

Abstract
Naloxone (17-allyl-4,5α-epoxy-3,14-dihydroxymorphinan-6-one HCl), a μ-opioid receptor antagonist, is administered intranasally to reverse an opioid overdose but its short half-life may necessitate subsequent doses. The addition of naltrexone [17-(cyclopropylmethyl)-4,5α-epoxy-3,14-dihydroxymorphinan-6-one], another μ-receptor antagonist, which has a reported half-life of 3 1/2 hours, may extend the available time to receive medical treatment. In a phase 1 pharmacokinetic study, healthy adults were administered naloxone and naltrexone intranasally, separately and in combination. When administered with naloxone, the Cmax value of naltrexone decreased 62% and the area under the concentration-time curve from time zero to infinity (AUC0–inf) decreased 38% compared with when it was given separately; lower concentrations of naltrexone were observed as early as 5 minutes postdose. In contrast, the Cmax and AUC0–inf values of naloxone decreased only 18% and 16%, respectively, when given with naltrexone. This apparent interaction was investigated further to determine if naloxone and naltrexone shared a transporter. Neither compound was a substrate for organic cation transporter (OCT) 1, OCT2, OCT3, OCTN1, or OCTN2. There was no evidence of the involvement of a transmembrane transporter when they were tested separately or in combination at concentrations of 10 and 500 µM using Madin-Darby canine kidney II cell monolayers at pH 7.4. The efflux ratios of naloxone and naltrexone increased to six or greater when the apical solution was pH 5.5, the approximate pH of the nasal cavity; there was no apparent interaction when the two were coincubated. The importance of understanding how opioid antagonists are absorbed by the nasal epithelium is magnified by the rise in overdose deaths attributed to long-lived synthetic opioids and the realization that better strategies are needed to treat opioid overdoses.
Introduction
Opioid overdose in the United States led to 750,000 emergency department visits and more than 49,000 opioid-related deaths in 2017 (https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates). The use of the antagonist naloxone (17-allyl-4,5α-epoxy-3,14-dihydroxymorphinan-6-one HCl) has been endorsed by multiple government agencies to limit opioid-induced fatalities (https://www.surgeongeneral.gov/priorities/opioid-overdose-prevention/naloxone-advisory.html). Improvised naloxone kits for intranasal administration have been promoted for use by nonmedical personnel and the general public to counteract opioid overdoses (Carpenter et al., 2016); however, approximately one-half of subjects in a human use study could not assemble and use the device without proper training (Edwards et al., 2015). In 2015, the U.S. Food and Drug Administration approved Narcan, an intranasal device that delivers 4 mg naloxone in a volume of 0.1 ml. Ninety percent of subjects were able to use it correctly without any training; it also produces plasma concentrations as rapidly as an intramuscular injection (Krieter et al., 2016). Due to its short half-life, naloxone may be effective for 1 hour or less, and depending on the quantity and nature of the opioid ingested the person could relapse into respiratory depression (Li et al., 2018) before trained medical personnel respond.
Naltrexone [17-(cyclopropylmethyl)-4,5α-epoxy-3,14-dihydroxymorphinan-6-one], a μ-opioid receptor antagonist, has a reported half-life of approximately 3 1/2 hours (Yuen et al., 1999) and has a 5-fold higher affinity for the receptor compared with naloxone (Cassel et al., 2005). While the duration of occupancy of naloxone on the μ-receptor has a half-life of 2 hours (Kim et al., 1997), naltrexone has a half-life duration of 72 hours (Lee et al., 1988). This is longer than the plasma half-life of naltrexone and its major metabolite 6β-naltrexol (Meyer et al., 1984), suggesting it remains on the receptor longer than indicated by the plasma concentrations. Combining it with naloxone may increase the window for response to an opioid overdose.
An initial study demonstrated that naltrexone can be absorbed after nasal administration of 2 mg in 1 ml of water; the Cmax value was 3.86 ng/ml at 0.38 hours (Brown et al., 2014). When a crushed extended-release oxycodone tablet containing 3.6 mg naltrexone was administered intranasally, the maximum concentration of the antagonist was 4.4 ng/ml at 0.3 hours (Setnik et al., 2015).
While naltrexone is indicated for the prevention of relapse to opioid dependence, it has not been evaluated for the reversal of an opioid overdose. A pilot study was conducted to assess the feasibility of combining naltrexone with naloxone as an intranasal formulation to lengthen the time for the reversal of opioid-induced respiratory depression in emergency situations. The results demonstrate reductions in plasma concentrations when the two drugs were combined relative to when they were given separately; the reduction in naltrexone concentrations was more pronounced than for naloxone. In vitro studies were conducted to determine the nature of the interaction. This report details the results from both the clinical and in vitro studies.
Materials and Methods
Pharmacokinetic Study
Study Participants.
The clinical study was conducted by Vince & Associates Clinical Research (Overland Park, KS). The study was approved by the MidLands Independent Review Board (Overland Park, KS); all subjects gave written, informed consent before participation. The study was carried out in accordance with the 1996 International Conference on Harmonization for Good Clinical Practices Guidelines (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf). The study was registered on ClinicalTrials.gov as NCT03851731.
Healthy male and female volunteers aged 18–55 years, with body mass index of 18–30 kg/m2, participated in the pharmacokinetic (PK) study. Participants were currently not taking either prescription or over-the-counter medications, and nonsmokers or those who smoked 20 or fewer cigarettes per day were enrolled. Screening procedures conducted within 21 days of study initiation included the following: medical history, physical examination, evidence of nasal irritation, 12-lead ECG, complete blood count, clinical chemistry, coagulation markers, hepatitis and human immunodeficiency screening, urinalysis, and urine drug screen. Female participants were tested for pregnancy at screening and admission to the clinic. Participants were excluded if they had abnormal nasal anatomy or symptoms (e.g., runny nose, nasal polyps), an upper respiratory tract infection, used opioid analgesics for pain relief within the previous 14 days, or in the judgment of the investigator had significant acute or chronic medical conditions. Participants were required to abstain from grapefruit juice and alcohol 72 hours prior to admission to the end of the last blood draw of the study. On days of dosing, a participant’s vital signs were required to be within the acceptable range before receiving naloxone, defined as: systolic blood pressure >90 and ≤140 mm Hg; diastolic blood pressure >55 and ≤90 mm Hg; resting heart rate >40 and ≤100 beats per minute; and respiratory rate >8 and ≤20 respirations per minute.
