

Summary
RNA splicing is related to many human diseases; however, lack of efficient genetic approaches to modulate splicing has prevented us from dissecting their functions in human diseases. Recently developed base editors (BEs) offer a new strategy to modulate RNA splicing by converting conservative splice sites, but it is limited by the editing precision and scope. To overcome the limitations of currently available BE-based tools, we combined SpCas9-NG with ABEmax to generate a new BE, ABEmax-NG. We demonstrated that ABEmax-NG performed precise A⋅T to G⋅C conversion with an expanded scope, thus covering many more splicing sites. Taking advantage of this tool, we precisely achieved A⋅T to G⋅C conversion exactly at the splice sites. We further modeled pathogenic RNA splicing in vitro and in vivo. Taken together, we successfully generated a versatile tool suitable for precise and broad editing at the splice sites.
Subject Areas: Biological Sciences, Molecular Biology, Biological Sciences Tools
Graphical Abstract
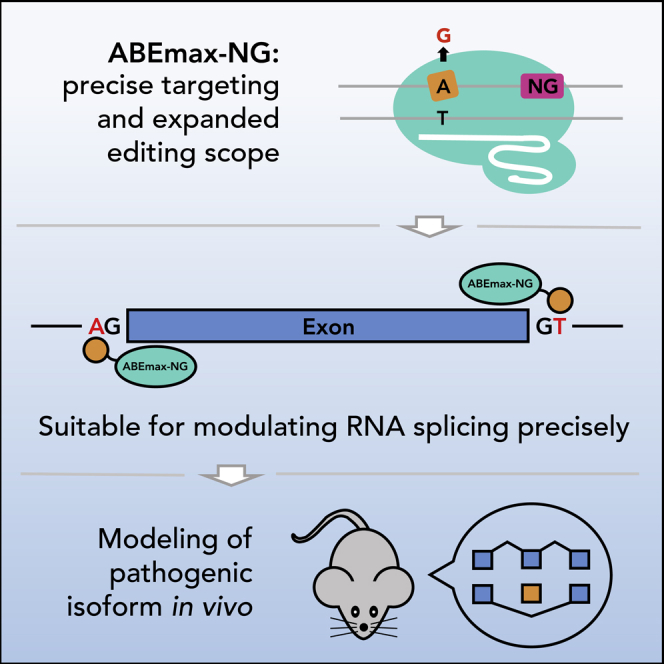
Highlights
-
•
ABEmax performs precise A⋅T to G⋅C conversion with an expanded scope
-
•
ABEmax-NG covers more splicing sites, resulting in precise RNA splicing modulation
-
•
ABEmax-NG efficiently and precisely models pathogenic RNA splicing in vitro and in vivo
Biological Sciences; Molecular Biology; Biological Sciences Tools
Introduction
As a critical biological process, RNA splicing plays many important roles in transcriptome diversity and development (Kalsotra and Cooper, 2011). Although more and more splicing isoforms have been increasingly identified with the development of new sequencing techniques, little is known about the functions of the vast majority of the splice isoforms due to lack of effective genetic approaches to modulate splicing.
Base editor (BE) systems have been recently developed that can induce single-nucleotide changes efficiently without formation of double-stranded DNA breaks (Rees and Liu, 2018). The controllability and precision of BEs make them widely used to induce or correct single point mutations (Liu et al., 2018b, Zeng et al., 2018). As splice sites are highly conserved in various species (Lim and Burge, 2001), BEs can be used to modulate RNA splicing by mutating splice sites. Indeed, recent studies have applied cytosine base editors (CBEs) to modulate multifarious RNA splicing by converting conserved guanine to adenine at either 5′ splice donor (SD) sites or 3′ splice acceptor (SA) sites (Gapinske et al., 2018, Liu et al., 2018a, Yuan et al., 2018). However, CBE-based splicing modulation is limited by the deficiency of CBE. First, CBE generates indels, although it is reduced by fusing uracil DNA glycosylase inhibitor (Wang et al., 2017). Second, CBE induces high proximal mutations, which makes it impossible to be applied to precise single-site editing (Lee et al., 2018). ABE (adenine base editor)-based splicing modulation has been successfully achieved in plant (Kang et al., 2018). Nevertheless, the requirement for the protospacer adjacent motif (PAM) to be adjacent to the target locus by current Cas9 variants limits the target sites of both ABE- and CBE-based splicing modulation. Therefore BEs suitable for splicing modulation are earnestly needed.
