

Abstract
The enteric nervous system (ENS) represents a vast network of neuronal and glial cell types that develops entirely from migratory neural crest (NC) progenitor cells. Significant improvements in our understanding of the molecular mechanisms underlying NC induction and regional specification have recently led to the development of a robust method to recreate the process in vitro using human pluripotent stem cells (hPSCs). Directing the fate of hPSCs towards the enteric NC presents an accessible and scalable in vitro model of ENS development. The application of hPSC-derived enteric neural lineages provides a powerful platform for ENS-related disease modeling and drug discovery. Here we present a detailed protocol for the induction of a regionally specific NC intermediate that occurs over the course of a 15 day interval and is an effective source for the in vitro derivation of functional enteric neurons from hPSCs. Additionally, we introduce a new and improved protocol that we have developed to optimize the protocol for future applications in regenerative medicine in which components of undefined activity have been replaced with fully defined culture conditions. This protocol provides access to a broad range of human ENS lineages within a 30 day period.
Keywords: Enteric nervous system (ENS), human pluripotent stem cells (hPSCs) differentiation, enteric neural crest (ENC), enteric neuron (EN), defined culture conditions
INTRODUCTION
During embryogenesis, NC induction occurs at the interface of the non-neuronal ectoderm and the folding neural plate as a result of bone morphogenic protein (BMP), fibroblast growth factor (FGF), and Wnt signaling pathway activity1. During neurulation, dorsally localized NC cells delaminate and migrate away from the newly formed neural tube. Migratory NC cells proliferate and act as progenitors for a remarkable diversity of cell types including various populations of peripheral neurons and glia, melanocytes, endocrine cells and mesenchymal precursor cells1–3. In the developing embryo, the neural crest shows an anterior-posterior spatial organization associated with the expression of regionally specific HOX genes. Distinct functional regions include the cranial NC, vagal NC, trunk NC and sacral NC located anteriorly to posteriorly respectively (Fig. 1).
Figure 1. Major subtypes of the embryonic NC along the anterior to-posterior axis.
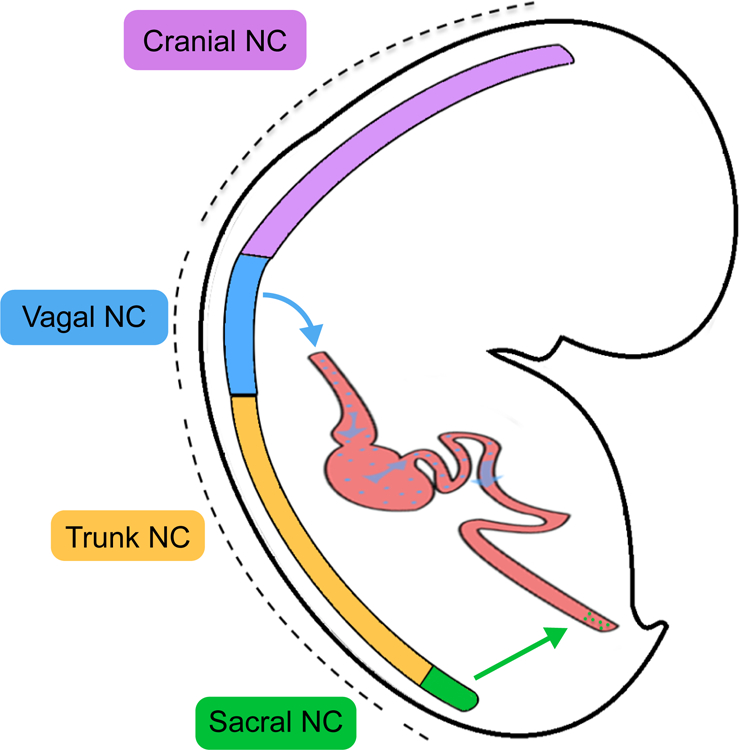
Migratory ENC progenitors are primarily derived from the vagal NC.
While the ENS is generated from both the vagal and sacral NC, vagal NC lineages positive for HOXB34 and HOXB55 migrate most extensively to colonize the entire length of the bowel6 (blue and green arrows in Fig. 1). Upon inclusion into the foregut, vagal NC cells display enteric neural crest (ENC) identity characterized by the expression of SOX10, PHOX2B, EDNRB, and ASCL1. Colonization of the intestinal tract by the ENC has been depicted as a rostrocaudally moving wave of proliferative multipotent ENS progenitors7. By week seven of embryogenesis in humans, migratory ENC cells reach the terminal hindgut8. Failure of ENC migration to the caudal regions of the bowel can result in congenital aganglionosis of the colon, a disorder known as Hirschsprung’s disease.
Post migratory ENC cells will commit to neuronal fates, a differentiation step associated with the downregulation of SOX10, sustained expression of EDNRB, ASCL1 and PHOX2B, and upregulation of pan neuronal markers such as TUJ19. ENC progenitors further differentiate to establish ganglia located between the circular and longitudinal layers of enteric smooth muscle, forming the myenteric plexus. Recent spatiotemporal analysis of the murine ENS has shown that ENC progenitors within the myenteric plexus proliferate along the serosa-mucosal axis to subsequently form the ganglia of the submucosal plexus10. Together, the myenteric and submucosal plexi establish the neuronal circuitry of the functional ENS.
Due to the capacity of the NC to undergo an extensive range of cell fate decisions, methods seeking to optimize NC induction and subtype specification from hPSCs have been an important focus of research11–13. hPSC-based protocols to derive NC commonly rely on a variation of the dual SMAD signaling inhibition protocol for neural induction, combined with the temporal activation of WNT signaling12,14. However, such methods often involve the use of poorly defined culture components such as serum, BSA fractions, and other animal-derived products, that may affect the reliability and reproducibility of the NC induction step. Accordingly, our group and others have recently reported protocols that use fully defined, xeno-free culture conditions for the reliable induction of cranial NC from hPSCs15,16. Here we present step-by-step instructions for both our previously published protocol14 (option A) and a similar, but fully defined differentiation protocol ( Option B) that integrates retinoic acid (RA) treatment to effectively transitioning the induction of cranial NC to a specific vagal NC regional identity14.
Applications
The spatial and temporal transience of the ENC has been a major factor limiting access to primary cells, particularly from human embryonic or fetal tissue samples. As a result, studying the developing ENS has largely relied upon studies in murine models. Work with such murine models resulted in the discovery of growth factors involved in the proliferation and differentiation of EN precursors, such as Neurotrophin-3 (NT-3) and glial cell line-derived neurotrophic factor (GDNF)17,18 among others. More recent single cell transcriptomics analysis of the developing murine ENS have revealed novel molecular states of lineally and functionally related ENS progenitors10. An appreciable conservation of the transcriptional processes underpinning ENS development across mammals19 supports the application of these factors to direct hPSC-derived ENC cells towards neurogenic commitments and may help further guide the identification, characterization and derivation of human enteric neuronal subtype lineages. A reliable protocol for the stepwise derivation of enteric neurons (ENs) from hPSCs provides the basis for modeling ENS development and the contribution of specific lineages to ENS disease.
The value of a reliable protocol for the stepwise derivation of ENS lineages from hPSCs relates to multiple research applications. The genetic contributions underpinning ENS pathogenesis can be probed by using induced pluripotent stem cell (iPSC) lines generated from patients suffering from enteric neuropathies20. Disease phenotypes can be modeled through in vitro differentiations and addressed via genetic or molecular perturbation strategies. Under the minimal, highly defined conditions of this protocol, it may be possible to further expand on our understanding of how precise perturbations impact cell fate commitments of EN progenitors and recapitulate disease phenotypes exhibited by EN lineages. Similarly, a scalable platform that produces unlimited numbers of hPSC-derived ENC cells or ENs on demand, enables high-throughput screening (HTS) assays that were previously unworkable. Therefore, such a protocol opens the door to testing the effects of large libraries of compounds or genes on fate commitment or the selective vulnerability of ENS lineages.