Study Design.
This was an inpatient, double-blind, randomized, three-period, three-treatment, six-sequence, crossover study. Participants were randomly assigned to one of six possible sequences. On the day after clinic admission, participants were administered the study drugs in randomized order with a 4-day washout period between doses. Participants remained in the clinic for 13 days until all three treatments were administered; they received a follow-up phone call 3–5 days after discharge. They fasted from midnight before each dosing day until 1 hour after dose administration. Participants refrained from smoking and caffeine-containing drinks for 1 hour before until 2 hours after dosing. They received one of the following three treatments in one nostril:
Treatment 1: Naltrexone (2 mg), intranasally; (one 0.1 ml spray of a 20 mg/ml formulation);
Treatment 2: Naloxone (4 mg), intranasally; (one 0.1 ml spray of a 40 mg/ml formulation); or
Treatment 3: Naltrexone (2 mg) and naloxone (4 mg), intranasally (one 0.1 ml spray of the 20 mg/ml naltrexone plus 40 mg/ml naloxone formulation).
The study drugs were administered in the supine position, and subjects remained in this position for approximately 1 hour after dosing. Participants were instructed not to breathe when the drug was administered to simulate an opioid overdose with a patient in respiratory arrest. Twelve-lead ECGs were collected predose and at 1 and 6 hours postdose. Venous blood samples (4 ml) were collected for the analyses of plasma naloxone and naltrexone concentrations at predose; 2.5, 5, 10, 15, 20, 30, 45, and 60 minutes; and 2, 3, 4, 6, 8, 12, 24, 30, 36, 48, 60, and 72 hours postdose using Vacutainer tubes containing sodium heparin. The plasma was stored at < −60°C until analyzed.
Study Drugs.
Naltrexone HCl and naloxone HCl powders were purchased from Mallinckrodt, Inc. (St. Louis, MO) and were current Good Manufacturing Practices grade (https://www.fda.gov/drugs/pharmaceutical-quality-resources/current-good-manufacturing-practice-cgmp-regulations). The formulations were made by the pharmacists at Vince & Associates Clinical Research; sterile water for injection was the vehicle for both compounds. The study drugs were administered using a LMA mucosal atomization device (Teleflex Medical Europe Ltd., Athione, Ireland) and a 1-ml disposable syringe. The syringes and devices were weighed before and after dose administration. Based on the dose analysis and weight of the dose administered, the mean ± S.D. of milligrams administered were the following: treatment A, 2.24 ± 0.03 mg naltrexone HCl; treatment B, 4.58 ± 0.05 mg naloxone HCl; and treatment C, 2.27 ± 0.07 mg naltrexone HCl plus 4.62 ± 0.15 mg naloxone HCl.
Analytical Methods.
Plasma naloxone concentrations were assayed as described previously (Krieter et al., 2016); the lower limit of quantitation was 0.01 ng/ml. The interday precision of the calibration curves and quality control samples ranged from 3.22% to 9.05% and the accuracy ranged between −3.14% and 5.33% during the analysis of the samples.
Plasma naltrexone and 6β-naltrexol concentrations were determined using a validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) assay by XenoBiotic Laboratories (Plainsboro, NJ). Plasma samples (0.15 ml) were mixed with 0.1 ml of 1% formic acid in water and 0.05 ml of acetonitrile:water (2:8) containing the internal standards (0.5 ng naltrexone-d3 and 0.25 ng 6β-naltrexol-d3) and added to individual wells of a preconditioned 96-well plate. The plate was washed sequentially with 1% formic acid in water, water, methanol:water (1:1), and methanol. The analytes were eluted using 4% ammonium hydroxide in methanol. After evaporation, the residue was dissolved in 0.15 ml methanol:0.1% formic acid (8:92) and submitted to LC-MS/MS analysis. The AB MDS Sciex API-5000 LC-MS/MS system (Framingham, MA) with an atmospheric pressure chemical ionization source was operated in the positive ion detection mode. The mobile phase consisted of a gradient from 93% mobile phase A (10 mM ammonium formate, pH 4.0)/7% mobile phase B [acetonitrile:methanol (2:8)] to 80% mobile phase A/20% mobile phase B over 1.7 minutes at a flow rate of 0.5 ml/min through a 2.1 × 50 mm Kinetex EVO C18 2.6 µm column (Phenomenex, Torrance, CA). Naltrexone eluted at approximately 1.45 minutes; ions monitored had mass-to-charge ratios (m/z) 342.2 and 324.2 for naltrexone and 345.2 and 327.3 for its internal standard. 6β-Naltrexol eluted at approximately 1.60 minutes; ions monitored had m/z 344.2 and 326.2 for 6β-naltrexol and 347.1 and 329.3 for its internal standard. The interday precision of the calibration curves and quality control samples for naltrexone ranged from 2.92% to 7.87%, and the accuracy ranged between −3.50% and 0.75% during the analysis of the samples. The interday precision of the calibration curves and quality control samples of 6β-naltrexol ranged from 2.89% to 7.38%, and the accuracy ranged between −7.13% and 2.00% during the analysis of the samples. The lower limit of quantification for both naltrexone and 6β-naltrexol was 0.02 ng/ml.
Data Analyses.
The safety population included all subjects who received at least one intranasal dose; the PK population included all participants who received at least one dose with sufficient data to calculate meaningful PK parameters. The PK parameters were calculated using standard noncompartmental methods and a validated installation of WinNonlin Phoenix, version 6.3 (Cetera, Princeton, NJ). Values of peak plasma concentrations (Cmax) and the time to reach Cmax were the observed values obtained directly from the concentration-time data. The terminal elimination half-life was estimated by linear regression analysis. The area under the concentration-time curve from time zero to the last quantifiable concentration was determined by the linear-up/log-down trapezoidal method and extrapolated to the area under the concentration-time curve from time zero to infinity (AUC0–inf) by adding the value of the last quantifiable concentration divided by the terminal rate constant. Since the extrapolated area under the plasma concentration-time curve was less than 20% for all participants, only AUC0–inf is reported. The apparent total body clearance was calculated as the dose divided by AUC0–inf. Within an ANOVA framework, comparisons of ln-transformed PK parameters were performed using a mixed-effects model, where sequence, period, and treatment were the independent factors. The 90% confidence intervals for the ratio of the geometric least-squares mean values of Cmax and AUC0–inf were constructed for comparison of the two drugs administered in combination versus separately. The 90% confidence intervals were obtained by exponentiation of the 90% confidence intervals for the differences between the least-squares mean values based on a ln scale. All analyses of demographic and safety data were performed using SAS statistical software, version 9.3 (SAS Institute, Inc., Cary, NC).