The application of ABE in splice site modulation in plants suggests the potential of ABE in RNA splicing modulation of animals. As ABE induces significantly few unwanted base conversions, indels, and proximal mutations (Lee et al., 2018), and a relaxed-PAM-recognized SpCas9 variant (SpCas9-NG) was developed recently (Nishimasu et al., 2018), we take advantage of them and try to combine the SpCas9-NG with ABEmax, which is the most efficient version of ABE (Koblan et al., 2018), to develop the NG-PAM-recognized adenine base editor (hereafter named ABEmax-NG). We demonstrated that ABEmax-NG has the character of precise single-base editing with expanded editing scope that is suitable for RNA splicing modulation. Furthermore, we have successfully modeled pathogenic splice site mutation in vivo.
Results
ABEmax-NG Is a Potential Tool with the Expanded Editing Scope that Is Suitable for Modulating RNA Splicing Precisely
Given splice sites only contain highly conserved two bases in various species (Lim and Burge, 2001), BE-based splicing modulation has to be very precise. ABE is considered more precise than CBE because it induces significantly few unwanted base conversions, indels, and proximal mutations (Lee et al., 2018). Also, ABE-mediated A⋅T to G⋅C conversion can be used to mutate the SA and SD sites; we hypothesize that ABE may be more suitable for modulating splicing than CBE. To demonstrate this hypothesis, we first explored the potential of ABEmax-NG, a combination of SpCas9-NG and ABEmax, in modulating RNA splicing over the human genome by bioinformatics analysis. As there are relatively conserved guanines near the SD and SA sites (Figure S1), we found that ABEs can precisely edit about twice as many splice sites as CBEs (Figure 1A), indicating that ABEs have advantages over CBEs not only in editing precision but also in the editing scope of the target splice sites. Then we further analyzed the editing scope among different PAMs over all validated ABE variants (Rees and Liu, 2018). As expected, ABEmax-NG can cover almost all editable sites, which is far beyond other variants (Figure 1B). With this, it has the most potential single guide RNAs (sgRNAs), and meanwhile, the most targeting genes (Figure 1C), making the ABEmax-NG a flexible tool to modulate various RNA splicing reactions. As expected, ABEmax-NG is also the most efficient tool that targets most human pathogenic splice sites (Figure 1D). This genome-wide analysis suggests that ABEmax-NG is a potential tool with expanded editing scope for precise modulation of RNA splicing.
Figure 1.

ABEmax-NG Has the Broadest Scope to Modulate RNA Splicing
(A) The proportion of human splice sites that can be edited precisely by CBE and ABE variants with distinct PAM specificities (NG, NGG, NGA, NGCG, NNGRRT, NNNRRT) and the corresponding editing windows. Human reference genome (hg38) and the annotation from GENCODE version 29 were used for analysis. The sgRNAs with a single target site in their editing windows were considered as precise editing sgRNAs and used for further analysis.
(B) The number of human splice sites can be edited precisely by ABEs. Distributions for distinct PAM specificities were shown.
(C) Cumulative percentage of genes whose splice sites can be edited precisely by ABEs. The horizontal axis shows the number of sgRNAs that can be designed per gene. Distributions for distinct PAM specificities were shown.
(D) The number of pathogenic human splice sites in the ClinVar database can be edited precisely by ABEs. Distributions for distinct PAM specificities were shown.