We have previously shown that hPSC-derived ENC cells can engraft within the murine host colon and differentiate into functional ENs14. Looking forward, several enteric neuropathies of the gastrointestinal tract have been described as potential targets for EN cell transplantation21. Option B sets a new standard in presenting a method to derive ENs from hPSCs under highly defined conditions as a step towards the production of clinical grade cells suitable for translational applications in the treatment of enteric neuropathies. Much remains to be understood in terms of the ideal cell type, state of differentiation, and optimal delivery method used to replace damaged or absent cells in enteric neuropathies, but such translational applications present a rational strategy for preclinical development and an exciting area of research.
Comparison to other methods
The protocol described here presents the evolution of our previously published work and stands amid a limited set of previously reported methods for the derivation of enteric neural progenitors from pluripotent stem cells22. Many labs in the stem cell field no longer rely on the support of feeder cells and have adopted the use of defined basal media, such as mTeSR™1 (Stemcell Tech, 85850) or Essential 8 (Life Technologies, A2858501) for the maintenance of hPSC lines. Nevertheless, previous ENC induction methods commonly involve media containing serum replacement factors, namely knockout serum replacement (KSR), as is also the case in Option A14,20. In an effort to reduce the inconsistencies and quality control measures that undefined conditions may introduce to a protocol, we optimized the ENC induction step under minimal, chemically defined conditions.
Recent studies have implemented alternative strategies for general NC induction using hPSCs, namely free floating embryoid body based approaches23,24. The migratory cells that come as a result of embryoid body and subsequent neural rosette formations have been shown to be positive for neural crest specific markers Sox10, TFAP2A, BRN3A, ISL1 and ASCL1, and a subset found to be positive for regionally specific vagal markers HOXB2 and HOXB5, even without the inclusion of RA23. Overall neural crest induction efficiency was assessed by FACS of p75 and HNK1 double positive cells, a strategy used to isolate NC cells in previous protocols (Lee et al 2007). Results showed >60% induction efficiency in ES cell line H9 and across independent hiPSC lines23. Enriched NC populations were then co-cultured with primary gut explants in a Transwell system to promote ENC identities enriched for HOXB2, HOXB3, HAND2 and EDNRB. Notably, this method incorporates brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), nerve growth factor (NGF), neurotrophin-3 (NT3) into the culture conditions. How these factors affect commitments of EN precursors, namely identities positive for VIP and calretinin23, remains an interesting point of inquiry. A similar embryoid body approach incorporated brief exposure to RA during NC induction before eventually combining hPSC-derived NC cells with hPSC-derived intestinal organoids (HIOs)24. In terms of ENC induction efficiency, data directly comparing monolayer and embryoid body strategies remains limited. Therefore, the optimal use of each strategy for a given application should be explored further.
Limitations
Despite the broad applicability and reliable outcomes of the protocol, there are limitations that should be considered. It has been shown that hPSC-derived ENs produced through both Option A and Option B comprise many of the neuronal subtypes resembling those found throughout the ENS in vivo14. However, the extent to which ENs derived in vitro accurately recapitulate those of the human ENS, both in terms of the developmental pathways taken by multipotent ENC progenitors and the connectivity established by post mitotic neurons, warrants further exploration. Likewise, molecular characterization of the various neuronal subtype lineages generated through this protocol have been limited to a catalogue of transcription factors and neurochemical identities6,25. It will be highly valuable to compare the detailed molecular features of hPSC-derived EN lineages to those of primary human cells such as by single cell transcriptomics to resolve how effectively they map onto the human ENS in vivo. Furthermore, the current protocol is focused on generating ENC lineages derived from the vagal NC, and it will be important to develop protocols to generate ENC lineages from hPSC-derived sacral NC.
Experimental Design
Here we present a protocol for the derivation of EN lineages from hPSCs. It is important to note that steps 1&2 of this protocol present two parallel options, Option A and Option B. The development and utility of Option A was previously established in Fattahi et. al., 2016 14. The adoption of Option B should be favored by users seeking to derive enteric neuron lineages following the most chemically defined and reliable method currently available.
The important points of difference between Option A and Option B are found in maintenance of hPSCs (Step 1) and during the ENC induction phase (Step 2). Adoption of Essential 8 (E8) for Option 2 offers a chemically defined basal media for the maintenance of hPSCs26, in place of the feeder cell and KSR media used in Option A. Transition from E8 to E6 basal media, in conjunction with precise combinations of BMP and Wnt signaling, and addition of RA, trigger the developmental cues required for ENC induction. Option A requires the gradual titration between relative amounts of basal media KSR and N2, while Option B utilizes a single defined basal media E6. Consequently, Option A involves dual SMAD inhibition using SB431542 and LDN-193189, while the conditions of Option B only demand the TGFβ signaling inhibition using SB431542. Interesting, as a result of replacing the KSR used in Option A, early activation of low levels of BMP signaling with BMP4 becomes necessary to induce NC specification under the conditions of Option B. For both options, CHIR 99021 is used to activate canonical Wnt signaling, though lower concentrations are necessary in the conditions of Option B, and for both options, retinoic acid is used to pattern NC cells towards the vagal ENC identity. A schematic illustration is provided outlining the induction conditions of Option A (Supplementary Fig 1) and Option B (Fig. 2A).
Figure 2. Induction of ENC cells from hPSCs.

a) Protocol (days 0–12) for ENC induction using option B. BMP4, Recombinant human bone morphogenetic protein-4; CHIR, CHIR 99021; RA, Retinoic Acid; SB, SB431542. b) Confluency of hPSCs on day 0 of differentiation. c) Phase contrast and SOX10::GFP reporter line GFP expression on day 2, day 6 and day 12. d) Representative image of FACS analysis of CD49D/SOX10::GFP positive ENC cells on day 12. e) Quantitative reverse transcriptase PCR (qRT–PCR) for vagal NC markers HOXB3, HOXB5, and ENC lineage marker PAX3 for ENC cells versus hPSCs. N=3 biological replicates. FC, fold change. Scale bars = 200 μm.
To ensure optimal induction efficiency, hPSCs must be cultured under Option A or Option B maintenance conditions for several passages before differentiation. A critical parameter that should be controlled and adjusted carefully is the density of hPSCs at the beginning of ENC induction. Independent batches of differentiated cells should be quality controlled to ensure their ENC identity based on gene expression and functional characteristics. It is also important to consider baseline variability among different hPSC line in terms of differentiation efficiency (see Anticipated Results). Cell line specific optimization of culture density may be required when using hPSCs with low baseline ENC differentiation capacity.
MATERIALS
REAGENTS - CELL CULTURE
• Human embryonic or induced pluripotent stem cell lines.
Critical: The quality of hPSC lines used in your differentiations should be verified by routine characterization of pluripotency including expression of markers such as NANOG and OCT4 and their ability to differentiate into endodermal, mesodermal and ectodermal lineages. The cell lines used in this manuscript are human ES cell line H9 (WA-09) H9-derivative SOX10::GFP (WiCell Research Institute, Memorial Sloan Kettering Cancer Center), human ES cell line UCSF4 (UCSF) and human iPS cell line WTC11 (Coriell Institute, UCSF).
Critical: Appropriate consent procedures and administrative regulations must be followed for work involving hESCs and hiPSCs. Please consult your institution to assure adherence with national and institutional guidelines and regulations.
Caution: The hPSC lines should be STR profiled to confirm their identity and ensure they are not cross contaminated. Regular karyotyping and frequent mycoplasma testing are necessary to monitor genomic stability and to avoid latent contamination.