In Vitro Transporter Studies
Methods.
Transporter studies with organic cation transporter (OCT) 1, OCT2, and OCT3 were conducted using human embryonic kidney 293 (HEK293) cells (ATCC, Manassas, VA) that had been transfected with vectors containing human transporter cDNA; control cells were transfected with the vector only. The culture medium was Dulbecco’s modified Eagle’s medium supplemented with 8.9% FBS, 0.89% antibiotic, and 1.79 mM L-glutamine. The incubation medium for the HEK293 cells was Hanks’ balanced salt solution containing 10 mM HEPES, pH 7.4. OCTN1 and OCTN2 transporter studies used S2 cells established by culturing microdissected S2 segments derived from transgenic mice harboring the temperature-sensitive simian virus 40 large T-antigen gene. Cells were transfected with vectors containing the human transporter cDNA. Control cells for all five transporters contained only the vector cDNA. The culture medium for the S2 cells was RITC80-7 supplemented with 4.7% FBS, 9.3 µg/ml epidermal growth factor, 0.08 U insulin/ml, and 9.5 mg/l transferrin. PBS containing 0.2% bovine serum albumin (D-PBS), pH 7.4, was used as the incubation medium for the S2 cells. All cells were cultured with 5% CO2 and 95% relative humidity at 33°C (S2 cells) or 37°C (HEK293 cells).
The medium was removed by aspiration, the cells were rinsed with either 1 ml of Hanks’ balanced salt solution or D-PBS (this was replaced with medium containing the control inhibitor or solvent control), and then the cells were preincubated. After preincubation, the medium was removed and replaced with 0.3 ml buffer containing either the test article or the positive control. The final concentration of naloxone and naltrexone was 1 µM in 0.2% DMSO, the solvent for the test articles and controls. Cells were incubated for 2, 5, 10, or 20 minutes at 37°C, at which time the solutions were removed. Positive controls were 10 µM [14C]metformin (OCT1 and OCT2), 5 µM [14C]triethylamine (OCT3 and OCTN1), and 0.03 µM [3H]carnitine (OCTN2). All incubations were done in triplicate. HEK293 cells were washed one time with 1 ml of ice-cold D-PBS and twice with 1 ml of ice-cold PBS; S2 cells were washed three times with ice-cold D-PBS. After removal of the medium, 0.5 ml of purified water was added to each well to lyse the cells and samples were collected for analysis of naloxone and naltrexone and then analyzed by LC-MS/MS. Samples were mixed with 20 µl water with the internal standard (nalmefene). An AB Sciex API-4000 mass spectrometer was operated in the positive mode with an Acquity UPLC BEH C18 analytical column (2.1 × 50 mm, 1.7 µm). The mobile phase consisted of a gradient from 95% mobile phase A (10 mM ammonium acetate)/5% mobile phase B (10 mM ammonium acetate in acetonitrile with 0.1% ammonium hydroxide) to 10% mobile phase A/90% mobile phase B in 2 minutes. The flow rate was 0.5 ml/min. The ions monitored had m/z 328.2 and 310.0 for naloxone, m/z 342.2 and 324.1 for naltrexone, and m/z 340.3 and 322.3 for the internal standard. Concentrations were calculated as the area under the curve compared with that of the internal standard using known concentrations of naloxone and naltrexone. For the positive controls, a 0.3 ml aliquot of the cell lysate was mixed with 5 ml of scintillation cocktail, and radioactivity was measured by liquid scintillation counting. Samples were collected for protein content using the BCA-Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). The uptake amount and cleared volume of naloxone and naltrexone were calculated as follows:
 |
For positive controls, disintegrations per minute were substituted for picomoles in the previous equations.
Transport studies were also conducted using wild-type Madin-Darby canine kidney II (MDCKII) cells that had been transfected with vectors containing human transporter cDNA (Netherlands Cancer Institute, Amsterdam, Netherlands). Cells were plated and maintained on 24-well Transwell plates (Corning, Corning, NY) for 3–5 days prior to the experiment. Culture medium was removed and incubation medium (Hanks’ balanced salt solution supplemented with 25 mM HEPES and 25 mM glucose) was added to the cells. The pH of the basolateral buffer was 7.4 and that of the apical buffer was either pH 7.4 or 5.5. Approximately 10 minutes after incubation medium was added, the transepithelial electrical resistance was recorded and cells were preincubated at 37°C for 30–60 minutes. After preincubation, the medium containing naloxone, naltrexone, or control compounds ([3H]mannitol and [14C]caffeine) was added to the donor chamber. Samples were collected from the receiver side at 15, 60, and 120 minutes and replaced by 0.1 ml of incubation medium. The transepithelial electrical resistance was also measured at the end of the incubation to determine if the cells were still confluent. Samples were mixed with 25 µl of methanol:water (1:1 v/v) and 75 µl of the internal standard (hydroxybuproprion-d6) in methanol:water (1:1 v/v) and analyzed by LC-MS/MS using an AB Sciex API-5500 mass spectrometer as described previously, except that the gradient changed from 70% mobile phase A/30% mobile phase B to 5% mobile phase A/95% mobile phase B over 3 minutes. Ions for the internal standard were monitored at m/z 262.0 and 244.0. Concentrations of radioactivity were determined as detailed previously.
The apparent permeability (Papp) was calculated as follows:
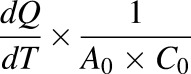 |
where dQ is the amount of test drug transported (in picomoles); dT is the incubation time (in seconds); A0 is the surface area of the membrane (in squared centimeters); and C0 is the initial concentration of the test drug in the donor chamber (in picomoles per cubic centimeter). The efflux ratio was calculated as the Papp basal-to-apical/Papp apical-to-basal ratio. The sex of the cell lines used in the experiments is unknown.
Results
Pharmacokinetic Study
Subject Characteristics.
All subjects initiating the study (Table 1) received at least one dose of naloxone and/or naltrexone; 11 subjects completed the study. One female subject withdrew during the first period due to a moderate headache with mild nausea that occurred 28 hours after administration of 2 mg naltrexone.