The ABEmax-NG Performs Efficient Base Editing in HEK293FT Cells
To demonstrate the potential of ABEmax-NG, we first constructed it by introducing R1335V/L1111R/D1135V/G1218R/E1219F/A1322R/T1337R mutations for converting SpCas9 nickase to SpCas9-NG nickase on ABEmax (Figure 2A). Then we tested its versatility by checking different NG(N) PAM recognition of ABEmax-NG in HEK293FT cells using an enhanced green fluorescent protein (EGFP) reporter, which was developed by mutating two bases of the EGFP-coding gene, one at the third position of the Thr-63 codon, which does not change the amino acid sequence of EGFP but provides multiple PAM sequences for ABEmax-NG recognition test, and another at the first position of the Gln-69 codon, converting the Gln-69 codon (CAG) into a stop codon (TAG), thus changing EGFP to stopped-EGFP (sEGFP). This sEGFP can be restored when the stop codon is corrected by ABEmax-NG (TAG to CAG), hence the editing efficiency of ABEmax-NG in different PAM sequence can be measured as the frequency of EGFP-expressing cells (Figure 2A). Then we designed an sgRNA to target sEGFP with the mutated T at position 7 of the protospacer, counted from the PAM sequence (Figure 2A; Table S1). The plasmids expressing sgRNA and ABEmax-NG were co-transfected with the EGFP reporter system.
Figure 2.

Verification of Versatile Base Editing of ABEmax-NG Using EGFP Reporter System
(A) ABEmax-NG construct and the schematic of the EGFP reporter system. The TAG stop codon was converted to glutamine by ABEmax-NG to restore EGFP protein production. The different third base in PAM does not change the original threonine of EGFP protein.
(B) ABEmax-NG-induced conversion of sEGFP to EGFP in HEK293FT cells. Scramble, non-targeting sgRNA served as negative control; sgRNA, sgRNA targeting sEGFP. Scale bars, 100 μm.
(C) Analysis of base editing by fluorescence-activated cell sorting. The percentage was calculated as the number of EGFP-positive cells/total transfected cells. Data are represented as the mean ± SEM (n = 3 from three independent experiments).
(D) The chromatograms of Sanger sequencing showing ABEmax-NG can induce sEGFP to EGFP conversion in different NG(N) PAM.
EGFP fluorescence could be observed in the cells 48 h after transfection targeting different PAM sequences (Figure 2B). Based on the analysis by flow cytometry, ABEmax-NG can recognize all types of NG(N) PAMs efficiently. In contrast, ABEmax, as expected, only recognized the NGG PAM with high efficiency and the NGA PAM with modest efficiency (Figure 2C). Sanger sequencing results further confirmed that ABEmax-NG induces the target base editing, resulting in the conversion of sEGFP to EGFP with different NG(N) PAMs (Figure 2D). These results demonstrated that ABEmax-NG is a versatile BE with expanded editing scope.
ABEmax-NG Can Induce Precise A-to-G Base Conversions In Vitro and In Vivo
To characterize this new tool, we comprehensively examined the editing of ABEmax-NG at 40 endogenous target sites with NG(N) PAMs in mouse-derived Neuro-2a (N2a) cells (Table S1). Compared with ABEmax that only induces A-to-G conversion at the NGG sites, ABEmax-NG performed efficient editing toward all types of PAMs (Figure S2). To further analyze the ABEmax-NG-mediated editing, we next chose 16 NG(N) sites for targeted deep sequencing. As expected, ABEmax-NG induced A-to-G conversion efficiently with few unwanted base conversions (the frequency: 0%–0.54%), indels (the frequency: 0%–2.79%), and proximal mutations (the frequency: 0%–0.32%) (Figures 3A, 3C, S3A, and S3B), indicating that ABEmax-NG is a precise BE.
Figure 3.

Efficient A-to-G Substitution in Mouse N2a Cells and Embryos by ABEmax-NG
(A) A-to-G editing by ABEmax and ABEmax-NG at 16 endogenous target sites in mouse N2a cells. The target base in the editing window was shown, counting the end distal to the protospacer adjacent motif (PAM) as position 1. Data are represented as the mean ± SEM (n = 3 from three independent experiments).
