived suppressor cells (M-MDSC and G-MDSC respectively) [49]. Most commonly, surface marker profiling based on expression of CD11b, F4/80, CD11c, Gr1, Ly6C, and Ly6G has been used to categorize the different tumor-infiltrating myeloid cells in experimental mouse tumor models [28, 48-50]. There is a plethora of myeloid-cell secreted mediators of angiogenesis that regulate various aspects during vessel formation including growth factors and cytokines (VEGF, FGF, TNF-α, TGF-β, PDGF-B, PIGF, Neuropilin-1, CXCL chemokines (e.g. CXCL-8,-12), semaphorins), as well as various proteases including matrix metalloproteinases (MMP-2,-7,-9, and -14) and cysteine cathepsin proteases [6, 27, 51-60]. There is also a wealth of experimental studies in various murine tumor models that have provided compelling evidence for the functional significance of myeloid cells in regulating tumor angiogenesis and subsequent tumor and metastasis progression of which some of the seminal and more recent studies will be highlighted below [24, 61, 62].
2.1.1. Tumor-associated macrophages (TAM)
One of the most prominent myeloid populations that can make up to 30% of intratumoral cells are tumor-associated macrophages (TAM) and are often associated with increased vessel density in tumors [28, 49, 63]. One of the first seminal studies underscoring the functional importance of TAMs and their production of VEGF in tumor angiogenesis were conducted in the endogenous mouse mammary virus-polyoma middle T-antigen (PyMT) tumor model, with similar results forthcoming from other experimental tumor systems [24, 64, 65]. In summary, VEGF-producing macrophages facilitated the angiogenic switch and subsequent progression to malignancy because impairing TAM activity by blocking their CSF1/CSF1R signaling pathway, or macrophage-specific deletion of VEGF, or even broadly depleting TAMs by clodronate liposomes delayed the angiogenic switch while restoration of macrophages rescued the tumor’s ability to promote angiogenesis [24, 64, 65]. Important to note is that VEGF loss in intratumoral TAMs caused are more aggressive tumor adaptation in multiple subcutaneous isograft models and the PyMT tumor model because it accelerated tumor growth due to reduced tumor apoptosis and intratumoral hypoxia. It was speculated that by just reducing angiogenesis to a certain level, more functional, normalized vessels with increased pericyte coverage had been generated that reduced intratumoral hypoxia and apoptosis, supported by the observation that myeloid-derived VEGF was sufficient to increase the susceptibility of these tumors to chemotherapeutic cytotoxicity; an effect that has been commonly attributed to the improved blood flow and oxygenation by intratumoral vessel normalization [11, 64]. Monocytes (TEMs) that express the angiopoietin receptor Tie 2 (TEK) represent a subset of macrophages that are often closely associated with blood vessels through endothelial cell secretion of the Tie2 ligand Angiopoetin-2 (Ang-2) [37]. Their relevance to tumor angiogenesis and subsequent tumor growth has been demonstrated by either selectively ablating TEM by virtue of Tie2 promoter-driven thymidine kinase expression or by antibody-mediated neutralization of the Tie2 ligand Ang2, in mammary, pancreatic neuroendocrine and brain tumor mouse model systems [66, 67]. Thereby, TEMs were found to not only instigate neovascularizarion during tumor progression but also in response to chemotherapy and radiation [68-70]. Notably, genetic deletion of Tie2 in these cells disabled their specific perivascular location and ability to promote angiogenesis [67]. As pharmacological or genetic inhibition of Tie2 in macrophages mimics the effects of Ang2 deletion in tumor endothelial cells further underscores the Ang2-Tie2 axis as a critical signaling node between TAMs and endothelial cells in promoting neovascularization [67, 71]. TAMs also express neuronal and vascular guidance molecules that regulate cell survival and migration [72, 73]. Among those, Semaphorin3A (Sema3A), like Ang2 induced by hypoxia in tumors, has also been shown to facilitate macrophage recruitment and TAM-directed angiogenesis by mediating Nrp-1-dependent signaling of a PlexinA1/PlexinA4/VEGFR1 complex. Upon activation of this holocomplex, TAMs migrated into hypoxic regions where they secreted various immunosuppressive and proangiogenic factors. However, as soon as TAMs were positioned in the hypoxic environment, Nrp1 expression was repressed by HIF2α-mediated activation of the NF-kB pathway, and the migratory response of TAMs to Sema3A terminated, trapping these cells in hypoxic areas. Congruently, loss of Nrp-1 in TAMs was sufficient to disable their infiltration into hypoxic regions and thus, their hypoxia-induced immunosuppressive conversion, impaired neovascularization and thus, delayed tumor growth and metastasis in mouse models of pancreatic, lung, breast cancer [74]. Similar findings were obtained in an experimental glioma model, where genetic and pharmacological depletion of Nrp1 in infiltrating macrophages and resident microglia exhibited antitumoral effects by their reprogramming to an immunosupportive phenotype [75].
2.1.2. Tumor -associated neutrophils (TAN) and MDSC
The other major myeloid cell types implicated in angiogenesis are neutrophils, granule-containing cells, which are the most abundant white blood cells and the first cells to be recruited to injuries. Neutrophils play a key role in the front-line defense against invading pathogens where they either phagocytose micro-organisms, release soluble antimicrobial molecules from their granules or generate neutrophil extracellular traps (NETs), web-like fibers that are composed of chromatin and serine proteases and trap and kill extracellular microbes [76, 77]. In tumors and metastases, neutrophils secrete proangiogenic factors and proteases similar to those of macrophages, most predominantly VEGF, FGF and MMPs [47, 78, 79]. Interestingly, neutrophils can carry VEGF-enriched granules which are released upon TNF stimulation and enable a quick route of VEGF accessibility to promote blood vessel growth [80]. In line with this observation, neutrophils have been shown to release MMP-9 to liberate sequestered VEGF from the extracellular matrix (ECM) in dysplastic pancreatic islet lesions of Rip1Tag 2 mice. Released VEGF triggered the onset of angiogenesis in these premalignant lesions and enabled malignant conversion and progression [59, 81]. This was indeed a crucial mechanism because neutrophil depletion with antibodies against Gr1 substantially reduced the angiogenic switch in these pancreatic islet lesions [81]. Conversely, G-CSF-stimulated neutrophils upregulated the expression of the angiogenic factor Bv8 (prokineticin-2) in several tumor models which stimulated endothelial cell survival, migration and proliferation and attracted more neutrophils thus, providing a positive feedback loop for neutrophil recruitment and activation [82].
Tumors also recruit myeloid-derived immunosuppressive cells (MDSC), immature myeloid cells that can be divided into granulocytic (G-MDSC) and monocytic (M-MDSC) cell populations and convey not only immunosuppressive but also angiogenic properties [28, 83]. MDSC had first been studied for their ability to suppress human CD3+ and mouse CD4+ or CD8+ T cells [84]. In context to angiogenesis, MDSC-produced VEGF was sufficient to suppress the expression of the adhesion molecules ICAM1 and VCAM1 in tumor-associated endothelial cells, hence limiting T cell adhesion and extravasation [85, 86]. Several factors including G-CSF, IL-1β and IL-6, recruit, activate and expand MDSCs in the tumor via STAT3 activation stimulating their pro-angiogenic properties while blocking their differentiation into neutrophils or macrophages respectively [87, 88]. It is therefore not surprising that their pro-angiogenic expression profile and functions largely overlap with those of mature macrophages and neutrophils [79, 89].