TABLE 1.
Pharmacokinetics of naloxone: subject demographics
| Demographics | All | Male | Female |
|---|---|---|---|
| Number | 12 | 6 | 6 |
| Mean age, y (range) | 36.0 (22.0–48.0) | 39.2 (26.0–48.0) | 32.8 (22.0–48.0) |
| Race | |||
| White | 4 | 1 | 3 |
| Black/African American | 8 | 5 | 3 |
| Ethnicity | |||
| Hispanic or Latino | 1 | 0 | 1 |
| Not Hispanic or Latino | 11 | 6 | 5 |
| Mean Weight, kg (range) | 74.7 (49.4–99.2) | 82.7 (71.8–99.2) | 66.6 (49.4–84.8) |
| Mean BMI, kg/m2 (range) | 25.0 (19.2–29.3) | 25.3 (23.6–27.2) | 24.6 (19.2–29.3) |
BMI, body mass index.
Pharmacokinetics.
The geometric mean Cmax and AUC0–inf values of naloxone following a 4-mg intranasal dose were 4.30 ng/ml and 8.13 ng·h/ml, respectively (Fig. 1; Table 2). The Cmax and AUC0–inf values decreased by approximately 18% and 16 %, respectively, when naloxone was administered in combination with 2 mg naltrexone (Table 3). The median time to reach Cmax value remained unchanged at 30 minutes, and the elimination half-life was also unchanged.
Fig. 1.

Mean (S.D.) concentrations of naloxone in healthy participants following intranasal administration of 4 mg naloxone alone and in combination with 2 mg naltrexone. Upper graph: concentrations to 12 hours postdose; Lower graph: concentrations to 1 hour postdose.
TABLE 2.
Geometric mean pharmacokinetic parameters (%CV) of naloxone, naltrexone, and 6β-naltrexol
| Variablea (U) | Naloxone |
Naltrexone |
6β-Naltrexol |
|||
|---|---|---|---|---|---|---|
| Alone (Treatment B) | Plus Naltrexone (Treatment C) | Alone (Treatment A) | Plus Naloxone (Treatment C) | Alone (Treatment A) | Plus Naloxone (Treatment C) | |
| N | 11 | 11 | 12 | 11 | 12 | 11 |
| Cmax (ng/ml) | 4.30 (47.5) | 3.60 (36.5) | 4.55 (80.0) | 1.71 (35.1) | 2.09 (32.5) | 2.09 (26.2) |
| Tmax (h) | 0.50 (0.25–0.75) | 0.50 (0.25–0.75) | 0.33 (0.17–1.0) | 0.75 (0.25–2.0) | 2.00 (0.75–4.0) | 2.00 (0.75–3.0) |
| AUC0–inf (ng·h/ml) | 8.13 (38.2) | 7.00 (32.5) | 9.61 (39.1) | 5.88 (25.2)b | 30.8 (32.7) | 28.3 (28.4) |
| λz (h−1) | 0.380 (32.1) | 0.355 (43.9) | 0.319 (18.2) | 0.322 (10.3)b | 0.0433 (31.6) | 0.0430 (24.5) |
| t1/2 (h) | 1.83 (32.1) | 1.95 (45.7) | 2.17 (18.2) | 2.15 (10.3)b | 16.0 (31.6) | 16.1 (24.5) |
| CL/F (l/min) | 6.71 (38.2) | 7.79 (43.9) | 3.12 (39.1) | 5.10 (25.2)b | NC | NC |
AUC0–inf, area under the plasma concentration-time curve from time zero to infinity; CL/F, apparent clearance; λz, terminal phase rate constant; NC, not calculated; t1/2, terminal phase half-life; Tmax, time to reach Cmax; Treatment A, 2 mg naltrexone, intranasally; Treatment B, 4 mg naloxone, intranasally; Treatment C, 2 mg naltrexone plus 4 mg naloxone, intranasally.
Geometric mean (%CV) for all except median (range) for Tmax.
N = 10.
TABLE 3.
Statistical summary of treatment comparisons
| Variable | Analyte | Comparison (Treatment C vs. Reference) | Geometric Mean Ratio (Treatment C/Reference) | 90% CI |
|---|---|---|---|---|
| Cmax (ng/ml) | Naloxone | C vs. B | 81.5 | 63.6–105 |
| Naltrexone | C vs. A | 38.4 | 25.7–57.3 | |
| 6β-Naltrexol | C vs. A | 101 | 92.7–110 | |
| AUC0–inf (ng·h/ml) | Naloxone | C vs. B | 84.6 | 70.3–102 |
| Naltrexone | C vs. A | 61.6 | 50.9–74.6 | |
| 6β-Naltrexol | C vs. A | 94.4 | 89.3–99.8 |
AUC0–inf, area under the plasma concentration-time curve from time zero to infinity; CI, confidence interval; Treatment A, 2 mg naltrexone, intranasally; Treatment B, 4 mg naloxone, intranasally; Treatment C, 2 mg naltrexone plus 4 mg naloxone, intranasally.
When naloxone was added to the naltrexone intranasal formulation, the Cmax value decreased from 4.55 to 1.71 ng/ml, a decline of approximately 62% (Fig. 2; Table 2), and the AUC0–inf value decreased approximately 38%. The median time to reach Cmax value increased from 0.33 to 0.75 hours when the combination formulation was administered compared with naltrexone alone. However, the half-life of naltrexone was unchanged.
Fig. 2.

Mean (S.D.) concentrations of naltrexone in healthy participants following intranasal administration of 2 mg naltrexone alone and in combination with 4 mg naloxone. Upper graph: concentrations to 12 hours postdose; Lower graph: concentrations to 1 hour postdose.
The decreased concentration of naltrexone concentrations in treatment C was evident even at 5 and 10 minutes after dose administration; the concentrations were 83%–86% lower than when naltrexone was dosed alone. The concentrations of naltrexone continued to be considerably lower even at 6 hours postdose. In contrast, there was no change in the PK values of 6β-naltrexol when naltrexone was administered with or without naloxone (Fig. 3; Tables 2 and 3). The two formulations were bioequivalent for 6β-naltrexol, based on the 90% confidence intervals of Cmax and AUC0–inf (Table 3).
Fig. 3.

Mean (S.D.) concentrations of 6β-naltrexol in healthy participants following intranasal administration of 2 mg naltrexone alone and in combination with 4 mg naloxone.