(B) A-to-G editing efficiency of ABEmax and ABEmax-NG at the four endogenous target sites in mouse embryos. Data are represented as the mean ± SEM (n = 3 for untreated, n = 8 for ABEmax, n = 8 for ABEmax-NG).
(C) Statistical analysis of the A-to-G editing frequency, unwanted base conversions, and indels induced by ABEmax-NG in (A). The median and interquartile range (IQR) are shown.
(D) Statistical analysis of the A-to-G editing frequency, unwanted base conversions, and indels induced by ABEmax-NG in (B). The median and IQR are shown.
Then we set out to determine whether ABEmax-NG can also work well in vivo. To this end, four sgRNAs covering all types of NG(N) PAMs have been subjected to test experiments in a total of 64 mouse embryos as previously described (Liu et al., 2018b). As expected, efficient base editing was achieved (Figures S5A and 3B). It is noteworthy that almost no unwanted base conversion, indel, and proximal mutation were detected (Figures 3D, S3C, and S3D), confirming that ABEmax-NG acts as a precise BE in vivo.
ABEmax-NG Can Serve as a Useful Modulator for RNA Splicing
The precision of ABEmax-NG suggests that ABEmax-NG could be a versatile tool for RNA splicing modulation, which is only limited to two editable bases for each SD and SA site. Thus, four splicing sites corresponding to human homologous pathogenic mutations were first tested in N2a cells (Table S1). Interestingly, the targeted deep sequencing showed that all the selected splice sites were edited by ABEmax-NG efficiently and precisely (Figures S4A and S4B). As expected, the corresponding changes to RNA splicing were detected by RT-PCR amplification of the respective cDNAs (Figure S4C). These results were further confirmed by Sanger sequencing (Figure S4D). Then we tested its performance in vivo. As expected, ABEmax-NG also worked well in mouse embryos except one embryo harboring few indels (the frequency is 3.35%; Figures 4A and 4B). Taken together, we demonstrated that ABEmax-NG performed efficient and precise splice site editing and is suitable for modulation of RNA splicing.
Figure 4.

ABEmax-NG Mediates Pathogenic Exon Skipping In Vivo
(A) A-to-G editing efficiency of ABEmax and ABEmax-NG at four splice sites in mouse embryos. Data are represented as the mean ± SEM (n = 3 for untreated, n = 8 for ABEmax, n = 8 for ABEmax-NG).
(B) Statistical analysis of the A-to-G editing frequency, unwanted base conversions, and indels induced by ABEmax-NG in (A). The median and interquartile range are shown.
(C) Schematic of ABEmax-NG-induced BBS2-long to BBS2-short switch. Top panel: Exon 4 is included in normal BBS2. Bottom panel: ABEmax-NG mutates the invariant A to G at the splice acceptor site, leading to the exclusion of Exon 4.
(D) The chromatogram of Sanger sequencing of tail confirmed heterozygous genotype of founder mouse M12.
(E) The individual fraction of each base induced by ABEmax-NG in BBS2 target splice site from different tissues of founder mouse M12 at 4 weeks. Data are analyzed by deep sequencing.
(F) Quantification of the rate of exon skipping of BBS2 Exon 4 from different tissues of founder mouse M12 at 4 weeks by deep sequencing of RT-PCR amplification of the cDNA.
The outperformance of ABEmax-NG encouraged us to model pathogenic RNA splicing in mouse. For this purpose, we focused on BBS2 gene, a member of the Bardet-Biedl syndrome (BBS) gene family. It is demonstrated that the splice site mutation of c.472-2A > G for this gene may cause human BBS, which is a developmental disorder that affects multiple systems (Innes et al., 2010). The ABEmax-NG-encoding mRNA together with sgBBS2 mRNA were co-injected into zygotes as previously (Liu et al., 2018b). A total of 40 embryos were transferred into two surrogate mothers, which generated 19 offspring (Figure S5B). Based on the results of deep sequencing, all 19 founder mice carried the expected target site mutation (Figure S6A).