2.2. Adaptive immune cells regulate angiogenesis
The implications of lymphocyte in tumor angiogenesis is poorly explored but recent results suggest that they have the ability to directly and indirectly regulate blood vessel growth and modulation. B-cells can directly promote angiogenesis by producing proangiogenic factors such as VEGF, FGF2, and MMP-9 [90], or indirectly by polarizing macrophages to an immunosuppressive and proangiogenic phenotype in an IgG -dependent manner [91]. T-cells on the other hand, appear to negatively or positively regulate tumor angiogenesis dependent on the T-cell type. CD8+ T-cells and CD4+ T-helper 1 cells produce IFNγ that restrains endothelial cell proliferation and induces the production of the angiostatic chemokines CXCL 9,10 and 11 in TAMs [47, 92]. In contrast, Treg cells suppress INFγ - expressing CD4+ Th1 cells and secrete VEGF via hypoxia-induced CCL28, which both contribute to a proangiogenic tumor environment [93]. Congruent with these results, intratumoral CD4+ and CD8+ T-cell depletion displayed an increase in dysfunctional tumor vessels and subsequent hypoxia which was reverted by CD8 influx and activity through checkpoint immunotherapy (anti-PD1 and/or anti-CTLA4) or adoptive TH1 transfer in models of murine tumors and patient-derived tumors inducing tumor vessel normalization and reduced both hypoxia and metastases [94].
3. Metabolic pathways in immune cells regulate angiogenesis
The tumor induces a major disturbance in tissue homeostasis and cellular metabolism by endorsing a hypoxic and acidic microenvironment that strongly affects metabolic availability not only for tumor cells but also other cell constituents within the tumor (Fig.2). Although it is well-established that intratumoral hypoxia induces the recruitment of immunosuppressive and angiogenic myeloid cells to the tumor site, it is less understood how subsequent changes in metabolite availability for immune cells affect their ability to support blood vessel growth. Exposure to low pH and hypoxia impacts immune cells in different ways dependent on the immune cell subtype and subsequently leads to escape of immunosurveillance, angiogenesis and cancer progression [95, 96]. T and NK cells appear to lose their antitumor function and become anergetic and apoptotic while regulatory T cells are engaged to block cytotoxic T-cell activity, and myeloid cells become immunosuppressive which all together sustains tumor growth. Under normoxia, glycolytic pyruvate enters the tricarboxylic acid (TCA) cycle for oxidative phosphorylation (OXPHOS), whereas lactate conversion from pyruvate is enhanced in anaerobic conditions. In cancer cells, pyruvate to lactate conversion occurs already in the presence of oxygen due to metabolic alterations producing metabolic acidosis with an intratumoral pH of 6.0–6.5 [97]. Under low oxygen conditions, tumor cells increase their glycolytic flux, resulting in a significant secretion of lactate which induces an acidic environment [95, 96]. There is increasing evidence that tumor-secreted lactate affects the behavior of intratumoral TAMs because it induces the expression of VEGF and Arginase-1 involved in suppressing T-cell responses; these are hallmarks of TAM polarization to an immunosuppressive and angiogenic phenotype [98]. Endothelial cells undergo metabolic changes when they become activated to form new vessels. Despite their exposure to high oxygen levels, endothelial cells, like tumor cells, divert the majority of pyruvate to lactate using aerobic glycolysis [99, 100]. Similarly, Teff cells predominantly use glycolysis for sufficient ATP synthesis. Consequently, the metabolic similarities between immune cells, endothelial cells and tumor cells potentiate a competition for several substrates (Fig.2).
Figure 2. Metabolic competition in the tumor microenvironment.
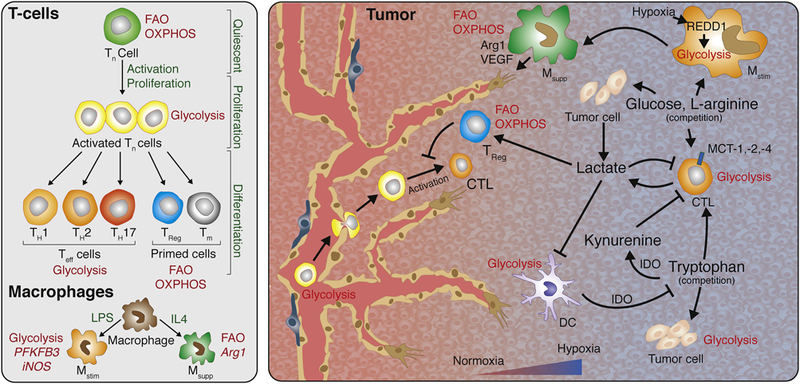
Left: Metabolic changes during T-cell development, activation, and differentiation. Quiescent Tn cells rely primarily on FAO and OXPHOS, but change to glycolysis for rapid proliferation when activated. Upon further differentiation TH1, TH2 and TH17 cells remain glycolytic, while Treg and Tm cells switch back to FAO and OXPHOS. Unlike LPS stimulated Mstim macrophages that are characterized by a primarily glycolytic metabolism, IL4 stimulated Msupp macrophages are characterized by an oxidative metabolism. Right: Intratumoral competition for metabolites. Immune cell function in the tumor microenvironment is strongly regulated by the oxygen and nutrient availability. Poorly perfused areas can induce a hypoxic response, stimulating glycolysis and lactate dependent acidification. These environmental changes affect macrophage polarization and immune cell function. Lactate as by-product of glycolysis directly suppresses CTLs and DCs, but can can be used as carbon source for Treg cells, promoting an immune suppressive tumor microenvironment. In addition to tumor cells, several immune cells including CTLs, Mstim macrophages (regulated by a REDD1 dependent hypoxia response), and DCs rely on glycolysis, making them compete for the available glucose. Other nutrients such as the amino acids L-arginine and tryptophan are also subjected to cellular competition. Several intratumoral cell types are L-arginine auxotroph, and both tumor cells and CTLs are depending on tryptophan for their function. Increased IDO activity in tumors results in tryptophan depletion and formation of the immune suppressive kynurenine, rendering the tumor microenvironment immunosuppressive. Arg1, arginase 1; CTL, cytotoxic T-lymphocyte; DC, dendritic cell; FAO, fatty acid oxidation; IDO, Indolamine 2,3- diogygenase; IL4, interleukin 4; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; Mstim, Immune stimulatory macrophage, Msupp, Immune suppressive macrophage OXPHOS, oxidative phosphorylation; PFKFB3, 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3; Teff, Effector T-cell; TH, T-helper cell; Tn, naive T-cell; Tm, mature T-cell; Treg, regulatory T-cell; VEGF, vascular endothelial growth factor.