DMEM/F-12, no glutamine (Life Technologies, 21331020)
Essential 8™ Flex Medium Kit (Life Technologies, A2858501)
Essential 6™ Medium (Life Technologies, A1516401)
Neurobasal™ Medium (Life Technologies, 21103049)
N-2 Supplement (CTS™, A1370701)
B-27™ Supplement, serum free (Life Technologies, 17504044)
MEM Nonessential Amino Acids (Corning, 25–025-CI)
Glutagro™ (Corning, 25–015-CI)
BSA, Bovine Serum Albumin (Sigma, A4503)
PBS, Phosphate-Buffered Saline, Ca2+- and Mg2+-free (Life Technologies, 10010023)
EDTA (Corning, MT-46034CI)
Accutase™ (Stemcell Technologies, 07920)
STEM-CELLBANKER® DMSO Free (Amsbio, 11897F)
• BMP-4, Recombinant Human BMP-4 Protein (R&D Systems, 314-BP) Critical: Stock aliquots should be at stored −80 °C. One aliquot should be kept at 4 °C to avoid multiple freeze/thaw cycles and used within 4 weeks.
CHIR 99021 (Tocris, 4423) Critical: Stock aliquots should be stored at −20 °C. One aliquot should be kept at 4 °C and used within 4 weeks.
FGF2, Recombinant Human FGF Basic (R&D Systems #233-FB) Critical: Stock aliquots should be stored at −80 °C. One aliquot should be kept at 4 °C to avoid multiple freeze/thaw cycles and used within 4 weeks.
GDNF, Recombinant Human Glial Derived Neurotrophic Factor (Peprotech, 450–10) Critical: Stock aliquots should be stored at −80 °C. One aliquot should be kept at 4 °C to avoid multiple freeze/thaw cycles and used within 4 weeks.
RA, Retinoic Acid (Sigma, R2625) Critical: Stock aliquots should be stored at −80 °C. One aliquot should be kept at 4 °C to avoid multiple freeze/thaw cycles and used within 4 weeks.
SB431542 (R&D Systems, 1614) Critical: Stock aliquots should be stored at 4 °C.
Y-27632 dihydrochloride ((Tocris Bioscience, 1254) Critical: Stock aliquots should be stored at −20 °C. One aliquot should be kept at 4 °C and used within 4 weeks.
Matrigel® hESC-Qualified Matrix, *LDEV-Free, (Corning, 354277)
Vitronectin XF (Stemcell Technologies, 07180)
FN, Fibronectin, Human (Corning, 356008) Critical: Stock aliquots should be stored at −80 °C. One aliquot should be kept at 4 °C and used within 4 weeks.
LM, Laminin I, Mouse (Cultrex, 3400–010) Critical: Stocks should be stored at −80 °C
PO, Poly-L-Ornithine Hydrobromide (Sigma, P3655) Critical: Stock aliquots should be stored at −80 °C. One aliquot should be kept at 4 °C and used within 4 weeks.
Trypan Blue Solution, 0.4% (Life Technologies, 15250061) Caution: Trypan Blue is a suspected carcinogen and should be handled with care. Collect all materials exposed to Trypan Blue for disposal according to institutional guidelines.
Gelatin, powder (Sigma, G9391)
MEF CF-1 mitomycin C–treated mouse embryonic fibroblasts (Applied StemCell, Inc., ASF-1223)
FBS, fetal bovine serum (Sciencell, 0025)
DMEM, Dulbecco’s modified Eagle medium (Life Technologies, 11965–118).
Collagenase IV (Life Technologies, 17104–019)
KSR, Knockout Serum Replacement (Life Technologies, 10828–028)
L-glutamine (Life Technologies, 25030–081)
Knockout DMEM (Life Technologies, 10829–018)
KSR, Knockout Serum Replacement (Life Technologies, 10828–028)
2-mercaptoethanol (Life Technologies, 21985–023)
DMEM/F12 powder (Life Technologies, 12500–062)
Glucose (Sigma, G7021)
Sodium bicarbonate (Sigma, S5761)
Putrescine (Sigma, cat. no. P5780)
Progesterone (Sigma, cat. no. P8783)
Sodium selenite (Bioshop Canada, SEL888)
Transferrin (Celliance/Millipore, 4452–01)
Insulin (Sigma, I6634)
EQUIPMENT
Horizontal Laminar Flow Hood
Cell culture centrifuge (i.e. Eppendorf 5810R)
Inverted microscope (i.e. Evos FL) with fluorescence equipment and digital imaging capture system.
CO2 incubator with controlling and monitoring system for CO2, humidity and temperature
Refrigerator 4 °C, freezer −20 °C, freezer −80 °C.
Cell culture disposables: Petri dishes, multiwell plates, conical tubes, pipettes, pipette tips, cell scrapers, etc.
Hemocytometer (i.e. Hausser Scientific)
qPCR System (i.e. 7900HT Fast Real-Time PCR System)
FACS Analyzer (i.e. BD LSRFortessa)
REAGENT SETUP
Critical: Concentrations listed below are final concentrations. Final and stock concentrations can be found in Tables 1 and 2.
Table 1.
List of growth Factors and small molecules
| Recombinant protein/ Small Molecule | Stock concentration | Dilution | Final concentration | Company | Cat. no. |
|---|---|---|---|---|---|
| BMP4 | 10 μg ml−1 | 10,000 | 1 ng ml−1 | R&D Systems | 314-BP |
| CHIR 99021 (Cocktail A) | 6 mM | 10,000 | 600 μM | Tocris | 4423 |
| CHIR 99021 (Cocktail B & C) | 6 mM | 4,000 | 1.5 μM | Tocris | 4423 |
| CHIR 99021 (NC-C) | 6 mM | 2,000 | 3 μM | Tocris | 4423 |
| FGF2 | 10 μg ml−1 | 1,000 | 10 ng ml−1 | R&D Systems | 233-FB/CF |
| Fibronectin | 1 mg ml−1 | 500 | 2 μg ml−1 | Corning | 356008 |
| GDNF | 10 μg ml−1 | 1,000 | 10 ng ml−1 | Peprotech | 450–10 |
| Laminin | 1 mg ml−1 | 500 | 2 μg ml−1 | Cultrex | 3400–010 |
| PO | 15 mg ml-1 | 1,000 | 15 μg ml−1 | Sigma Aldrich | P3655 |
| RA | 1 mM | 1,000 | 1 μM | Sigma Aldrich | R2625 |
| SB431542 | 10 mM | 1,000 | 10 μM | R&D Systems | 1614 |
| Y-27632 dihydrochloride | 10 mM | 1,000 | 10 μM | R&D Systems | 1254 |
Table 2.
List of supplements
| Medium Supplement | Stock Volume | Final concentration | Company | Cat. no. |
| B-27 minus vitamin A | 10 ml | 20 μl ml−1 | Life Technologies | 12587010 |
| Glutagro | 100 ml | 10 μl ml−1 | Corning | 25–015-CI |
| MEM NEAAs | 100 ml | 10 μl ml−1 | Corning | 25–025-CI |
| N2 | 5 ml | 10 μl ml−1 | CTS | A1370701 |
Critical: All solutions, including stock solutions, should be prepared under sterile conditions in a laminar flow hood to avoid contamination.
Critical: Avoid prolonged light exposure to culture mediums.
ES medium, hPSC medium for maintenance in Option A
Combine 100 ml of KSR to 400 ml DMEM/F12, no glutamine. Add 5 ml of 200 mM L-glutamine, and 5 ml of MEM Nonessential Amino Acids. Filter sterilize, then add 10 ng/ml of recombinant FGF2. Store at 4ºC (use within 2 weeks).
MEF medium, MEF culture medium for Option A
Combine 100 ml FBS to 900 ml of DMEM. Filter sterilize before use. Store at 4ºC (use within 3 weeks).
KSR medium, early ENC differentiation medium in Option A
Combine 410 ml of Knockout DMEM, 75 ml of KSR, 5 ml of 200 mM L-glutamine), 5 ml of MEM non-essential amino acids, and 500 μl of 2-mercaptoethanol. Store at 4ºC (use within 3 weeks).