It is known that the target mutation causes skipping of Exon 4 of the BBS2 gene (Figure 4C). Thus, we chose the founder mouse M12 harboring a heterozygous target mutation (Figures 4D and S6A), to further characterize the genotype and RNA splicing in different tissues (heart, liver, spleen, lung, kidney, brain, testis and intestine) at 4 weeks. As expected, all examined tissues carried the heterozygous target mutation and the isoform of the BBS2 RNA transcript skipping Exon 4 (Figures 4E, 4F, S6B, and S6C). These results demonstrated that ABEmax-NG can serve as a useful RNA splicing modulator in vivo.
Off-Target Analysis by WGS
The increased off-target possibility of SpCas9-NG prompted us to comprehensively investigate the off-target effects of ABEmax-NG carefully (Nishimasu et al., 2018). We performed whole-genome sequencing (WGS) on genomic DNA samples isolated from the founder mouse M12 and a wild-type mouse (WT) at depth of 33× and 30×, respectively (Figure 5D). After filtering out mouse dbSNPs, a total of 353,868 and 330,821 single nucleotide polymorphisms were detected over the genomes of WT and M12, respectively. The mutation frequency did not seem to increase significantly in M12 (Figure 5A). The heterozygous target mutation was also confirmed by WGS (Figure 5B). Given the mismatch tolerance of SpCas9 (Cho et al., 2014), we use CasOT (Xiao et al., 2014) to explore potential off-target sites. We used the criteria that up to a mismatch of 2 bp may be included in the seed region and a mismatch of 5 bp in the non-seed region with NG PAM. Among 24,297 potential off-target sites (Figure 5C), two off-target sites that are located in the intronic and intergenic regions were detected (Figures 5E, S7A, and S7B). For further demonstration, we performed Sanger sequencing of the two off-target sites, confirming the off-target mutagenesis (Figure S7C).
Figure 5.

Whole-Genome Analysis of BBS2 Mutant (M12) and Wild-Type (WT) Mice
(A) Summary of single nucleotide polymorphism (SNP) analysis. After filtering out naturally occurring variants in the mouse SNP database, 330,821 SNPs were obtained over the M12 genome. The number of A/T conversions were shown.
(B) Confirmation of the on-target base editing by analyzing the whole-genome sequencing results of M12.
(C) Summary of on-/off-target sites. A total of 24,298 sites, including 1 on-target site and 24,297 off-target sites were analyzed.
(D) Summary of the whole-genome sequencing.
(E) Summary of off-target analysis.
Discussion
In this study, taking advantage of ABE with few unwanted base conversions, indels, and proximal mutations (Lee et al., 2018), and SpCas9-NG with relaxed PAM (Nishimasu et al., 2018), we introduced R1335V/L1111R/D1135V/G1218R/E1219F/A1322R/T1337R mutations into ABEmax and developed ABEmax-NG to induce A-to-G conversion with expanded editing scope and high precision. The versatility of ABEmax-NG confers it as a useful RNA splicing modulator, which is currently the most suitable tool available targeting most human pathogenic splice sites.
More and more splice isoforms have been identified with the development of sequencing technique. It is important to explore the function of the different splicing isoforms because of their potential important biological roles (Kalsotra and Cooper, 2011). The application of BE in splice site modulation opens the potential of BE in studying RNA splicing (Gapinske et al., 2018, Kang et al., 2018, Liu et al., 2018a, Yuan et al., 2018). Nevertheless, the current BE-based splicing modulation is limited. Here we found that ABEs can precisely edit about twice as many splice sites as CBEs (Figure 1A), demonstrating that ABEs have more potential for studying splicing isoform. Moreover, with the application of the relaxed-PAM-recognized SpCas9-NG, the ABEmax-NG has the broadest editing scope to modulate RNA splicing.
Considering that both SD and SA sites contain only two bases, the precision of the base editing is critical for splicing modulation. As ABE performs precise base editing (Lee et al., 2018), as expected, ABEmax-NG induced almost no unwanted base conversion and indel when targeted at the splice sites (Figures S4A, S4B, 4A, and 4B). Compared with CBE, which induced unwanted base editing around splice sites (Lee et al., 2018, Yuan et al., 2018), ABEmax-NG obviously is a better tool for modulation of RNA splicing.