A metabolic competition between TAMs and endothelial cells was elegantly demonstrated in a recent study. Hypoxic TAMs were shown to express very high levels of REDD1, a negative regulator of mTOR, which blocked glycolysis in TAMs and thus made more glucose available for endothelial cells to form new blood vessels. Remarkably, genetic deletion of REDD1 in TAMs increased their glycolytic rate to a level that it competed with neighboring endothelial cells for glucose and suppressed their angiogenic activity. Indeed, competition over glucose induced vessel normalization and improved oxygenation in tumors resulting in growth reduction [101]. In support of a reciprocal regulation between TAMs and endothelial cells, tumor endothelial cells can sustain the angiogenic/immunosuppressive phenotype in TAMs by generating a positive metabolic feedback loop in cancer that keeps tumor vessels in an angiogenic mode. In an experimental model of glioblastoma (GBM), EC-secreted IL6 and a more diffuse release of CSF1 promoted the conversion to angiogenic/immunosuppressive TAMs in a HIF-2α-dependent manner [102]. Both, IL6 and CSF1 signaling in TAMs activated peroxisome proliferator-activated receptor gamma (PPAR-γ), a key transcriptional factor involved in the control of lipid uptake and glucose metabolism, via HIF-2α induction [103]. Endothelial-specific deletion of the IL6 gene was sufficient to reduce microvascular proliferation in the tumor, decrease the expression of the immunosuppressive marker arginase-1 in TAMs and increase the survival of GBM-bearing mice [102]. Besides changes in glucose metabolism, amino acid metabolites can also be dysregulated in tumors and affect immune cell behavior. Increased IDO activity in tumors has been shown to deplete tryptophan and accumulate kynurenine and its derivatives, thereby inhibiting the activation and proliferation of immune cells and rendering the tumor microenvironment immunosuppressive and angiogenic. A variety of tumor cells also lack the enzyme argininoscuccinate synthetase-1, the enzyme responsible for L-arginine synthesis, which makes tumor cells completely dependent on TAM-supplied L-arginine [104, 105]. The activity of two the L-arginine consuming enzymes nitric oxide synthase (NOS) and Arginase is strongly upregulated in several tumor types [106-109]. These enzymes generate reactive nitrogen species which are shown to impair T-cell mediated immune responses [110].
Another important metabolic pathway in macrophages entails the amino acid arginine. While the arginine-degrading enzyme arginase-1 is more abundantly expressed in angiogenic/immunosuppressive TAMs and degrades arginine into ornithine and urea, inducible nitric oxide synthetized (iNOS, or Nos2) is enriched in antitumoral TAMs and generates citrullin and nitric oxide from arginine and NADPH. Arginine depletion by macrophage-expressing arginase-1 has been linked to the inhibition of anti-tumor T cell responses [111, 112]. Similarly, nitric oxide release by hypoxic TAMs is immunosuppressive [113]. Important to note however is that local γ-irradiation of patient and murine pancreatic tumors induced iNOS in TAMs which in turn normalized tumor blood vessels, recruited host T cells as well as adoptively transferred cytotoxic T cells [114]. These results underscore the direct and indirect positive antitumor effects of macrophage-secreted nitric oxide on blood vessels and T-cell influx and activation [114] which is in contrast to the immunosuppressive function of nitric oxide released by hypoxic TAMs in avascular areas of the tumor [113]. All together, these results reiterate the impact of metabolic changes in regulating the pleiotrophic functions of immune cells in angiogenesis and immunosuppression [115].
4. Immune cells facilitate resistance to antiangiogenic therapy
Compelling data from multiple laboratories have provided a convincing rationale for the development of VEGF and VEGF receptor inhibitors to block intratumoral vascular growth and subsequent tumor propagation. Subsequently, bevacizumab (Avastin, Genentech/Roche), a monoclonal antibody against VEGF, as well as sorafenib (Nexavar, Bayer) and sunitinib (Sutent, Pfizer), both kinase inhibitors that target VEGF receptor (VEGFR) were the first FDA-approved angiogenic inhibitors followed by other angiogenic tyrosine kinases, aflibercept, another VEGF-Trap antibody (Regeneron) and, more recently Ramucirumab, an antibody against VEGFR2 (ImClone/Lilly) [116]. Although encouraging effects of these inhibitors were observed in a subset of cancer patients, antiangiogenic therapy has revealed very limited and transient beneficial effects by improving progression-free-survival and quality-of-life but not overall survival, and only in a fraction of cancer patients [11, 117, 118]. Interestingly, however, experimental studies in which the treatment was beneficial, discovered that albeit tumor vessels were pruned, they also exhibited enhanced pericyte coverage concomitant with a more functional morphology and subsequent improvement in blood flow and oxygenation which was coined by Rakesh Jain as “vessel normalization” [119]. Importantly, such vascular changes were also observed by functional MRI analysis in cancer patients undergoing anti-VEGFR therapy. Normalization of tumor vessels during VEGF blockade has shown to improve drug and oxygen delivery thereby reducing intratumoral hypoxia which in turn contributed to a more immunosupporting tumor environment associated with increased CD8 cytotoxic T cell influx [120] (Fig.1). Anti-VEGF- induced vascular normalization is however transient and continuous treatment will result in further pruning and restoration of hypoxia leading to various adaptation mechanisms to overcome vascular growth restrictions (Fig.1). These imply at least two major relapse categories, in which tumors either reinstate growth by revascularization or alter their growth behavior without obligate neovascularization [121]. Adaptation to vascular growth restrictions also involve the recruitment of myeloid cells instigated by therapy-induced hypoxia which like during tumor progression, induce similar factors that mobilize innate immune cells from the bone marrow and attract them to the tumor site [27, 67]. Here, myeloid cells sustain vascular growth by stimulating VEGF-independent pathways. In response to angiogenic inhibition, TAMs, TEMs and neutrophils enhance the expression of various other angiogenic molecules besides VEGF like Ang2, FGF-1,2, chemokines, and MMP9 [122-124]. Specifically, neutrophils and MDSC (often only referred to as Gr1+/Ly6G+ immune cells) were recruited in several tumor model systems relapsing from antiangiogenic therapy and thereby endorsed resistance to VEGF blockade by secreting increased levels of proangiogenic molecules including Bv8 which exerts direct proangiogenic effects on endothelial cells [89, 125]. One of the underpinning mechanisms that lead to enhanced neutrophil infiltration by VEGF inhibition was facilitated by TH17A T-cell-induced IL17 which in turn enhanced intratumoral levels of the neutrophil attractant CSF-3 (GM-CSF) [126]. A more recent study described the implication of CXCL5-secreting Ly6Clo monocytes in the recruitment of neutrophils during VEGF inhibition [127]. Interestingly, CX3CR1+ Ly6Clo monocytes also entered the tumor when VEGF signaling was blocked due to upregulation of CX3CL1 on tumor endothelial cells [127]. Besides neutrophils, macrophages also possess the capacity to protect tumors from the deleterious effects of antiangiogenic therapy. Upregulation of the hypoxia-regulated factor Ang2 activated the Ang2-Tie2 axis and enabled enhanced infiltration of TIE2-expressing macrophages (TEMs) in pancreatic and breast tumor models undergoing VEGFR2 inhibition. Conversely, dual ANG2/VEGFR2 blockade targeted both Tie2+TEMs and VEGFR2 on endothelial cells, and thus, suppressed revascularization and progression in tumors [122]. Importantly, dual Ang2-VEGFR2 blockade prolonged vessel normalization and CD8 influx in comparison to monotherapies thereby enhancing survival in different mouse tumor models of brain, pancreas and breast [41]. TEMs also protected spontaneous MMTV-PyMT mammary tumors treated with the vascular-disrupting agent combrestatin A4 phosphate (CA4P) that induced substantial intratumoral hypoxia due to tumor vessel destruction which in turn enhanced CXCL12 expression and subsequent infiltration of CXCR4+ TEMs. Conversely, combined CA4P treatment and CXCR4 inhibition restrained TEM influx, enhanced CA4P-induced tumor necrosis, and thus sustained response and further reduced tumor burden [128]. It is important to reiterate that the different intratumoral myeloid cells express a redundant profile of multiple angiogenic factors that enables them to compensate for each other if necessary. Indeed, depletion of TAMs lead to an increase of neutrophils and targeting TANs cause an enhanced influx of TAMs, an oscillating pattern between these immune cell populations that disabled the efficacy of antiangiogenic therapy in the pancreatic Rip1Tag2 tumor model [89]. While most tumor model systems will respond to antiangiogenic therapy by slowing down tumor growth, PNETs in Rip1Tag2 mice first responded very well to VEGF signaling blockade accompanied by tumor growth inhibition and vessel normalization, but then exhibited an pro-angiogenic relapse within weeks dependent on the specific drug regimen [124, 129, 130]. During the response phase, intratumoral myeloid cells in PNET tumors became angiostatic and immunostimulating which was associated with the upregulation of CXCL14 and other angiostatic chemokines leading to an influx of cytotoxic CD8 cells. In relapsing tumors, however, myeloid cells converted back into an immunosuppressive and angiogenic phenotype and CD8 influx stopped. The underlying mechanism is based on the activation of PIA3Kγ signaling in myeloid cells by hypoxia-induced factors like CXCL12 and IL6 that rendered the cells insensitive to VEGF/R inhibition. PI3Kγ activated myeloid cells promoted reneovascularization even in the presence of VEGF/R inhibitors and caused a proangiogenic tumor relapse [89]. In support of these results, myeloid PI3Kγ signaling was shown to inhibit NFκB while stimulating C/EBPβ activation, thereby inducing a transcriptional program that promoted immunosuppression [131]. Thus, pharmacological inhibition of myeloid PI3Kγ/d improved and sustained the tumor response to antiangiogenic therapy by converting all innate immune cells to an angiostatic and immunostimulatory state associated with enhanced cytotoxic T cell infiltration and activity [89].