There were minor differences in the PK parameters between males and females (Table 4). However, the small sample size of this pilot study precludes any definitive conclusions regarding sex-related differences following intranasal administration of either drug.
TABLE 4.
Geometric mean pharmacokinetics parameters (%CV) of naltrexone and naloxone in female and male participants
| Variable (U)a | Naloxone (Treatment B) |
Naloxone (Treatment C) |
Naltexone (Treatment A) |
Naltrexone (Treatment C) |
||||
|---|---|---|---|---|---|---|---|---|
| Female | Male | Female | Male | Female | Male | Female | Male | |
| N | 5 | 6 | 5 | 6 | 6 | 6 | 5 | 6 |
| Cmax (ng/ml) | 4.75 (62.4) | 3.96 (36.6) | 3.41 (42.6) | 3.77 (34.2) | 5.45 (50.8) | 3.80 (109) | 1.71 (43.2) | 1.71 (31.6) |
| Tmax (h) | 0.75 (0.25–0.75) | 0.42 (0.27–0.75) | 0.50 (0.33–0.50) | 0.50 (0.25–0.75) | 0.42 (0.17–1.0) | 0.33 (0.25–0.75) | 1.0 (0.50–2.0) | 0.75 (0.25–1.0) |
| AUC0–inf (ng·h/ml) | 8.90 (46.3) | 7.55 (32.7) | 7.33 (47.9) | 6.74 (17.9) | 11.9 (19.2) | 7.75 (42.6) | 7.07 (29.7)b | 5.20 (12.3) |
| λz (h−1) | 0.412 (30.0) | 0.355 (34.9) | 0.407 (32.6) | 0.317 (51.8) | 0.309 (24.0) | 0.329 (11.6) | 0.314 (14.0)b | 0.327 (8.08) |
| t1/2 (h) | 1.68 (30.0) | 1.95 (34.9) | 1.70 (32.6) | 2.19 (51.8) | 2.25 (24.0) | 2.10 (11.6) | 2.21 (14.0)b | 2.12 (8.08) |
| CL/F (l/min) | 6.13 (46.3) | 7.23 (32.7) | 7.44 (47.9) | 8.10 (17.9) | 2.52 (19.2) | 3.87 (42.6) | 4.25 (29.7)b | 5.76 (12.3) |
AUC0–inf, area under the plasma concentration-time curve from time zero to infinity; CL/F, apparent clearance; t1/2, terminal phase half-life; λz, terminal phase rate constant; Tmax, time to reach Cmax; Treatment A, 2 mg naltrexone, intranasally; Treatment B, 4 mg naloxone, intranasally; Treatment C, 2 mg naltrexone plus 4 mg naloxone, intranasally.
Geometric mean (%CV) for all except median (range) for Tmax.
N = 4.
Safety.
Five subjects experienced at least one adverse event of any grade or attribution that was judged to be related to the test drugs; all were mild in severity. Headache was the single most frequent adverse event (three events in each of three subjects). There was one drug-related incident of mild inflamed mucosa (score of 1) that occurred 24 hours after dosing with 2 mg naltrexone. Vital signs, ECG, and clinical laboratory parameters did not reveal any clinically significant changes after any of the doses.
In Vitro Transporters
The ratios of naloxone and naltrexone uptake by the five transporter-expressing cell lines compared with the control cells were all less than 2, indicating that neither compound was a substrate for OCT1, OCT2, OCT3, OCTN1, or OCTN2 (Table 5). Positive controls had ratios of uptake that ranged from 6.4 for OCT3 to 75.4 for OCTN2 and demonstrated inhibition of uptake by their respective inhibitor.
TABLE 5.
Uptake of naloxone and naltrexone into transport-expressing and control cells
| Transporter |
Compound |
Concentration |
Inhibitor |
Incubation Time |
Cleared Volumea |
Test/Control Ratio |
|
|---|---|---|---|---|---|---|---|
| Control Cells |
Plus Transporter |
||||||
| µM | min | µl/mg protein | µl/mg protein | ||||
| OCT1 | Naloxone | 1 | — | 10 | 31.4 ± 0.4 | 28.7 ± 1.8 | 0.9 |
| Naltrexone | 1 | — | 10 | 65.7 ± 3.8 | 70.3 ± 6.6 | 1.1 | |
| [14C]Metformin | 10 | — | 5 | 0.94 ± 0.113 | 13.1 ± 0.2 | 13.9 | |
| [14C]Metformin | 10 | 100 µM quinidine | 5 | 0.158 ± 0.023 | 0.609 ± 0.001 | 3.9 | |
| OCT2 | Naloxone | 1 | — | 10 | 37.1 ± 4.7 | 37.3 ± 6.2 | 1.0 |
| Naltrexone | 1 | — | 10 | 78.0 ± 11.3 | 71.0 ± 6.4 | 0.9 | |
| [14C]Metformin | 10 | — | 2 | 0.609 ± 0.080 | 46.4 ± 0.2 | 76.2 | |
| [14C]Metformin | 10 | 300 µM quinidine | 2 | 0.240 ± 0.031 | 0.652 ± 0.225 | 2.7 | |
| OCT3 | Naloxone | 1 | — | 20 | 30.6 ± 4.2 | 35.9 ± 2.5 | 1.2 |
| Naltrexone | 1 | — | 20 | 88.9 ± 7.5 | 80.2 ± 5.9 | 0.9 | |
| [14C]TEA | 5 | — | 20 | 3.29 ± 0.31 | 20.9 ± 0.6 | 6.4 | |
| [14C]TEA | 5 | 100 µM verapamil | 20 | 0.946 ± 0.143 | 1.22 ± 0.32 | 1.3 | |
| OCTN1 | Naloxone | 1 | — | 10 | 197 ± 4 | 134 ± 25 | 0.7 |
| Naltrexone | 1 | — | 10 | 179 ± 24 | 190 ± 56 | 1.1 | |
| [14C]TEA | 5 | — | 5 | 0.998 ± 0.259 | 12.1 ± 1.4 | 12.1 | |
| [14C]TEA | 5 | 100 µM verapamil | 5 | 0.579 ± 0.148 | 0.877 ± 0.228 | 1.5 | |
| OCTN2 | Naloxone | 1 | — | 10 | 179 ± 16 | 161 ± 6 | 0.9 |
| Naltrexone | 1 | — | 10 | 209 ± 19 | 209 ± 46 | 1.0 | |
| [3H]Carnitine | 0.03 | — | 2 | 1.32 ± 0.04 | 99.5 ± 3.8 | 75.4 | |
| [3H]Carnitine | 0.03 | 30 µM verapamil | 2 | 0.953 ± 0.109 | 31.4 ± 2.2 | 32.9 | |
TEA, triethylamine.