N2 medium, late ENC differentiation medium in Option A
Dissolve one bag of DMEM/F12 powder in 550 ml of distilled water. Add: 1.55 g of glucose, 2.00 g of sodium bicarbonate, 16.1 μg putrescine, 32 μg progesterone, 5.2 μg sodium selenite, 100 mg transferrin, 25 mg insulin (dissolved in 10 ml of 5 mM NaOH). Add double-distilled water (with a resistance of 18.2 M) to a final volume of 1000 ml. Filter sterilize and store at 4ºC (use within 3 weeks).
E8-C, hPSC medium for maintenance in Option B
Combine Essential 8-Flex supplement (20 μl ml−1) with Essential 8™ Flex Medium. Store at 4ºC (use within 2 weeks).
Cocktail A, first ENC differentiation medium in Option B
Combine BMP4 (1 ng ml−1), SB431542 (10 μM), CHIR 99021 (600 nM), with Essential 6™ Medium. Store at 4ºC (use within 2 weeks).
Cocktail B, second ENC differentiation medium in Option B
Combine SB431542 (10 μM), CHIR 99021 (1.5 μM), with Essential 6™ medium. Store at 4ºC (use within 2 weeks).
Cocktail C, third ENC differentiation medium in Option B
Combine SB431542 (10 μM), CHIR 99021 (1.5 μM), Retinoic Acid (1 μM), with Essential 6™ medium. Store at 4ºC (use within 2 weeks).
NC-C, ENC medium for spheroid maintenance for both options
Combine FGF2 (10 ng ml−1), CHIR 99021 (3 μM), N2 Supplement (10 μl ml−1 ), B27 Supplement (20 μl ml−1), Glutagro (10 μl ml−1), MEM Nonessential Amino Acids (10 μl ml−1), with Neurobasal® Medium. Store at 4ºC (use within 2 weeks).
EN-C, EN medium for differentiation and maintenance for both options
Combine GDNF (10 ng ml−1), Ascorbic Acid (100 μM), N2 Supplement (10 μl ml−1), B27 Supplement (20 μl ml−1), Glutagro (10 μl ml−1), MEM Nonessential Amino Acids (10 μl ml−1), with Neurobasal® Medium. Store at 4ºC (use within 2 weeks).
EDTA 1x for passaging hESCs
Combine EDTA (500 μM) with PBS.
Matrigel (500 μl aliquots)
Thaw frozen vial of Matrigel overnight at 4 °C. Prepare 500 μl aliquots in 50 ml conical tubes using chilled pipette tips and keep frozen at −20 °C.
Critical: Matrigel must be kept cold to prevent gelatinization. Chill 50 ml centrifuge tubes at 4 °C for 1 hour before making aliquots of Matrigel.
EQUIPMENT SETUP
Critical: All equipment should be prepared under sterile conditions in a laminar flow hood to avoid contamination.
MEF-coated dishes for use in Option A
Prepare MEF coated 10-cm dish at least one day before hPSC passaging by coating culture surface with 0.1% gelatin dissolved in PBS (5 ml). Incubate at room temperature (20−25°C) for 10 minutes. Thaw vial of mitomycin-C treated MEFs in a 37°C water bath and resuspend cells in MEF medium (100,000 cells ml−1). Aspirate 0.1% gelatin and add ~1.2 ×106 MEFs to 10-cm dish (15,000 cells/ cm2 well surface area). Culture MEFs overnight in a 37°C incubator. MEF coated dishes may be left cultured for up to 3 days before plating hPSCs.
Matrigel-coated plates for use in both options
Dilute a 500 μl frozen aliquot of Matrigel in 50 ml of cold DMEM:F12. Pipette up and down vigorously with a 25 ml or 50 ml serological pipette to break frozen Matrigel pellet. Coat wells with the diluted Matrigel solution (100 µl/ cm2 well surface area) and let stand in a 37 °C incubator overnight. Aspirate the Matrigel solution before plating hPSCs.
Critical: To achieve a fully defined system, Matrigel-coated plates should be substituted with Vitronectin-coated plates.
Vitronectin-coated plates for use in Option B
Dilute Vitronectin (10 μl ml−1) with PBS and mix thoroughly. Coat wells with diluted Vitronectin solution (100 µl/ cm2 well surface area) and let plates stand in a 37°C incubator overnight. Aspirate the Vitronectin solution before plating hPSCs.
PO/Lam/FN-coated plates for both options
Combine PO (15 μg ml−1) with PBS. Coat wells with PO/PBS solution (100 µl/ cm2 well surface area) and let stand in 37 °C incubator overnight. The following day, combine FN (2 μg ml−1) and Laminin (2 μg ml−1) with PBS. Aspirate PO/PBS and coat well with FN/LM/PBS solution (100 µl/ cm2 well surface area). Let plates stand in 37 °C incubator for a minimum of 2 hours. Aspirate FN/LM/PBS solution before plating cells.
PROCEDURE
Thawing frozen hPSCs
Critical: Frozen stocks of hPSCs should be kept in a liquid nitrogen cryogenic storage system at −1560 C. Frozen cells should be thawed and plated immediately.
Critical: For hPSCs lines that were previously maintained in mTESR1, first establish the line in mTESR1 for the initial passage, before transitioning the cultures to KSR based hES medium (option A) or E8 medium (option B). The cultures should be passaged at least twice in new medium before continuing the protocol.
- Remove vial of hPSCs from liquid nitrogen and transfer vial to a 37 °C water bath.
-
iCaution: Cryogenic gloves and safety glasses should be worn when working with liquid nitrogen.
-
i
Keep hold of the top of the sealed vial, and gently swirl around the water bath to ensure even thawing of frozen cells. Once only a small pellet of ice remains, remove the vial from water bath, spray the sealed vial with 70% ethanol, and transfer to laminar flow hood.
Add 0.5– 1 ml of E8-C directly into vial and gently mix by pipetting up and down 1–2 times. Transfer cell suspension to a conical tube.
Centrifuge the conical tube at 1200rpm (290x g) for 1 minute.
-
Carefully aspirate supernatant with a sterile pipette tip while avoiding contact with the pellet. Resuspend the pellet with 2 ml of hESC-medium (option A) or E8-C (option B) and plate suspension into a single well of a 6-well MEF-coated (option A) or Matrigel-coated or Vitronectin-coated plate (option B).
Critical: A ROCK (Rho kinase) inhibitor such as Y-27632 dihydrochloride may be included in the initial E8-C medium conditions to enhance recovery and prevent excess cell death27. Combine Y-27632 dihydrochloride (10 μM) with E8-C in a separate conical tube. Use this medium to break cell pellet after centrifugation and initial plating. Aspirate Y-27632 dihydrochloride supplemented medium from wells 3–5 hours after plating, and replace with fresh E8-C. Prolonged ROCK inhibition may adversely affect pluripotency and differentiation28.
Proceed by expanding colonies as described in the next stage of the protocol.
Maintaining hPSCs cultures
Critical: Before attempting induction of ENC cells, it is important to first become comfortable in maintaining cultures of hPSCs (Step 1), and newly thawed lines should be passaged several times prior to differentiation.
-
2
Passage hPSC several times prior to differentiation by using our previously published procedure (option A) or fully defined conditions (option B)Critical: After the first 2–3 passages after thawing, several wells of hPSCs should be refrozen to maintain back-up stocks as described in Amsbio Stem-CellBanker® protocol.
Option A
i) On the day of passaging, aspirate human ES cell medium from hPSC culture and add PBS (10 ml/ 10-cm dish). Gently rock the dish to wash cultures and aspirate off PBS.Ii_ Add collagenase IV (2 ml/ 10-cm dish) and incubate at room temperature for 10 min.
Iii) Aspirate collagenase IV and add PBS (10 ml/ 10-cm dish). Gently rock the dish to wash colonies and aspirate off PBS.
Iv) Use a cell scraper to displace colonies from the culture surface.