5. Antiangiogenic Therapy meets Immunotherapy
Various studies including those mentioned above, have provided compelling evidence that antiangiogenic therapy is most efficacious when an immunostimulatory environment is generated. This is in agreement with the concept of vascular normalization to reduce hypoxia and polarize innate immune cells to an immunosupportive phenotype. Emerging from these studies is the proposition that angiogenesis and inflammation are reciprocally regulated and that immune cells play a pivotal role in regulating both processes. It also underscores the tumor’s vigilance to conserve an immunosuppressive and angiogenic milieu to overcome growth restrictions and escape immune surveillance. Current clinical immunotherapies, most notably those using immune checkpoint inhibitors (ICI) that reactivate exhausted cytotoxic T-cells, have shown unprecedented outcomes in patients with melanoma and non-small lung cancers which lead to the accelerated FDA-approval of ipilimumab, an inhibitory antibody against the immune checkpoint receptor CTLA4 and pembrolizumab, an antibody against the PD1 receptor [132-136]. The beneficial effects by ICIs are however only observed in 15-40% of patients and there is currently no clear explanation for the differing responses. Besides insufficient abundance of tumor antigens and redundancy of negative checkpoint regulators, lack of sufficient cytotoxic T-cell-infiltration into the tumor has been suggested as a major hurdle for several tumor types. As anti-angiogenic therapy modulates the tumor vasculature and thus can enhance T-cell infiltration as well as generate myeloid cells with immunosupporting features, combining antiangiogenic therapies with ICI or immunotherapies that modulate the immune system are attractive options to increase the percentage and endurance of efficacy in cancer patients. In support, combination of the VEGFR inhibitor axitinib and anti-CTLA4 but not monotherapies substantially prolonged survival in a melanoma mouse model by increasing effector T-cell influx and dendritic cell maturation and reducing intratumoral MDSC [137]. The rationale of antiangiogenic immunotherapy is further supported by recent studies in which tumors relapsing from antiangiogenic therapy using an anti-VEGF or dual anti-VEGF-Ang2 antibody, upregulated the negative immune checkpoint regulator PD-L1 in tumor and stromal cells [38, 41]. This lead to immunosuppression triggered by PD-L1 binding PD-1 on the surface of activated T-cells to produce T-cell anergy or exhaustion. Combining immunotherapy using anti-PD-L1 with antiangiogenic therapy (either anti-VEGF or anti-VEGF/Ang2) had reciprocal beneficial effects in that immunotherapy targeted evasion from antiangiogenic therapy, while vascular normalization elicited by antiangiogenic treatment could increase lymphocyte infiltration and activation [38, 41]. In addition, antiangiogenic immunotherapy induced a specialized form of blood vessels in treated tumors that benefited from the therapy reminiscent of high endothelial venules (HEVs). HEVs are specialized blood vessels typically found in secondary lymphoid organs where they facilitate lymphocyte trafficking [138, 139]. They also have been found to sporadically appear in tumors and metastasis of patients forming tertiary lymphoid structures where they correlated with good prognosis and patient survival [138]. Congruently, intratumoral HEVs in these tumor models substantially enhanced CTL infiltration, activity and tumor cell destruction leading to improved outcome [38]. Little is known about the mechanistic underpinnings for HEV induction in tumors but it is likely to be dependent on the LIGHT/lymphotoxin-β receptor (LTβR) signaling axis in tumor-associated endothelial cells which was induced in HEV+ tumors [38, 140, 141]. In support, vessel-directed targeting of LIGHT peptide in experimental tumors normalized blood vessels and induced HEV formation and enhanced intratumoral lymphocyte infiltration [140]. Moreover, LTβR activation could also sensitize tumors to antiangiogenic immunotherapy by enhancing T-cell influx suggesting that treatments that induce not only vessel normalization but also intratumoral HEVs may enhance the therapeutic effects with immunotherapy [38, 141]. These preclinical studies support the notion that anti-angiogenic therapy can improve immunotherapy by supporting vascular changes such as vessel normalization and HEV formation but also demonstrated that immune checkpoint inhibitors can sensitize and prolong efficacy of VEGF signaling blockade. In line with this concept, a recent study demonstrated T-lymphocyte infiltration promoted blood vessel normalization in tumors [94, 142]. Mice depleted of CD4 and CD8 T-cells developed tumors that exhibited more abnormal tumor vessels and hypoxic areas than those of wildtype mice, while activation of cytotoxic T-cells through checkpoint immunotherapy (anti-PD1 and/or anti-CTLA4) or adoptive TH1 transfer in models of murine tumors and patient-derived tumors induced blood vessel normalization and reduced both hypoxia and metastases. Indeed, transcriptional profiling of patient tumors revealed a “blood vessel normalization” signature associated with good prognosis that also implemented the T-cell receptor signaling pathway [94, 142]. These studies provide strong evidence in human tumors that blood vessel normalization and T-lymphocyte infiltration can amplify the positive effects conferred by each individual component. What all of these studies reveal is the notion that tumor vessels need to be remodeled to enable enhanced T-cell infiltration and improve immunotherapies while immunotherapies themselves can contribute to a more functional tumor vasculature providing a strong positive feedback loop that can help to restrain and eradicate tumor growth.