N = 3, Mean ± S.D.
—, inhibitor was not added to the incubation.
Permeability of naloxone and naltrexone across a polarized cell layer was tested using control MDCKII cells. Concentrations on the donor side were either 10 or 500 µM. Transporter studies normally use buffers that are pH 7.4 on both the apical and basolateral sides. Since the pH of the nasal passage is approximately pH 5.5–6.5, the studies were conducted also with the apical buffer at pH 5.5, while the basolateral buffer remained at pH 7.4.
When the pH of the buffer was pH 7.4 on both sides, the efflux ratios of both naloxone and naltrexone were less than 2 at concentrations of 10 and 500 µM (Tables 6 and 7). The addition of 50-fold higher concentration of naloxone to both the lower and higher naltrexone donor solutions did not reduce the efflux ratio to an appreciable amount. Similar results were observed when the higher concentration of naltrexone was added to the naloxone solutions.
TABLE 6.
Bidirectional permeability of naltrexone across MDCKII cells
The Papp values are mean (%CV), n = 3. 3H-Mannitol Papp = 0.49–1.37 × 10−6 cm/s; 14C-caffeine Papp = 31.2–34.3 × 10−6 cm/s. The efflux ratio represents the Papp of the basolateral-to-apical direction divided by the Papp of the apical-to-basolateral direction.
| Concentration |
Papp (×10−6 cm/s) |
Efflux Ratio | |
|---|---|---|---|
| A to B | B to A | ||
| Apical and basolateral (pH = 7.4) | |||
| Naltrexone (10 μM) | 24.3 (9.9) | 36.1 (6.9) | 1.49 |
| Naltrexone (10 μM)/naloxone (500 μM) | 27.9 (5.0) | 33.3 (5.7) | 1.19 |
| Naltrexone (500 μM) | 36.2 (8.2) | 39.1 (2.8) | 1.08 |
| Naltrexone (500 μM)/naloxone (500 μM) | 26.7 (7.1) | 41.4 (6.0) | 1.55 |
| Apical (pH = 5.5)/basolateral (pH = 7.4) | |||
| Naltrexone (10 μM) | 5.13 (1.6) | 62.3 (6.1) | 12.1 |
| Naltrexone (10 μM)/naloxone (500 μM) | 6.92 (24.1) | 122 (4.1) | 17.6 |
| Naltrexone (500 μM) | 7.48 (7.0) | 136 (2.9) | 18.2 |
| Naltrexone (500 μM)/naloxone (500 μM) | 7.51 (6.7) | 107 (2.8) | 14.2 |
A to B, apical to basolateral; B to A, basolateral to apical.
TABLE 7.
Bidirectional permeability of naloxone across MDCKII cells
The Papp values are mean (%CV), n = 3. 3H-Mannitol Papp = 0.41–1.11 × 10−6 cm/s; 14C-caffeine Papp = 15.5–53.5 × 10−6 cm/s. The efflux ratio represents the Papp of the basolateral-to-apical direction divided by the Papp of the apical-to-basolateral direction.
| Concentration |
Papp (×10−6 cm/s) |
Efflux Ratio | |
|---|---|---|---|
| A to B | B to A | ||
| Apical and basolateral (pH = 7.4) | |||
| Naloxone (10 µM) | 30.6 (5.2) | 31.5 (10.8) | 1.03 |
| Naloxone (10 µM)/naltrexone (500 µM) | 39.6 (10.9) | 23.7 (3.8) | 0.60 |
| Naloxone (500 µM) | 25.3 (9.5) | 48.3 (3.1) | 1.91 |
| Naloxone (500 µM)/naltrexone (500 µM) | 19.7 (9.1) | 37.3 (0.8) | 1.90 |
| Apical (pH = 5.5)/basolateral (pH = 7.4) | |||
| Naloxone (10 µM) | 6.48 (25.6) | 43.3 (4.6) | 6.69 |
| Naloxone (10 µM)/naltrexone (500 µM) | 5.83 (8.7) | 62.5 (1.1) | 10.7 |
| Naloxone (500 µM) | 3.29 (6.7) | 174 (6.3) | 52.9 |
| Naloxone (500 µM)/naltrexone (500 µM) | 5.08 (4.9) | 134 (6.0) | 26.3 |
A to B, apical to basolateral; B to A, basolateral to apical.
Lowering the pH of the apical buffer to pH 5.5, while maintaining the basolateral at pH 7.4, caused a 3- to 5-fold decrease in Papp values of naltrexone in the A-to-B ratio and 2- to 4-fold increase in the efflux direction (Table 6). The efflux ratios increased to between 12.1 and 18.2. Similar results were observed using naloxone (Table 7). The Papp values changed considerably regardless of whether naloxone and naltrexone were tested separately or in combination.
The mean transepithelial electrical resistance values were above 100 Ω × cm2 both pre- and postdose in all of the MDCKII studies. The Papp values of [3H]mannitol were in the range of 0.41–1.37 × 10–6 cm/s in the A-to-B and B-to-A directions, while they ranged between 15.5 and 53.5 × 10–6 cm/s for [14C]caffeine in all of the incubations.
Discussion
Delivery of naloxone by the nasal route has been recognized by the medical community and public officials as an effective way to reverse opioid overdoses (Ryan and Dunne, 2018). However, the short half-life of naloxone and the increased incidence of overdoses linked to synthetic opioids with a longer duration of action may require more than one dose to be administered to prevent renarcotization (Klebacher et al., 2017). Therefore, the addition of a longer-acting opioid antagonist to the naloxone formulation was initially hypothesized as a means to increase the time to obtain proper medical attention.
The large decrease in the nasal absorption of naltrexone in the presence of naloxone was unexpected. It was observed as early as 5 minutes following administration of both opioid antagonists but was far more pronounced for naltrexone. The Cmax value of naltrexone was reduced to 1.7 ng/ml when the drugs were combined, i.e., less than 2 ng/ml, which is generally regarded as a concentration that is sufficient to adequately block the effects of opioid agonists (Comer et al., 2006). The combination product of naloxone and naltrexone was not pursued further.