V) Resuspend detached colonies in 1 ml of human ES cell medium and pipet up and down to disassociate larger colonies.
Vi) Add appropriate volume of colony suspension with enough human ES cell medium for replating. The optimal volume of colony susupension will depend on the confluency of cultures and passaging ratio. For new users, a passaging ratio of 1:6 is advised. Add this volume of cell suspension to 10 ml of ES cell medium for each new plate (e.g., add 160 μl of cell suspension to 10 ml of ES cell medium for each new 10-cm dish)
Vii) Aspirate MEF medium from cultured MEF dish and add ES cell suspension.
-
Viii) Label plate with cell line, date, and new passage number. Incubate at 5% CO2 and 37 °C.
iX) hPSC cultures should be passaged once a week by repeating steps i-vii when they reach ~80% confluency. For continued maintenance, passaging ratios generally vary between 1:6 and 1:12 (i.e resuspend the pellet of cells collected from 1 well at ~80% confluency with 12 ml of fresh hESC medium. Add 2 ml of this suspension to each well of a new 6 well plate).
Option B
i) Aspirate old E8-C medium from the corner of well using a sterile pipette tip. Add fresh E8-C (200 µl/ cm2 well surface area). Replace medium with fresh E8-C every other day.
Ii) When colonies are ~80% confluent, begin passage by aspirating E8-C from the corner of a single well.
Iii) Add PBS (100 µl/ cm2 well surface area) and gently rock plate to wash off loose debris. Aspirate PBS using a sterile pipette tip.
Iv) Add EDTA 1x (100 µl/ cm2 well surface area). Replace lid of plate and watch for detachment of edges of colonies from well surface through an inverted microscope (2–4 minutes).
-
V) Use a P1000 micropipette or a 5 ml serological pipette to mechanically harvest colonies from the well. Transfer EDTA 1x cell suspension to a 15 ml conical tube.
i. Critical: Pipetting too vigorously may lead to excessive colony dissociation and adversely affect cell viability. Total time in EDTA 1x and pipetting technique should be optimized to maintain cell viability.
-
Vi) Centrifuge the conical tube at 1200rpm (290x g) for 1 minute.
vii) Carefully aspirate supernatant with a sterile pipette tip while avoiding contact with the pellet. Resuspend the pellet with final volume of E8-C and plate suspension in new Matrigel-coated or Vitronectin-coated 6-well plate.
viii) Label plate with cell line, date, and new passage number. Incubate at 5% CO2 and 37 °C.
iX) hPSC cultures should be passaged by repeating steps i-viii once every 5 days when they reach ~80% confluency. For continued maintenance, passaging ratios generally vary between 1:12 and 1:18 (i.e resuspend the pellet of cells collected from 1 well at ~80% confluency with 2– 3 ml of E8-C and transfer 1 ml of this suspension to a new 15 ml conical tube. Add fresh E8-C to the new tube to bring the total volume to 12 ml. Add 2 ml of this suspension to each well of a new 6 well plate).
ENC Induction (Day 0 – 12)
-
3
Once a consistent base of hPSCs maintenance technique has been established, initate ENC induction by replating cells and replacing maintenance media with differentiation media as described in option A for our published method or option B using defined inducers. Induction proceeds gradually over an 11- to 12-day period of stepwise transitions in culture conditions (Fig. 2a).
Option A
Day −1: Replating hPSCs for differentiation. On the day before the start of ENC induction, remove human ES cell medium from hPSC colonies and add PBS (10 ml/10-cm dish). Replace plate lid and gently rock the dish to wash colonies and aspirate the PBS.
Add 0.05% trypsin (2 ml/ 10-cm dish) and vigorously shake back and forth for 1 to 2 minutes to detach MEFs. MEFs should detach before hPSC colonies. Aspirate medium containing MEFs, leaving hPSC colonies attached. Let dish stand without medium for 1 minute at room temperature.
Add human ES cell medium supplemented with Y-27632 (10 μM) and mechanically detach colonies by pipetting up and down using a P1000 pipet. More dissociation is necessary compared to hPSC maintenance passaging and cells should be separated into single cells or small clusters of 5–10 cells.
Aspirate Matrigel solution from coated plates and add fresh human ES cell medium supplemented with Y-27632. Plate ~100,000 cells/cm2 onto Matrigel coated plates containing human ES cell medium supplemented with Y-27632. Incubate overnight at 37 °C and 5% CO2.
Day 0: Beginning of ENC induction. When monolayer is ~70% confluent (generally on the day after step iv is carried out), aspirate human ES cell medium from dish and add fresh KSR medium supplemented with SB431542 (10 μM) and LDN-193189 (1 μM). Incubate cells for 24h.
Day 2. Aspirate old medium and add fresh KSR medium supplemented with SB431542 (10 μM), LDN-193189 (1 μM), and CHIR-99021 (3 μM). Incubate cells for 48h.
Day 4. Aspirate old medium and add a mixture of 75% KSR and 25% N2 medium supplemented with SB431542 (10 μM), LDN-193189 (1 μM), and CHIR-99021 (3 μM). Incubate cells for 48h.
Day 6. Aspirate old medium and add a mixture of 50% KSR and 50% N2 medium supplemented with SB431542 (10 μM), LDN-193189 (1 μM), CHIR-99021 (3 μM), and Retinoic Acid (1 μM). Incubate cells for 48h.
Day 8. Aspirate old medium and add a mixture of 25% KSR and 75% N2 medium supplemented with SB431542 (10 μM), LDN-193189 (1 μM), CHIR-99021 (3 μM), and Retinoic Acid (1 μM). Incubate cells for 48h.
Day 10. Aspirate old medium and add N2 medium supplemented with SB431542 (10 μM), LDN-193189 (1 μM), CHIR-99021 (3 μM), and Retinoic Acid (1 μM). Incubate cells for 24–48h.
Day 11/12. Check cells. ENC cells are ready to be assayed or further differentiated. The proportion of SOX10+ ENC cells should be expected to be ~50% to 60%. Pure populations of ENC cells can be obtained across cells lines by FACS for CD49D+ cells.
Critical: As confluency continues to increase over the course of NC induction, cells may detach from the underlying monolayer. Avoid excess loss of cells by tipping the plate and gently adding fresh media to corner and side of well.
Option B
-
i
Day −2: Replating hPSCs for differentiation. Two days before ENC induction, aspirate E8-C from hPSC cultures and use the same passage technique as described in Option B, Step 2 but use a 5:6 passaging ratio. (i.e all cells from 5 wells to a new 6-well plate) and leave in EDTA for 3–5 minutes for increased cell separation.
-
ii
Feed cells with E8-C. Cells will continue to propagate and after 2 days the culture should become nearly confluent as a monolayer (Fig 2b) while maintaining typical hPSC morphology (Supplementary Fig 2).
-
iii
Day 0: Beginning of ENC induction. Aspirate old E8-C medium from corner of well using a sterile pipette tip. Add Cocktail A (200 μl/ cm2 well surface area). Record date of day 0 of ENC differentiation. Incubate at 5% CO2 and 37 °C for 48h.
-
iv
Day 2. Aspirate Cocktail A from corner of well using a sterile pipette tip. Add Cocktail B (200 μl/ cm2 well surface area). Incubate at 5% CO2 and 37 °C for 48h.
-
v
Day 4. Aspirate old Cocktail B using a sterile pipette tip and add fresh Cocktail B (200 µl/ cm2 well surface area). Incubate at 5% CO2 and 37 °C for 48h.
-
vi
Day 6. Aspirate Cocktail B using a sterile pipette tip. Add Cocktail C (400 μl/ cm2 well surface area). Incubate at 5% CO2 and 37 °C for 48h. At ~day 6, SOX10::GFP+ cells begin to cluster within the monolayer, indicating SOX10+ ENC lineage identity. GFP+ cluster size and prevalence continue to increase over the remaining ENC differentiation (Fig 2c).