In summary, we have developed ABEmax-NG, a versatile BE that is the best available tool to successfully model human pathogenic splice site mutations in vitro and in vivo.
Limitations of the Study
Although ABEmax-NG covers the majority of splice sites, there are still some isoforms that cannot be modulated.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank members of Huang lab, Sun lab, and Chen lab for helpful discussions. This work is supported by the National Key R&D Program (2016YFC0905901, 2016YFC1000307), National Natural Science Foundation of China (81830004), National Postdoctoral Program for Innovative Talents (BX201700266), and Local Grants (16JC1420200, 17JC1420103).
Author Contributions
X.H., Q.S., and X.M. conceived, designed, and supervised the project. S.H., Z.L., and X.L. performed most experiments with the help of G.L., J.L., Z.L., G.Y., and Y.Z. Z.L. and X.L. provided expert technical assistance. S.H. and X.H. wrote the paper with inputs from all authors. X.L. edited the manuscript. X.H., Q.S., and X.M. managed the project.
Declaration of Interests
The authors declare no competing interests.
Published: May 31, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.05.008.
Contributor Information
Xu Ma, Email: nfpcc_ma@163.com.
Qiang Sun, Email: qsun@ion.ac.cn.
Xingxu Huang, Email: huangxx@shanghaitech.edu.cn.
Supplemental Information
References
- Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cho, S.W., Kim, S., Kim, Y., Kweon, J., Kim, H.S., Bae, S., and Kim, J.S. (2014). Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24, 132-141. [DOI] [PMC free article] [PubMed]
- Gapinske M., Luu A., Winter J., Woods W.S., Kostan K.A., Shiva N., Song J.S., Perez-Pinera P. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol. 2018;19:107. doi: 10.1186/s13059-018-1482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gapinske, M., Luu, A., Winter, J., Woods, W.S., Kostan, K.A., Shiva, N., Song, J.S., and Perez-Pinera, P. (2018). CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol. 19, 107. [DOI] [PMC free article] [PubMed]
- Innes A.M., Boycott K.M., Puffenberger E.G., Redl D., MacDonald I.M., Chudley A.E., Beaulieu C., Perrier R., Gillan T., Wade A. A founder mutation in BBS2 is responsible for Bardet-Biedl syndrome in the Hutterite population: utility of SNP arrays in genetically heterogeneous disorders. Clin. Genet. 2010;78:424–431. doi: 10.1111/j.1399-0004.2010.01481.x. [DOI] [PubMed] [Google Scholar]; Innes, A.M., Boycott, K.M., Puffenberger, E.G., Redl, D., MacDonald, I.M., Chudley, A.E., Beaulieu, C., Perrier, R., Gillan, T., Wade, A., et al. (2010). A founder mutation in BBS2 is responsible for Bardet-Biedl syndrome in the Hutterite population: utility of SNP arrays in genetically heterogeneous disorders. Clin. Genet. 78, 424-431. [DOI] [PubMed]
- Kalsotra A., Cooper T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011;12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kalsotra, A., and Cooper, T.A. (2011). Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715-729. [DOI] [PMC free article] [PubMed]
- Kang B.C., Yun J.Y., Kim S.T., Shin Y., Ryu J., Choi M., Woo J.W., Kim J.S. Precision genome engineering through adenine base editing in plants. Nat. Plants. 2018;4:427–431. doi: 10.1038/s41477-018-0178-x. [DOI] [PubMed] [Google Scholar]; Kang, B.C., Yun, J.Y., Kim, S.T., Shin, Y., Ryu, J., Choi, M., Woo, J.W., and Kim, J.S. (2018). Precision genome engineering through adenine base editing in plants. Nat. Plants 4, 427-431. [DOI] [PubMed]
- Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]; Koblan, L.W., Doman, J.L., Wilson, C., Levy, J.M., Tay, T., Newby, G.A., Maianti, J.P., Raguram, A., and Liu, D.R. (2018). Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843-846. [DOI] [PMC free article] [PubMed]