Although the concentration of naltrexone was below the target of 2 ng/ml, direct absorption into the brain via the olfactory nerves that protrude through the cribiform plate in the olfactory epithelium may lead to a higher concentration at the site of action (Illum, 2000). The cerebral spinal fluid/plasma ratio of zidovudine was higher after intranasal administration compared with an intravenous infusion at 15 minutes postdose in rats (Seki et al., 1994). Similar results were observed in the rat using cephalexin (Sakane et al., 1994). However, the olfactory epithelium accounts for only 3%–5% of the nasal cavity’s total surface area, which may limit its role in the efficient transfer of drugs directly to the central nervous system (Grassin-Delyle et al., 2012).
Rabiner et al. (2011) hypothesized that 6β-naltrexol contributed to the long occupancy of the μ-opioid receptor due to its long plasma half-life. Due to this possibility, the Food and Drug Administration draft guidance on new formulations of naltrexone hydrochloride requires the analysis of 6β-naltrexol (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM194641.pdf). This metabolite, while approximately 2-fold less potent than the parent compound on the μ-receptor, is 100-fold less potent than naltrexone in vivo in nonhuman primates (Ko et al., 2006) and has no effect on either morphine-induced analgesia or pupil constriction in humans (Yancey-Wrona et al., 2011). Thus, 6β-naltrexol is peripherally restricted; therefore, its involvement in the central nervous system effects of naltrexone is minimal.
While naloxone could have been administered using the Food and Drug Administration–approved device with naltrexone delivered using a mucosal atomization device, the decision was made to use cGMP-grade material for both compounds and the mucosal atomization device for delivering all three formulations. Administering both using the mucosal atomization device eliminated any time lag that would occur if naloxone and naltrexone were administered in sequence. It also eliminated variability due to the use of two different delivery devices for naloxone for treatments B and C and kept the administered volume at 0.1 ml for all three phases of the study.
In this pilot study, sterile water was used as the vehicle. The marketed intranasal formulation of naloxone contains benzalkonium chloride, sodium EDTA, NaCl, and HCl for pH control (https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=208411). The results from the pivotal clinical study (Krieter et al., 2016) and a pilot study using sterile water were very similar (results not shown). Since the results using water versus saline with a preservative and stabilizer were similar, a simpler vehicle was used in the present study. In addition, Vanky et al. (2017) used water for injection with NaCl as their vehicle for naloxone and produced very similar values as those reported in Krieter et al. (2016). The formulation for the marketed naloxone autoinjector consists of saline and HCl for pH adjustment (https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=209862).
The PK interaction of the two compounds, especially the dramatic effect on naltrexone, remains unexplained. Both compounds have pKa values of approximately 8.0 (Wermeling, 2013) and would be predominantly ionized at pH 5.5–6.5, the pH of the nasal epithelium (England et al., 1999). If absorbed passively, there should not be any significant interaction. The early and large effects on naltrexone suggest that they share a transport mechanism.
Human nasal epithelial cells have appreciable levels of OCT3, OCTN1, and OCTN2 with a minor amount of OCT1 (Shao et al., 2013). They also express P-glycoprotein, several members of the multidrug resistance-associated family, and organic anion and peptide transporters (Al-Ghabeish et al., 2015). Neither naloxone nor naltrexone is a substrate for P-glycoprotein (Mahar Doan et al., 2002; Kanaan et al., 2009). Naloxone inhibited the transport of the OATP-A substrate deltorphin II into cRNA-injected oocytes, but it was not tested itself using the system (Gao et al., 2000). None of the five cation transporters tested in the present study demonstrated activity toward either naloxone or naltrexone.
The concentrations of naloxone and naltrexone tested in the OCT and OCTN assays were in the low micromolar range, such that the transporters were not saturated. However, the concentrations of both compounds in the nasal formulation (53 mM naltrexone and 110 mM naloxone) were much higher to keep the administered volume at 0.1–0.15 ml (Grassin-Delyle et al., 2012). Further in vitro studies were conducted using MDCKII cells to determine potential interactions between the two compounds. The wild-type cell line contains canine MDR1, MRP2, MRP4, and OCTN2 (Gartzke and Fricker, 2014). When both the apical and basolateral solutions were at pH 7.4, there was no indication that either compound interacted with a transporter or with each other. Their apparent permeabilities were greater than 20 × 10−6 cm/s, the same range as caffeine. The efflux ratio was less than 2 when their concentrations were 10 and 500 µM. The addition of 500 µM naloxone did not appreciably affect the permeability of naltrexone at either concentration; similar results were demonstrated when naloxone was tested. The permeability constant was similar between naltrexone and naloxone in both the A-to-B and the B-to-A directions.
In additional experiments, the pH of the apical buffer was lowered to 5.5, while the basolateral buffer was maintained at pH 7.4 to mimic the difference in the pH between the two sides of the nasal epithelium (England et al., 1999). In contrast to the initial experiments, the permeability constant of naltrexone in the A-to-B direction decreased 3- to 5-fold, while it increased 2- to 4-fold in the opposite direction; the efflux ratio was above 12 in all four conditions tested. The Papp values did not change appreciably when 500 µM naloxone was added to the apical buffer, suggesting that naltrexone and naloxone were not interacting either with a transporter or each other. Similar changes were measured when naloxone was tested. While there was some variability, the Papp values were similar between the two compounds. The increased efflux ratio may reflect a trapping of the ionized form of naltrexone and naloxone, both weak bases, in the acidic milieu of the apical compartment. This trapping phenomenon has been previously described with other drugs in an acidic environment (Kazmi et al., 2013). Horvath et al. (2007) showed that the permeability of the substrate for OCTN1 and OCTN2, cationic fluorophore 4-[4-(dimethylamino)-styryl]-N-methylpyridinium, decreased when the pH of the apical buffer decreased from pH 7.4 to 5.7. However, these investigators did not measure the Papp value in the efflux direction.
In a study subsequent to the present one, the permeability enhancer dodecyl maltopyranoside (Maggio and Pillion, 2013) was added to the solution used to dose 4 mg naltrexone by intranasal administration (Krieter et al., 2019). Compared with the control solution, the addition of dodecyl maltopyranoside resulted in an almost 3-fold increase in the Cmax value of naltrexone, a 54% increase in the AUC0–inf, and a decrease in the median time to reach Cmax from 30 to 10 minutes. When examined using MDCKII monolayer cultures, the increased exposure corresponded to a decrease in the transepithelial electrical resistance of approximately 50%. Future studies may explore further the mechanism and clinical relevance of this interaction for other opioid antagonists.