-
vii
Day 8. Aspirate old Cocktail C using a sterile pipette tip and add fresh Cocktail C (400 μl/ cm2 well surface area). Incubate at 5% CO2 and 37 °C for 48h.
Critical: As confluency continues to increase over the course of NC induction, cells may detach from the underlying monolayer. Avoid excess loss of cells by tipping the plate and gently adding fresh media to corner and side of well.
-
viii
Day 10. Aspirate old Cocktail C using a sterile pipette tip and add fresh Cocktail C, increasing volume to 600 μl/ cm2 of well surface area. Incubate at 5% CO2 and 37 °C for 24–48h.
-
ix
iX. Day 11/12. Check appearance of ENC cells. Move to next step on day 11 if SOX10::GFP+ clusters are detaching from monolayer. Otherwise, wait until day 12. Day 11 or 12 is the end of the ENC induction period.. ENC cells are characterized by co-expression of SOX10::GFP and CD49D (Fig 2d). ENC lineages are confirmed by the expression of HoxB2, HoxB5, and PAX3 (Fig 2e). Optional purification of ENC populations can be prepared by FACS using CD49D surface marker staining (Box 1).
BOX 1:Purification of NC by FACS.
After 12 days of ENC induction under Option B (Step 8), fluorescence activated cell sorting (FACS) can be used to prepare purified populations of NC cells. Previous NC induction protocols have suggested using p75/HNK1 marker staining for FACS analysis11,13. However, p75 expression is found outside of the ENC and a portion of p75/HNK1 double positive cells have been shown to be SOX10::GFP-12. We have demonstrated that CD49D (α 4 integrin) is a specific marker for SOX10+ hPSC-derived NC lineages16. Here we present a procedure for the purification of ENC cells by FACS using CD49D. FACS purification is particularly recommended for experiments and assays that involve early ENC progenitors (day 11). The maintenance of 3D spheroid culture conditions is generally sufficient to enhance the purity of NC cells and neurons in the later stages of differentiation without the need for further FACS purification (Fig 7).
ENC Spheroid formation (Day 12 – 15)
CRITICAL Following 11- to 12-days of ENC induction phase (Step 8), both option paths converge at the ENC spheroid phase. ENC monolayers are detached from the well surface and transferred to ultra-low attachment plates to form free floating 3D spheroids. Spheroids are maintained in NC-C medium for 3–4 days as part of a NC maintenance process (Fig 3a).
Figure 3. ENC spheroid culture.

a) Protocol (days 12–15) for ENC spheroid formation. NB+N2+B27; NB/N2/B27, Neurobasal medium with N2 and B27 supplement; FGF2, Recombinant Human FGF Basic; CHIR, CHIR 99021. b) Phase contrast and SOX10::GFP reporter line GFP expression of 3D spheroids on day 14. Scale bar = 200 μm.
-
4
On day 11- or 12, aspirate Cocktail C from ENC induction phase plate using a sterile pipette tip. Add Accutase (100 µl/ cm2 well surface area). Incubate for 30 minutes at 37 °C and 5% CO2.
-
5
DO NOT ASPIRATE Accutase. Add NC-C (100 µl/ cm2 well surface area). Use a serological pipette to mechanically harvest cells from the surface of well. Add the cell suspension to a 15 ml conical tube.
-
6
Centrifuge the conical tube at 1200rpm (290x g) for 1 minute.
-
7
With a sterile pipette tip, carefully aspirate as much supernatant as possible while avoiding the cell pellet.
-
8
Resuspend the pellet with the appropriate volume of NC-C and transfer the cell suspension to an ultra-low attachment 6-well plate (2 ml/ well). 10 cm2 of ENC monolayer is transferred to 1 well of an ultra-low attachment 6 well plate (i.e. A 6-well ENC induction plate corresponds to a 6 well ultra-low attachment plate). Incubate at 37 °C and 5% CO2 for 2 days). 3D spheroids will continue to congregate during this period.
-
9
After 2 day incubation period (day 13–14), 3D spheroids should be observable. Gently swirl ultra-low attachment plates to group the free-floating spheroids into the center of each well. Using a P1000 micropipette, slowly aspirate the old NC-C by moving around the circumference of well, actively avoiding any removal of spheroids.
-
10
Add 2 ml of fresh NC-C to each ultra-low attachment plate well. Incubate at 37 °C and 5% CO2 for an additional 24h. Examples of 3D spheres at day 14 are shown in Fig 3b.
EN Induction Phase (Day 15→)
Critical. After the ENC spheroid phase (Steps 9–15) and 15 total days from the start of ENC differentiation, ENC spheroids are dissociated with Accutase treatment and replated on PO/LM/FN-coated wells. This stage marks the final replating of the protocol and the beginning of EN induction (Fig. 4a).
Figure 4. Characterization of hPSC-derived ENC and enteric neurons.

a) Protocol for neuronal differentiation and maturation of ENC precursors, b) Flow cytometry analysis of CD49D positive ENC cells from hESC line UCSF4 and hiPSC line WTC11 on day 12. c) Flow cytometry analysis of CD49D positive ENC cells from hESC line UCSF4 and hiPSC line WTC11 after ENC spheroid enrichment on day 15. d) Immunofluorescence staining of TUJ1/TRKC on day 30 of EN induction. e) Flow cytometry analysis of TUJ1 and TRKC expression in EN cells on day 20, day 40 and day 55. f) Immunofluorescence images of CHAT, 5HT, NOS1, and GABA stained ENs on day 50. g) Flow cytometry analyses of CHAT, 5HT, NOS1, and GABA on ENS at day 75. AA, ascorbic acid; GDNF, Recombinant Human Glial Derived Neurotrophic Factor, F647, Alexa Fluor™ 647. Scale bars = 100 μm in c, f and 20 μm in e. o
-
11
On day 15, gently swirl ultra-low attachment plates to group the free-floating spheroids into center of well. Using a P1000 micropipette, slowly remove the old NC-C from the circumference of well while actively avoiding any removal of spheroids.
-
12
Add Accutase (1 ml) to each well and incubate for 30 minutes at 37 °C and 5%
-
13
Use a 5 ml serological pipette to gently dissociate the remaining spheroids by 2–3 rounds of pipetting. Transfer the cell suspension to a 50 ml conical tube.
Critical: Dissociation of spheroids using a P1000 micropipette adds an element of shear stress and may lead to excessive cell death. The use a serological pipette is recommended due to the larger diameter of the tip opening.
-
14
Centrifuge the conical tube at 1200rpm (290x g) for 1 minute.
-
15
Carefully aspirate supernatant using a sterile pipette tip while avoiding contact with the cell pellet.
-
16
Resuspend the pellet in 10 ml of EN-C.
-
17
Determine the viable cell concentration using a hemocytometer and Trypan Blue.
-
18
Add the remaining volume of EN-C necessary to replate the cell suspension at ~100,000 cells/ cm2 of surface area to the conical tube.
-
19
Aspirate the FN/Laminin/PBS solution from wells using a sterile pipette tip.
-
20
Add the EN-C cell suspension to center of the well or dish.
-
21
Incubate at 37 °C and 5% CO2. Move EN plates in a north/south/east/west direction upon returning to incubator shelf to insure even distribution of cell attachment.
-
22
Replace EN-C medium (200 µl/ cm2 well surface area) every other day until 30-to 40-days after the start of ENC induction.
Critical: After 30 - to 40-days of differentiation, reduce EN-C medium replacement to 1- to 2-times a week but increase volume to 400 µl/ cm2. If cultures begin detaching from the surface of the well, supplement EN-C with FN (2 μg ml−1) and LM (2 μg ml−1).