Conclusions
Currently, various combinatorial treatment modalities of angiogenic inhibitors, specifically those targeting the VEGF/R and Ang2/Tie axis with immune checkpoint inhibitors and other immunotherapies are evaluated in the clinical setting [116]. These trials have been initiated based on compelling evidence from mouse tumor model systems that such a combination modulates both the tumor vasculature and the immune system to foster an immunosupportive environment and thus improves and endures efficacy in cancer patients. While these clinical trials are still ongoing, the first randomized study (IMmotion150 (NCT01984242) was just published which evaluated the clinical activity of the combination of anti-PD-L1 (atezolizumab) with or without bevacizumab, against the standard- of-care angiogenic tyrosine kinase inhibitor, sunitinib, in patients with untreated renal cell cancer (mRCC) [143]. The combination of anti-PD-L1 plus bevacizumab produced encouraging and superior efficacy versus sunitinib, specifically in the subgroup of patients with tumors expressing PD-L1. While tumor mutation and neoantigen burden was not associated with progression-free survival (PFS), angiogenesis, T-effector/IFN-γ response, and myeloid inflammatory gene expression signatures were strongly and differentially associated with PFS within and across the treatments suggestive of using those as potential biomarkers [143] [144]. Notable is also the identification of myeloid inflammation as a potential resistance mechanism to anti-PD-L1 monotherapy in mRCC which may be overcome by the addition of bevacizumab. These first clinical results are encouraging and indeed confirm some of the results obtained in the preclinical setting. Results from other clinical trials will hopefully soon be available to validate the potency of antiangiogenic immunotherapy cocktails in improving the efficacy and survival of cancer patients.
Acknowledgments
Sources of funding
This work was supported by grants from the ERC CoG ImmunoFIT Methusalem (to MM) and from the Flamish government FWO (G066515N) (to MM) and FWO (G0A0818N) (to GB) and the Belgian Association Against Cancer for the project 2014-197 (to MM) and from the National Institute of Health NIH/NCI (to GB).
Abbreviations
- VEGF
vascular endothelial growth factor
- TNF-α
tumor-necrosis-factor a
- PDGFB
platelet-derived growth factor B
- PIGF
placental growth factor
- Sema3A
Semaphorin3A
- TAM
Tumor-associated macrophages
- TEM
Tie2-expressing macrophages
- TAN
Tumor-associated neutrophils
- PD-L1
programmed cell death 1 ligand 1
- PD-1
programmed cell death receptor 1
- PyMT
virus-polyoma middle T- antigen
- ICI
immune checkpoint inhibitors
- (ICB)
immune checkpoint blockade
- VCAM
vascular cell adhesion molecule
- ICAM
intercellular adhesion molecule
- iDC
immature dendritic cell
- DC
dendritic cell
- CTL
cytotoxic T-cell
- Treg
regulatory T-cell
- ROS
reactive oxygen species
- NO
nitric oxide
- iNOS
inducible nitric oxide synthase
- Arg1
arginase-1
- IL-10, -4, -13, -12, -23, -1b, -8
interleukin-10, -4, -13, -12, -23, -1b, -8
- TGFβ1
transforming growth factor-β1
- GM-CSF
granulocyte/monocyte-colony stimulating factor
- CSF-1
colony stimulating factor-1
- G- or M- MDSC
granulocytic or monocytic-myeloid derived suppressor cell
- /Th2M2-TAM
M2-polarized macrophage
- FGF-1, -2, -13
fibroblast growth factor-1, -2, -13
- PDGF
platelet-derived growth factor
- TNFα
tumor necrosis factor-α
- NK-cell
natural killer cell
- Th1
T-helper 1
- FAO
fatty acid oxidation
- IDO
Indolamine 2,3-diogygenase
- LPS
lipopolysaccharide
- Mstim
Immune stimulatory macrophage
- Msupp
Immune suppressive macrophage
- OXPFIOS
oxidative phosphorylation
- PFKFB3
6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3
- Teff
Effector T-cell
- TH
T-helper cell
- Tn
naïve T-cell
- Tm
mature T-cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
RM, MM and GB declare no competing interests
References
- 1.Bergers G & Benjamin LE (2003) Tumorigenesis and the angiogenic switch, Nat Rev Cancer. 3, 401–10. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J (2007) Is angiogenesis an organizing principle in biology and medicine?, J Pediatr Surg. 42, 1–11. [DOI] [PubMed] [Google Scholar]
- 3.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A & Gilbertson RJ (2007) A perivascular niche for brain tumor stem cells, Cancer Cell. 11, 69–82. [DOI] [PubMed] [Google Scholar]
- 4.Hambardzumyan D & Bergers G (2015) Glioblastoma: Defining Tumor Niches, Trends Cancer. 1, 252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmeliet P (2005) Angiogenesis in life, disease and medicine, Nature. 438, 932–6. [DOI] [PubMed] [Google Scholar]
- 6.Adams RH & Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis, Nat Rev Mol Cell Biol. 8, 464–78. [DOI] [PubMed] [Google Scholar]
- 7.Rivera L, Pandika M & Bergers G (2014) Escape mechanisms from antiangiogenic therapy: an immune cell's perspective, Adv Exp Med Biol. 772, 83–99. [DOI] [PubMed] [Google Scholar]
- 8.Santoni M, Bracarda S, Nabissi M, Massari F, Conti A, Bria E, Tortora G, Santoni G & Cascinu S (2014) CXC and CC chemokines as angiogenic modulators in nonhaematological tumors, Biomed Res Int. 2014, 768758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Folkman J (2000) Tumor angiogenesis in Cancer Medicine (al., H. e., ed), Decker BC, Hamilton, Ontario. [Google Scholar]
- 10.Carmeliet P & Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis, Nature. 473, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy, Science. 307, 58–62. [DOI] [PubMed] [Google Scholar]
- 12.Jain RK (2013) Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers, J Clin Oncol. 31, 2205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baluk P, Hashizume H & McDonald DM (2005) Cellular abnormalities of blood vessels as targets in cancer, Curr Opin Genet Dev. 15, 102–11. [DOI] [PubMed] [Google Scholar]
- 14.Liao D, Corle C, Seagroves TN & Johnson RS (2007) Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression, Cancer Res. 67, 563–72. [DOI] [PubMed] [Google Scholar]
- 15.Palazon A, Goldrath AW, Nizet V & Johnson RS (2014) HIF transcription factors, inflammation, and immunity, Immunity. 41, 518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza GL (2011) Oxygen sensing, homeostasis, and disease, N Engl J Med. 365, 537–47. [DOI] [PubMed] [Google Scholar]
- 17.Keith B, Johnson RS & Simon MC (2011) HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression, Nat Rev Cancer. 12, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeCouter J, Kowalski J, Foster J, Hass P, Zhang Z, Dillard-Telm L, Frantz G, Rangell L, DeGuzman L, Keller GA, Peale F, Gurney A, Hillan KJ & Ferrara N (2001) Identification of an angiogenic mitogen selective for endocrine gland endothelium, Nature. 412, 877–84. [DOI] [PubMed] [Google Scholar]
- 19.Simon MP, Tournaire R & Pouyssegur J (2008) The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1, J Cell Physiol. 217, 809–18. [DOI] [PubMed] [Google Scholar]
- 20.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD & Semenza GL (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1, Mol Cell Biol. 16, 4604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA & Semenza GL (2003) Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1, Circ Res. 93, 1074–81. [DOI] [PubMed] [Google Scholar]
- 22.Coussens LM, Tinkle CL, Hanahan D & Werb Z (2000) MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis, Cell. 103, 481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giraudo E, Inoue M & Hanahan D (2004) An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis, J Clin Invest. 114, 623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN & Pollard JW (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer, Cancer Res. 66, 11238–46. [DOI] [PubMed] [Google Scholar]