- Lee H.K., Willi M., Miller S.M., Kim S., Liu C., Liu D.R., Hennighausen L. Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nat. Commun. 2018;9:4804. doi: 10.1038/s41467-018-07322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lee, H.K., Willi, M., Miller, S.M., Kim, S., Liu, C., Liu, D.R., and Hennighausen, L. (2018). Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nat. Commun. 9, 4804. [DOI] [PMC free article] [PubMed]
- Lim L.P., Burge C.B. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. U S A. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lim, L.P., and Burge, C.B. (2001). A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. U S A 98, 11193-11198. [DOI] [PMC free article] [PubMed]
- Liu Z., Chen M., Chen S., Deng J., Song Y., Lai L., Li Z. Highly efficient RNA-guided base editing in rabbit. Nat. Commun. 2018;9:2717. doi: 10.1038/s41467-018-05232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu, Z., Chen, M., Chen, S., Deng, J., Song, Y., Lai, L., and Li, Z. (2018a). Highly efficient RNA-guided base editing in rabbit. Nat. Commun. 9, 2717. [DOI] [PMC free article] [PubMed]
- Liu Z., Lu Z., Yang G., Huang S., Li G., Feng S., Liu Y., Li J., Yu W., Zhang Y. Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat. Commun. 2018;9:2338. doi: 10.1038/s41467-018-04768-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu, Z., Lu, Z., Yang, G., Huang, S., Li, G., Feng, S., Liu, Y., Li, J., Yu, W., Zhang, Y., et al. (2018b). Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat. Commun. 9, 2338. [DOI] [PMC free article] [PubMed]
- Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nishimasu, H., Shi, X., Ishiguro, S., Gao, L., Hirano, S., Okazaki, S., Noda, T., Abudayyeh, O.O., Gootenberg, J.S., Mori, H., et al. (2018). Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259-1262. [DOI] [PMC free article] [PubMed]
- Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rees, H.A., and Liu, D.R. (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770-788. [DOI] [PMC free article] [PubMed]
- Wang L., Xue W., Yan L., Li X., Wei J., Chen M., Wu J., Yang B., Yang L., Chen J. Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res. 2017;27:1289–1292. doi: 10.1038/cr.2017.111. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang, L., Xue, W., Yan, L., Li, X., Wei, J., Chen, M., Wu, J., Yang, B., Yang, L., and Chen, J. (2017). Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res. 27, 1289-1292. [DOI] [PMC free article] [PubMed]
- Xiao A., Cheng Z., Kong L., Zhu Z., Lin S., Gao G., Zhang B. CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics. 2014;30:1180–1182. doi: 10.1093/bioinformatics/btt764. [DOI] [PubMed] [Google Scholar]; Xiao, A., Cheng, Z., Kong, L., Zhu, Z., Lin, S., Gao, G., and Zhang, B. (2014). CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30, 1180-1182. [DOI] [PubMed]
- Yuan J., Ma Y., Huang T., Chen Y., Peng Y., Li B., Li J., Zhang Y., Song B., Sun X. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell. 2018;72:380–394.e7. doi: 10.1016/j.molcel.2018.09.002. [DOI] [PubMed] [Google Scholar]; Yuan, J., Ma, Y., Huang, T., Chen, Y., Peng, Y., Li, B., Li, J., Zhang, Y., Song, B., Sun, X., et al. (2018). Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell 72, 380-394.e7. [DOI] [PubMed]
- Zeng Y., Li J., Li G., Huang S., Yu W., Zhang Y., Chen D., Chen J., Liu J., Huang X. Correction of the marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol. Ther. 2018;26:2631–2637. doi: 10.1016/j.ymthe.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zeng, Y., Li, J., Li, G., Huang, S., Yu, W., Zhang, Y., Chen, D., Chen, J., Liu, J., and Huang, X. (2018). Correction of the marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol. Ther. 26, 2631-2637. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.