ANTICIPATED RESULTS
This protocol is expected to reliably produce populations of hPSC-derived ENs under chemically defined conditions (Option B). Proportions of cells positive for EN identities may vary between cell lines, as well as between differentiations of a given cell line. Regardless, cells possessing a neuronal morphology should emerge by 20 days after the start of hPSC differentiation (Supplementary Fig. 3a, b) and stay viable for several weeks (Supplementary Fig. 3c,e). A major factor in maintaining long-term neuronal cultures is reducing the stress associated with the replacement of culture medium. For cultures above 50 days, users should wait until culture medium turns yellow, and carefully replace medium with FN/LM supplemented medium. Cultures lasting longer than 100 days can be achieved using this protocol. Neuronal identity should be confirmed through marker expression and relative gene expression analysis by qRT-PCR. Reducing the emergence of contaminating, non-neuronal cell types can be a challenge and continued optimization of the protocol should reduce this.
The identification of CD49D (α4 integrin) as a reliable surface marker of SOX10+ NC lineages16, enables the assessment of the ENC induction efficiency and their prospective isolation. We analyzed CD49D expression after 12 and 15 days of differentiation under Option B for two additional hPSC lines (hESC-UCSF4 and hiPSC-WTC11) (Fig. 4 bc). The results demonstrated initial variation in ENC induction efficiency between cell lines and validated the ENC spheroid phase (day12-day15) as necessary for the enrichment of CD49D+ enteric neuron precursors (Fig. 4c). After EN induction, neuronal identity is verified based on co-expression of pan-neuronal marker TUJ1 and enteric neuron precursor specific marker TRKC (Fig. 6 c,d). Expression of additional neuronal subtype specific markers include the cholinergic neuronal marker Choline Acetyl Transferase (CHAT), serotonin (5-HT), gamma-Aminobutyric acid (GABA) and neuronal nitric oxide synthase (nNOS) which labels nitric oxide (NO) producing neurons (Fig. 4 f,g). Co-expression analysis of CHAT and NOS1 reveals separate population of cholinergic and nitrergic neurons in the differentiated culture (Supplementary Fig. 4). Glial cells expressing glial fibrillary acidic protein (GFAP) and SOX10, also emerge in differentiated cultures at the later stages of EN induction step (Fig. 7).
Figure 6. Gene expression analysis of hPSC-derived enteric neurons.

a-f) Quantitative reverse transcriptase PCR (qRT–PCR) of ENS lineage markers PHOX2B, EDNRB, ASCL1, TUJ1, CHAT and GFAP for EN populations versus hPSCs. N=3 biological replicates. FC, fold change.
Figure 7. FACS purification of ENC lineages.

Time course flow cytometry analysis of CD49D expression in unsorted differentiated cultures (a) and populations sorted at day 11 for CD49D (b). FSC, forward scatter; SSC, side scatter.
Comparisons of relative gene expression between samples collected from separate time-points during differentiation reveal population level transitions in gene expression that are supported by previous descriptions of the transcriptional processes of in vivo ENS development29. High expression levels of ENC-derived progenitor markers PHOX2B, ASCL1, and EDNRB during the transition to EN induction reveal the presence of enteric precursors (Fig. 6a–c). The synchronous downregulation of precursor markers with upregulation of TUJ1 and CHAT illustrates neuronal commitments and maturity taking place over the course of EN induction (Fig. 6d, e). Additionally, the delayed emergence of enteric glia is seen by the increased expression of glial marker GFAP in the later stages of EN induction phase (Fig. 6f).
The primary contaminating cell types found within differentiations are NC-derived flat myofibroblast-like cells identifiable by expression of smooth muscle actin (SMA) (Supplementary Fig. 5). These contaminants are particularly unfavorable to the longevity of mature enteric neurons by catalyzing the eventual detachment of neurons from the well surface and resulting apoptosis. Minimizing the number cells expressing SMA presents a straightforward approach to further optimize the protocol and maximize the overall durability of enteric neuron populations.
REAGENTS
DMEM/F-12, no glutamine (Life Technologies Corporation, 21331020)
BSA, Bovine Serum Albumin (Sigma, A4503)
Anti-human CD49D antibody (Biolegend, 304314)
DAPI (Sigma, D9542)
Normocin, Antimicrobial Reagent (InvivoGen, ant-nr-1)
EQUIPMENT
5 ml Round Bottom Polystyrene Test Tube, w/ Cell Strainer Cap (Falcon 352235)
5 ml Round Bottom Polystyrene Test Tube, w/ Snap Cap (Falcon 352003)
FACS Analyzer (i.e BD LSRFortessa)
REAGENT SETUP
Staining medium
Dissolve BSA (0.02 mg ml−1) with DMEM/F-12, no glutamine. Add Pe/Cy7 anti-human CD49D antibody (1.25 μl ml−1). Prepare 2.4 ml per 6-well plate of ENC differentiations (400 μl per well).
Sorting medium
Dissolve BSA (0.02 mg ml−1) with DMEM/F-12, no glutamine.
PROCEDURE
On day 12 of ENC induction, aspirate Cocktail C from ENC induction plate using a sterile pipette tip. Add Accutase (100 μl/ cm2 well surface area). Incubate at 5% CO2 and 37 °C for 30 minutes.
DO NOT ASPIRATE Accutase. Use a serological pipet to mechanically harvest cells from the surface of well. Add cell suspension to a 15 ml conical tube.
Centrifuge the conical tube at 1200rpm (290x g) for 1 minute. With a sterile pipet tip, carefully aspirate as much supernatant as possible while avoiding contact with the cell pellet.
Resuspend the pellet with freshly prepared staining medium (400 μl for every well of a 6-well plate harvested).
Place the conical tube of cell suspension in ice for 20 minutes.
After 20 minutes, centrifuge the conical tube at 1200rpm (290x g) for 1 minute. With a sterile pipet tip, carefully aspirate as much supernatant as possible while avoiding contact with the cell pellet.
Resuspend the pellet with freshly prepared sorting medium (~1 ml total). Add DAPI (1 μl ml−1).
Transfer the stained cell suspension through the cell strainer cap to a 5 ml round bottom test tube for FACS.
FACS settings may vary per user. Collect CD49D+ population in a sterile 5 ml round bottom test tube and cap. An example of gating strategy is provided in Supplementary Fig. 6.
Centrifuge the test tube at 1200rpm (290x g) for 1 minute. With a sterile pipet tip, carefully aspirate as much supernatant as possible while avoiding contact with the cell pellet.
Resuspend the pellet with NC-C (1 ml/ 106 cells) and transfer suspension to an ultra-low attachment 6-well plate (2 ml/ well). Incubate at 37 °C and 5% CO2.
Resume protocol Step 9.
Due to the increased risk of microbial contamination during the optional FACS purification step, collected cells may be fed with NC-C supplemented with Normocin (1 μl ml−1). Antimicrobial supplemented medium should be used for a minimum of two days.
Troubleshooting
See Table 3 for troubleshooting guidance.
Table 3.
Troubleshooting
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 8 (d-2, i) | hPSCs are not dissociated into small clusters or single cells. | Cells have not been exposed to EDTA for enough time. | Avoid additional pipetting and increase the duration of EDTA treatment. |
| 8 (d-2, ii) | Confluency of hPSCs is lower than 80%. | Not enough hPSCs were collected from maintenance plates or passaging involved excessive cell death. | Increase passaging ratio to 5:4 (i.e all cells from 5 wells of maintenance plate to 4 wells of a new 6-well plate) |
| 8 (d8, vii) | Monolayer is detaching from the well surface. | Forceful feeding has separated monolayer cells from the well surface. | When adding fresh medium, tilt plate and gently add medium to the lower side and corner of the well. |
| 8 (d10, viii) | NC cells are detached from underlying monolayer and have spontaneously formed free-floating spheroids. | Accelerated and efficient ENC induction. | Since free-floating spheroids contain the desired ENC cells, do not aspirate away these cells with old medium. Collect these cells with a P1000 micropipette and add them back into well once Accutase has been added. |
| 15 (ENC spheroid phase) | ENC free-floating spheroids have conjoined to form large, irregularly shaped bodies. | Elevated confluency of ENC added to low-attachment well. | Gently separate large, irregularly shaped bodies into smaller spheroids by pipetting them up and down 2–3 times with a P1000 micropipette. |
Supplementary Material
Supplementary Table 1. List of primers
Supplementary Table 2. List of antibodies
Figure 5. Expression of glial lineage markers hPSC-derived EN population.