- 25.Bingle L, Lewis CE, Corke KP, Reed MW & Brown NJ (2006) Macrophages promote angiogenesis in human breast tumour spheroids in vivo, Br J Cancer. 94, 101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP & Ferrara N (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells, Nat Biotechnol. 25, 911–20. [DOI] [PubMed] [Google Scholar]
- 27.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z & Bergers G (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion, Cancer Cell. 13, 206–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murdoch C, Muthana M, Coffelt SB & Lewis CE (2008) The role of myeloid cells in the promotion of tumour angiogenesis, Nat Rev Cancer. 8, 618–31. [DOI] [PubMed] [Google Scholar]
- 29.Reinke JM & Sorg H (2012) Wound repair and regeneration, Eur Surg Res. 49, 35–43. [DOI] [PubMed] [Google Scholar]
- 30.Kasuya A & Tokura Y (2014) Attempts to accelerate wound healing, J Dermatol Sci. 76, 169–72. [DOI] [PubMed] [Google Scholar]
- 31.Dvorak HF (2015) Tumors: wounds that do not heal-redux, Cancer Immunol Res. 3, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motz GT & Coukos G (2011) The parallel lives of angiogenesis and immunosuppression: cancer and other tales, Nat Rev Immunol. 11, 702–11. [DOI] [PubMed] [Google Scholar]
- 33.Rivera LB & Bergers G (2015) Intertwined regulation of angiogenesis and immunity by myeloid cells, Trends Immunol. 36, 240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Augustin HG, Koh GY, Thurston G & Alitalo K (2009) Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system, Nat Rev Mol Cell Biol. 10, 165–77. [DOI] [PubMed] [Google Scholar]
- 35.Saharinen P, Eklund L, Pulkki K, Bono P & Alitalo K (2011) VEGF and angiopoietin signaling in tumor angiogenesis and metastasis, Trends Mol Med. 17, 347–62. [DOI] [PubMed] [Google Scholar]
- 36.De Palma M, Biziato D & Petrova TV (2017) Microenvironmental regulation of tumour angiogenesis, Nat Rev Cancer. 17, 457–474. [DOI] [PubMed] [Google Scholar]
- 37.De Palma M, Murdoch C, Venneri MA, Naldini L & Lewis CE (2007) Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications, Trends Immunol. 28, 519–24. [DOI] [PubMed] [Google Scholar]
- 38.Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, Feyen K, Tawney J, Hanahan D, Michael IP & Bergers G (2017) Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation, Sci Transl Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouzin C, Brouet A, De Vriese J, Dewever J & Feron O (2007) Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy, J Immunol. 178, 1505–11. [DOI] [PubMed] [Google Scholar]
- 40.Griffioen AW, Damen CA, Martinotti S, Blijham GH & Groenewegen G (1996) Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors, Cancer Res. 56, 1111–17. [PubMed] [Google Scholar]
- 41.Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, Kienast Y, Mueller HJ, Ooi CH, Laoui D & De Palma M (2017) Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade, Sci Transl Med. 9. [DOI] [PubMed] [Google Scholar]
- 42.Motz GT & Coukos G (2013) Deciphering and reversing tumor immune suppression, Immunity. 39, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaaf MB, Garg AD & Agostinis P (2018) Defining the role of the tumor vasculature in antitumor immunity and immunotherapy, Cell Death Dis. 9, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D & Carbone DP (1996) Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells, Nat Med. 2, 1096–103. [DOI] [PubMed] [Google Scholar]
- 45.Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, Schadendorf D, Buer J & Helfrich I (2012) Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth, J Exp Med. 209, 2001–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, Stockmann C, Combe P, Berger A, Zinzindohoue F, Yagita H, Tartour E, Taieb J & Terme M (2015) VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors, J Exp Med. 212, 139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mantovani A (2010) Molecular pathways linking inflammation and cancer, Curr Mol Med. 10, 369–73. [DOI] [PubMed] [Google Scholar]
- 48.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS & Albelda SM (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN, Cancer Cell. 16, 183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Talmadge JE & Gabrilovich DI (2013) History of myeloid-derived suppressor cells, Nat Rev Cancer. 13, 739–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wynn TA, Chawla A & Pollard JW (2013) Macrophage biology in development, homeostasis and disease, Nature. 496, 445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Palma M & Coussens LM (2008) Immune Cells and Inflammatory Mediators as Regulators of Tumor Angiogenesis in Angiogenesis: An Integrative Approach from Science to Medicine (Figg WD & Folkman J, eds), Springer Science + Business Media, LLC, New York. [Google Scholar]
- 52.Potente M, Gerhardt H & Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis, Cell. 146, 873–87. [DOI] [PubMed] [Google Scholar]
- 53.Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, Squadrito ML, Segura I, Li XJ, Knevels E, Costa S, Vinckier S, Dresselaer T, Akerud P, De Mol M, Salomaki H, Phillipson M, Wyns S, Larsson E, Buysschaert I, Botling J, Himmelreich U, Van Ginderachter JA, De Palma M, Dewerchin M, Claesson-Welsh L & Carmeliet P (2011) HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization through Downregulation of PIGF, Cancer Cell. 19, 31–44. [DOI] [PubMed] [Google Scholar]
- 54.Betsholtz C (2004) Insight into the physiological functions of PDGF through genetic studies in mice, Cytokine Growth Factor Rev. 15, 215–28. [DOI] [PubMed] [Google Scholar]
- 55.Joyce JA & Hanahan D (2004) Multiple roles for cysteine cathepsins in cancer, Cell Cycle. 3, 1516–619. [DOI] [PubMed] [Google Scholar]
- 56.Ferrara N, Gerber HP & LeCouter J (2003) The biology of VEGF and its receptors, Nat Med. 9, 669–76. [DOI] [PubMed] [Google Scholar]
- 57.Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M & Christofori G (2000) Fibroblast growth factors are required for efficient tumor angiogenesis, Cancer Res. 60, 7163–9. [PubMed] [Google Scholar]
- 58.De Falco E, Porcelli D, Torella AR, Straino S, Iachininoto MG, Orlandi A, Truffa S, Biglioli P, Napolitano M, Capogrossi MC & Pesce M (2004) SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells, Blood. 104, 3472–82. [DOI] [PubMed] [Google Scholar]
- 59.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z & Hanahan D (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis, Nat Cell Biol. 2, 737–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Le P, Liang C, Chan J, Kiewlich D, Miller T, Harris D, Sun L, Rice A, Vasile S, Blake RA, Howlett AR, Patel N, McMahon G & Lipson KE (2003) Potent and selective inhibitors of the Met [hepatocyte growth factor/scatter factor (HGF/SF) receptor] tyrosine kinase block HGF/SF-induced tumor cell growth and invasion, Mol Cancer Ther. 2, 1085–92. [PubMed] [Google Scholar]
- 61.Lin EY, Nguyen AV, Russell RG & Pollard JW (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy, J Exp Med. 193, 727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K & Schwendener RA (2006) Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach, Br J Cancer. 95, 272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J & Harris AL (1996) Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma, Cancer Res. 56, 4625–9. [PubMed] [Google Scholar]
- 64.Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA & Johnson RS (2008) Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis, Nature. 456, 814–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, Johnson M, Lusis AJ, Cohen DA, Iruela-Arispe ML & Wu L (2010) Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy, Blood. ll5, 1461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M & Naldini L (2005) Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors, Cancer Cell. 8, 211–26. [DOI] [PubMed] [Google Scholar]
- 67.Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L & De Palma M (2011) Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells, Cancer Cell. 19, 512–26. [DOI] [PubMed] [Google Scholar]
- 68.Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, Tazzyman S, Danson S, Addison C, Clemons M, Gonzalez-Angulo AM, Joyce JA, De Palma M, Pollard JW & Lewis CE (2015) Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy, Cancer Res. 75, 3479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK & Duda DG (2010) Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation, Cancer Res. 70, 5679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen L, Li J, Wang F, Dai C, Wu F, Liu X, Li T, Glauben R, Zhang Y, Nie G, He Y & Qin Z (2016) Tie2 Expression on Macrophages Is Required for Blood Vessel Reconstruction and Tumor Relapse after Chemotherapy, Cancer Res. 76, 6828–6838. [DOI] [PubMed] [Google Scholar]
- 71.Lewis CE, Harney AS & Pollard JW (2016) The Multifaceted Role of Perivascular Macrophages in Tumors, Cancer Cell. 30, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tamagnone L & Mazzone M (2011) Semaphorin Signals on the Road of Endothelial Tip Cells, Developmental Cell. 21, 189–190. [DOI] [PubMed] [Google Scholar]
- 73.Tamagnone L (2012) Emerging role of semaphorins as major regulatory signals and potential therapeutic targets in cancer, Cancer Cell. 22, 145–52. [DOI] [PubMed] [Google Scholar]
- 74.Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L & Mazzone M (2013) Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity, Cancer Cell. 24, 695–709. [DOI] [PubMed] [Google Scholar]
- 75.Miyauchi JT, Caponegro MD, Chen D, Choi MK, Li M & Tsirka SE (2018) Deletion of Neuropilin 1 from Microglia or Bone Marrow-Derived Macrophages Slows Glioma Progression, Cancer Res. 78, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Papapetropoulos A, Garcia-Cardena G, Dengler TJ, Maisonpierre PC, Yancopoulos GD & Sessa WC (1999) Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors, Lab Invest. 79, 213–23. [PubMed] [Google Scholar]
- 77.Bird L (2016) Tumour immunology: Neutrophils help tumours spread, Nat Rev Immunol. 16, 74–5. [DOI] [PubMed] [Google Scholar]
- 78.Liang W & Ferrara N (2016) The Complex Role of Neutrophils in Tumor Angiogenesis and Metastasis, Cancer Immunol Res. 4, 83–91. [DOI] [PubMed] [Google Scholar]
- 79.Coffelt SB & de Visser KE (2015) Immune-mediated mechanisms influencing the efficacy of anticancer therapies, Trends Immunol. 36, 198–216. [DOI] [PubMed] [Google Scholar]
- 80.Gaudry M, Bregerie O, Andrieu V, El Benna J, Pocidalo MA & Hakim J (1997) Intracellular pool of vascular endothelial growth factor in human neutrophils, Blood. 90, 4153–61. [PubMed] [Google Scholar]
- 81.Nozawa H, Chiu C & Hanahan D (2006) Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis, Proc Natl Acad Sci U S A. 103, 12493–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, Carano RA, Meng YG & Ferrara N (2007) Bv8 regulates myeloid-cell-dependent tumour angiogenesis, Nature. 450, 825–31. [DOI] [PubMed] [Google Scholar]
- 83.Chung AS, Lee J & Ferrara N (2010) Targeting the tumour vasculature: insights from physiological angiogenesis, Nat Rev Cancer. 10, 505–14. [DOI] [PubMed] [Google Scholar]
- 84.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P & Van Ginderachter JA (2008) Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity, Blood. 111, 4233–44. [DOI] [PubMed] [Google Scholar]
- 85.Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, Wagstaff J & Griffioen AW (2003) Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression, Cancer Res. 63, 2322–9. [PubMed] [Google Scholar]
- 86.Khan KA & Kerbel RS (2018) Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa, Nat Rev Clin Oncol. 15, 310–324. [DOI] [PubMed] [Google Scholar]
- 87.Kumar V & Gabrilovich DI (2014) Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment, Immunology. 143, 512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gabrilovich DI (2007) Molecular mechanisms and therapeutic reversal of immune suppression in cancer, Curr Cancer Drug Targets. 7, 1. [PubMed] [Google Scholar]
- 89.Rivera LB, Meyronet D, Hervieu V, Frederick MJ, Bergsland E & Bergers G (2015) Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy, Cell Rep. 11, 577–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W, Hoon DS, Wakabayashi M, Forman S & Yu H (2013) B cells promote tumor progression via STAT3 regulated-angiogenesis, PLoS One. 8, e64159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, Korets L, Lam J, Tawfik D, DeNardo DG, Naldini L, de Visser KE, De Palma M & Coussens LM (2010) FcRgamma activation regulates inflammation-associated squamous carcinogenesis, Cancer Cell. 17, 121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N & Coussens LM (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages, Cancer Cell. 16, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L & Coukos G (2011) Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells, Nature. 475, 226–30. [DOI] [PubMed] [Google Scholar]
- 94.Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, Sheng K, Dobrolecki LE, Zhang X, Putluri N, Phung TL, Mani SA, Stossi F, Sreekumar A, Mancini MA, Decker WK, Zong C, Lewis MT & Zhang XH (2017) Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming, Nature. 544, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nizet V & Johnson RS (2009) Interdependence of hypoxic and innate immune responses, Nat Rev Immunol. 9, 609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, Tuccitto A, Tagliabue E, Castelli C & Rivoltini L (2017) Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation, Semin Cancer Biol. 43, 74–89. [DOI] [PubMed] [Google Scholar]
- 97.Hsu PP & Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond, Cell. 134, 703–7. [DOI] [PubMed] [Google Scholar]
- 98.Colegio OR (2016) Lactic acid polarizes macrophages to a tumor-promoting state, Oncoimmunology. 5, e1014774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.De Bock K, Georgiadou M & Carmeliet P (2013) Role of endothelial cell metabolism in vessel sprouting, Cell Metab. 18, 634–47. [DOI] [PubMed] [Google Scholar]
- 100.Rivera LB & Bergers G (2014) Angiogenesis. Targeting vascular sprouts, Science. 344, 1449–50. [DOI] [PubMed] [Google Scholar]
- 101.Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J, Prenen H, Ghesquiere B, Carmeliet P & Mazzone M (2016) Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis, Cell Metab. 24, 701–715. [DOI] [PubMed] [Google Scholar]
- 102.Wang Q, He Z, Huang M, Liu T, Wang Y, Xu H, Duan H, Ma P, Zhang L, Zamvil SS, Hidalgo J, Zhang Z, O'Rourke DM, Dahmane N, Brem S, Mou Y, Gong Y & Fan Y (2018) Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha, Nat Commun. 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takeda N, O'Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A & Johnson RS (2010) Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis, Genes Dev. 24, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS & Clark MA (2004) Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation, Cancer. 100, 826–33. [DOI] [PubMed] [Google Scholar]
- 105.Siska PJ & Rathmell JC (2015) T cell metabolic fitness in antitumor immunity, Trends Immunol. 36, 257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tschugguel W, Schneeberger C, Unfried G, Czerwenka K, Weninger W, Mildner M, Gruber DM, Sator MO, Waldhor T & Huber JC (1999) Expression of inducible nitric oxide synthase in human breast cancer depends on tumor grade, Breast Cancer Res Treat. 56, 145–51. [DOI] [PubMed] [Google Scholar]