a) Immunofluorescence image of TUJ1/GFAP stained differentiated cultures on day 55. b) Flow cytometry analysis of SOX10 and GFAP expression on day 75 of differentiation. AF647, Alexa Fluor™ 647; AF488, Alexa Fluor™ 488.
Timing of procedures.
Steps 1–6, thawing frozen hPSCs: thawing and plating frozen cells: 15 minutes.
Step 7, maintaining hPSC cultures: passaging hPSCs for maintenance: 15 minutes.
Steps 8I-IV (Option A) Steps 8I-II (Option B), ENC induction phase: passaging hPSCs for ENC induction: 15 minutes.
Steps 8V-X (Option A) Steps 8III-IX (Option B), ENC induction phase: ENC induction medium changes: 5 minutes.
Steps 9–13, ENC spheroid phase: Passaging ENC cells to spheroids: 45 minutes.
Steps 14–15, ENC spheroid phase: Replacing to NC-C medium: 10 minutes.
Steps 16–21, EN induction phase: Passaging spheroids for EN induction: 45 minutes.
Steps 22, EN induction phase: Viable cell count with hemocytometer: 15 minutes.
Steps 23–26, EN induction phase: Plating cells for EN induction: 15 minutes.
Steps 27, EN induction phase: Replacing EN-C medium: 5 minutes.
Acknowledgements
We thank Claudia Bispo and Ashley Carlos (UCSF Flow Cytometry core facility) for excellent technical assistance. The work was supported by the UCSF Program for Breakthrough Biomedical Research and Sandler Foundation, and March of Dimes Grant No. 1-FY18–394 to F.F. and by a grant from the National Institutes of Neurological Disorders and Stroke (NINDS) 5R01NS099270.
Footnotes
EDITORIAL SUMMARY Human pluripotent stem cells are first differentiated into enteric neural crest cells and then into functional enteric neurons using a defined culture system.
TWEET Differentiation of hPSC to functional enteric neurons.
COVER TEASER In vitro derivation of human enteric neurons
RELATED LINKS
Key reference(s) using this protocol
Fattahi et al., 2015 Curr Protoc in Stem Cell Biol., DOI:10.1002/9780470151808.sc01h09s33
Fattahi et al., 2016 Nature. DOI: 10.1038/nature16951
Tchieu et al., 2017 Cell Stem Cell : DOI: 10.1016/j.stem.2017.08.015
Competing interests
The Memorial Sloan-Kettering Cancer Center has filed a patent application (CA3009509A1) on human pluripotent stem cell derived enteric neural crest lineages for use in Hirschsprung’s disease with LS and FF as inventors. LS is a co-founder and consultant of BlueRock Therapeutics Inc.
Data availability
The data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
- 1.Shyamala K, Yanduri S, Girish HC & Murgod S Neural crest: The fourth germ layer. J. Oral Maxillofac. Pathol 19, 221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crane JF & Trainor PA Neural Crest Stem and Progenitor Cells. Annu. Rev. Cell Dev. Biol 22, 267–286 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Thomas S et al. Human neural crest cells display molecular and phenotypic hallmarks of stem cells. Hum. Mol. Genet 17, 3411–3425 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan KK et al. Hoxb3 vagal neural crest-specific enhancer element for controlling enteric nervous system development. Dev. Dyn 233, 473–483 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Fu M, Chi Hang Lui V, Har Sham M, Nga Yin Cheung A & Kwong Hang Tam P HOXB5 expression is spatially and temporarily regulated in human embryonic gut during neural crest cell colonization and differentiation of enteric neuroblasts. Dev. Dyn 228, 1–10 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Heanue TA & Pachnis V Enteric nervous system development and Hirschsprung’s disease: advances in genetic and stem cell studies. Nat. Rev. Neurosci 8, 466 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Cheeseman BL, Zhang D, Binder BJ, Newgreen DF & Landman KA Cell lineage tracing in the developing enteric nervous system: superstars revealed by experiment and simulation. J. R. Soc. Interface 11, 20130815 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace AS & Burns AJ Development of the enteric nervous system, smooth muscle and interstitial cells of Cajal in the human gastrointestinal tract. Cell Tissue Res 319, 367–382 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Lo L, Dormand E & Anderson DJ SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron 38, 17–31 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Lasrado R et al. Lineage-dependent spatial and functional organization of the mammalian enteric nervous system. Science 356, 722–726 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Lee G, Chambers SM, Tomishima MJ & Studer L Derivation of neural crest cells from human pluripotent stem cells. Nat. Protoc 5, 688–701 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Mica Y, Lee G, Chambers SM, Tomishima MJ & Studer L Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep 3, 1140–1152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fattahi F, Studer L & Tomishima MJ Neural Crest Cells from Dual SMAD Inhibition. Curr. Protoc. Stem Cell Biol 33, 1H.9.1–9 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Fattahi F et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 531, 105–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tchieu J et al. A Modular Platform for Differentiation of Human PSCs into All Major Ectodermal Lineages. Cell Stem Cell 21, 399–410.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hackland JOS et al. Top-Down Inhibition of BMP Signaling Enables Robust Induction of hPSCs Into Neural Crest in Fully Defined, Xeno-free Conditions. Stem Cell Rep 9, 1043– 1052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chalazonitis A et al. Neurotrophin-3 induces neural crest-derived cells from fetal rat gut to develop in vitro as neurons or glia. J. Neurosci. Off. J. Soc. Neurosci 14, 6571–6584 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalazonitis A, Rothman TP, Chen J & Gershon MD Age-dependent differences in the effects of GDNF and NT-3 on the development of neurons and glia from neural crest-derived precursors immunoselected from the fetal rat gut: expression of GFRalpha-1 in vitro and in vivo. Dev. Biol 204, 385–406 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Memic F et al. Transcription and Signaling Regulators in Developing Neuronal Subtypes of Mouse and Human Enteric Nervous System. Gastroenterology (2017). doi: 10.1053/j.gastro.2017.10.005 [DOI] [PMC free article] [PubMed]
- 20.Lai FP-L et al. Correction of Hirschsprung-Associated Mutations in Human Induced Pluripotent Stem Cells Via Clustered Regularly Interspaced Short Palindromic Repeats/Cas9, Restores Neural Crest Cell Function. Gastroenterology 153, 139–153.e8 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Burns AJ et al. White paper on guidelines concerning enteric nervous system stem cell therapy for enteric neuropathies. Dev. Biol 417, 229–251 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stamp LA Cell therapy for GI motility disorders: comparison of cell sources and proposed steps for treating Hirschsprung disease. Am. J. Physiol.-Gastrointest. Liver Physiol 312, G348–G354 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Li W et al. Characterization and transplantation of enteric neural crest cells from human induced pluripotent stem cells. Mol. Psychiatry 23, 499–508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Workman MJ et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med 23, 49–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gershon MD & Ratcliffe EM Developmental biology of the enteric nervous system: Pathogenesis of Hirschsprung’s disease and other congenital dysmotilities. Semin. Pediatr. Surg 13, 224–235 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen G et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 8, 424–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe K et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol 25, 681–686 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Maldonado M, Luu RJ, Ramos MEP & Nam J ROCK inhibitor primes human induced pluripotent stem cells to selectively differentiate towards mesendodermal lineage via epithelial-mesenchymal transition-like modulation. Stem Cell Res 17, 222–227 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Sasselli V, Pachnis V & Burns AJ The enteric nervous system. Dev. Biol 366, 64–73 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. List of primers
Supplementary Table 2. List of antibodies