- 107.Thomsen LL, Lawton FG, Knowles RG, Beesley JE, Riveros-Moreno V & Moncada S (1994) Nitric oxide synthase activity in human gynecological cancer, Cancer Res. 54, 1352–4. [PubMed] [Google Scholar]
- 108.Zafirellis K, Zachaki A, Agrogiannis G & Gravani K (2010) Inducible nitric oxide synthase expression and its prognostic significance in colorectal cancer, APMIS. 118, 115–24. [DOI] [PubMed] [Google Scholar]
- 109.Mumenthaler SM, Yu H, Tze S, Cederbaum SD, Pegg AE, Seligson DB & Grody WW (2008) Expression of arginase II in prostate cancer, Int J Oncol. 32, 357–65. [PubMed] [Google Scholar]
- 110.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB & Ochoa AC (2004) Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses, Cancer Res. 64, 5839–49. [DOI] [PubMed] [Google Scholar]
- 111.Chang CI, Liao JC & Kuo L (2001) Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity, Cancer Res. 61, 1100–6. [PubMed] [Google Scholar]
- 112.Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S & Mazzone M (2014) Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment, Oncogene. 33, 1743–54. [DOI] [PubMed] [Google Scholar]
- 113.Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW & Johnson RS (2010) Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression, Cancer Res. 70, 7465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, Klapproth K, Schakel K, Garbi N, Jager D, Weitz J, Schmitz-Winnenthal H, Hammerling GJ & Beckhove P (2013) Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy, Cancer Cell. 24, 589–602. [DOI] [PubMed] [Google Scholar]
- 115.Rivera LB & Bergers G (2013) Location, location, location: macrophage positioning within tumors determines pro- or antitumor activity, Cancer Cell. 24, 687–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fukumura D, Kloepper J, Amoozgar Z, Duda DG & Jain RK (2018) Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges, Nat Rev Clin Oncol. 15, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, Batchelor TT & Sorensen AG (2009) Biomarkers of response and resistance to antiangiogenic therapy, Nat Rev Clin Oncol. 6, 327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ebos JM & Kerbel RS (2011) Antiangiogenic therapy: impact on invasion, disease progression, and metastasis, Nat Rev Clin Oncol. 8, 210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jain RK (2001) Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy, Nat Med. 7, 987–9. [DOI] [PubMed] [Google Scholar]
- 120.Jain RK (2014) Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia, Cancer Cell. 26, 605–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bergers G & Hanahan D (2008) Modes of resistance to anti-angiogenic therapy, Nat Rev Cancer. 8, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC & De Palma M (2014) Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade, Cell Rep. 8, 696–706. [DOI] [PubMed] [Google Scholar]
- 123.Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, Autiero M, Wyns S, Plaisance S, Moons L, van Rooijen N, Giacca M, Stassen JM, Dewerchin M, Collen D & Carmeliet P (2007) Anti-PIGF inhibits growth of VEGF(R)-Inhibitor-Resistant tumors without affecting healthy vessels, Cell. 131, 463–475. [DOI] [PubMed] [Google Scholar]
- 124.Casanovas O, Hicklin DJ, Bergers G & Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors, Cancer Cell. 8, 299–309. [DOI] [PubMed] [Google Scholar]
- 125.Shojaei F, Zhong C, Wu X, Yu L & Ferrara N (2008) Role of myeloid cells in tumor angiogenesis and growth, Trends Cell Biol. 18, 372–8. [DOI] [PubMed] [Google Scholar]
- 126.Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, Ouyang W & Ferrara N (2013) An interleukin-17- mediated paracrine network promotes tumor resistance to anti-angiogenic therapy, Nat Med. 19, 1114–23. [DOI] [PubMed] [Google Scholar]
- 127.Jung K, Heishi T, Khan OF, Kowalski PS, Incio J, Rahbari NN, Chung E, Clark JW, Willett CG, Luster AD, Yun SH, Langer R, Anderson DG, Padera TP, Jain RK & Fukumura D (2017) Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy, J Clin Invest. 127, 3039–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, Di Serio C, Naldini L, De Palma M, Tozer GM & Lewis CE (2011) TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice, J Clin Invest. 121, 1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pietras K & Hanahan D (2005) A multitargeted, metronomic, and maximum-tolerated dose "chemo-switch" regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer, J Clin Oncol. 23, 939–52. [DOI] [PubMed] [Google Scholar]
- 130.Allen E, Walters IB & Hanahan D (2011) Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition, Clin Cancer Res. 17, 5299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EE & Varner JA (2016) PI3Kgamma is a molecular switch that controls immune suppression, Nature. 539, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JA, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A & Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma, N Engl J Med. 363, 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM & Wolchok JD (2015) Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma, J Clin Oncol. 33, 1889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A & Wigginton JM (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer, N Engl J Med. 366, 2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM & Sznol M (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer, N Engl J Med. 366, 2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Topalian SL, Drake CG & Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy, Cancer Cell. 27, 450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Du Four S, Maenhout SK, Niclou SP, Thielemans K, Neyns B & Aerts JL (2016) Combined VEGFR and CTLA-4 blockade increases the antigen-presenting function of intratumoral DCs and reduces the suppressive capacity of intratumoral MDSCs, Am J Cancer Res. 6, 2514–2531. [PMC free article] [PubMed] [Google Scholar]
- 138.Ager A & May MJ (2015) Understanding high endothelial venules: Lessons for cancer immunology, Oncoimmunology. 4, e1008791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Girard JP, Moussion C & Forster R (2012) HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes, Nat Rev Immunol. 12, 762–73. [DOI] [PubMed] [Google Scholar]
- 140.Johansson-Percival A, He B, Li ZJ, Kjellen A, Russell K, Li J, Larma I & Ganss R (2017) De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors, Nat Immunol. 18, 1207–1217. [DOI] [PubMed] [Google Scholar]
- 141.He B, Jabouille A, Steri V, Johansson-Percival A, Michael IP, Kotamraju VR, Junckerstorff R, Nowak AK, Hamzah J, Lee G, Bergers G & Ganss R (2018) Vascular targeting of LIGHT normalizes blood vessels in primary brain cancer and induces intratumoural high endothelial venules, J Pathol. 245, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thienpont B & Lambrechts D (2017) It's T Time for Normal Blood Vessels, Dev Cell. 41, 125–126. [DOI] [PubMed] [Google Scholar]
- 143.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S, Powles T, Donskov F, Neiman V, Kollmannsberger CK, Salman P, Gurney H, Hawkins R, Ravaud A, Grimm MO, Bracarda S, Barrios CH, Tomita Y, Castellano D, Rini BI, Chen AC, Mekan S, McHenry MB, Wind-Rotolo M, Doan J, Sharma P, Hammers HJ, Escudier B & CheckMate I (2018) Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma, N Engl J Med. 378, 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DB, Brahmer JR, Carvajal RD, Hammers HJ, Puzanov I, Hodi FS, Kluger HM, Topalian SL, Pardoll DM, Wigginton JM, Kollia GD, Gupta A, McDonald D, Sankar V, Sosman JA & Atkins MB (2015) Survival, Durable Response, and Long-Term Safety in Patients With Previously Treated Advanced Renal Cell Carcinoma Receiving Nivolumab, J Clin Oncol. 33, 2013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]