The underlying explanation for the interaction between naltrexone and naloxone when they are administered together intranasally is unclear. While drug-drug interactions have been extensively studied following oral and intravenous dosing (Giacomini et al., 2010), few studies have focused on nasal administration. This route of administration has been advocated for an increasing number of drugs (Costantino et al., 2007; Grassin-Delyle et al., 2012). The importance of understanding how opioid antagonists like naloxone and naltrexone are absorbed by the nasal epithelium is magnified by the dramatic rise in overdose deaths attributed to high potency, long-lived synthetic opioids (e.g., fentanyl), and the realization that better strategies are needed to treat opioid overdose.
Abbreviations
- AUC0–inf
area under the concentration-time curve from time zero to infinity
- D-PBS
PBS containing 0.2% bovine serum albumin
- HEK293
human embryonic kidney 293
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MDCKII
Madin-Darby canine kidney II
- m/z
mass-to-charge ratio
- naloxone
17-allyl-4,5α-epoxy-3,14-dihydroxymorphinan-6-one HCl
- naltrexone
17-(cyclopropylmethyl)-4,5α-epoxy-3,14-dihydroxymorphinan-6-one
- OCT
organic cation transporter
- Papp
apparent permeability
- PK
pharmacokinetic
Authorship Contributions
Participated in research design: Krieter, Chiang, Gyaw, Skolnick, Snyder.
Performed data analysis: Snyder.
Wrote or contributed to the writing of the manuscript: Krieter, Skolnick, Gyaw, Snyder.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Contracts N01DA-12-8905, N01DA-13-8920, and N01DA-14-8914].
P.K. and S.G. are employees of the National Institutes of Health (NIH). C.N.C. is a former NIH employee. P.S. is an employee of Opiant Pharmaceuticals; he was employed by the NIH when this study was designed and executed. R.S. is an employee of Sekisui XenoTech LLC.
References
- Al-Ghabeish M, Scheetz T, Assem M, Donovan MD. (2015) Microarray determination of the expression of drug transporters in humans and animal species used for the investigation of nasal absorption. Mol Pharm 12:2742–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DC, Montgomery J, and Gertz B (2014) inventors, 3B Pharmaceuticals, Inc., assignee. Intranasal naltrexone. U.S. patent 2014/0249172 A1. 2014 Sep 4.
- Carpenter JE, Wong K, Ndje P, Ezeonyebuchi UI. (2016) Opioid overdose and naloxone delivery devices in the community setting. Am J Pharm Benefits 8:e96–e100. [Google Scholar]
- Cassel JA, Daubert JD, DeHaven RN. (2005) [3H]Alvimopan binding to the μ opioid receptor: comparative binding kinetics of opioid antagonists. Eur J Pharmacol 520:29–36. [DOI] [PubMed] [Google Scholar]
- Comer SD, Sullivan MA, Yu E, Rothenberg JL, Kleber HD, Kampman K, Dackis C, O’Brien CP, Chiang CN, Hawks RL. (2006) Injectable, sustained-release naltrexone for the treatment of opioid dependence: a randomized, placebo-controlled trial. Arch Gen Psychiatry 63:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino HR, Illum L, Brandt G, Johnson PH, Quay SC. (2007) Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm 337:1–24. [DOI] [PubMed] [Google Scholar]
- Edwards ET, Edwards ES, Davis E, Mulcare M, Wiklund M, Kelley G. (2015) Comparative usability study of a novel auto-injector and an intranasal system for naloxone delivery. Pain Ther 4:89–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England RJ, Homer JJ, Knight LC, Ell SR. (1999) Nasal pH measurement: a reliable and repeatable parameter. Clin Otolaryngol Allied Sci 24:67–68. [DOI] [PubMed] [Google Scholar]
- Gao B, Hagenbuch B, Kullak-Ublick GA, Benke D, Aguzzi A, Meier PJ. (2000) Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J Pharmacol Exp Ther 294:73–79. [PubMed] [Google Scholar]
- Gartzke D, Fricker G. (2014) Establishment of optimized MDCK cell lines for reliable efflux transport studies. J Pharm Sci 103:1298–1304. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassin-Delyle S, Buenestado A, Naline E, Faisy C, Blouquit-Laye S, Couderc LJ, Le Guen M, Fischler M, Devillier P. (2012) Intranasal drug delivery: an efficient and non-invasive route for systemic administration: focus on opioids. Pharmacol Ther 134:366–379. [DOI] [PubMed] [Google Scholar]
- Horvath G, Schmid N, Fragoso MA, Schmid A, Conner GE, Salathe M, Wanner A. (2007) Epithelial organic cation transporters ensure pH-dependent drug absorption in the airway. Am J Respir Cell Mol Biol 36:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illum L. (2000) Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci 11:1–18. [DOI] [PubMed] [Google Scholar]
- Kanaan M, Daali Y, Dayer P, Desmeules J. (2009) P-glycoprotein is not involved in the differential oral potency of naloxone and naltrexone. Fundam Clin Pharmacol 23:543–548. [DOI] [PubMed] [Google Scholar]
- Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, Parkinson A. (2013) Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab Dispos 41:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Wagner HN, Jr, Villemagne VL, Kao PF, Dannals RF, Ravert HT, Joh T, Dixon RB, Civelek AC. (1997) Longer occupancy of opioid receptors by nalmefene compared to naloxone as measured in vivo by a dual-detector system. J Nucl Med 38:1726–1731. [PubMed] [Google Scholar]
- Klebacher R, Harris MI, Ariyaprakai N, Tagore A, Robbins V, Dudley LS, Bauter R, Koneru S, Hill RD, Wasserman E, et al. (2017) Incidence of naloxone redosing in the age of the new opioid epidemic. Prehosp Emerg Care 21:682–687. [DOI] [PubMed] [Google Scholar]
- Ko MC, Divin MF, Lee H, Woods JH, Traynor JR. (2006) Differential in vivo potencies of naltrexone and 6β-naltrexol in the monkey. J Pharmacol Exp Ther 316:772–779. [DOI] [PubMed] [Google Scholar]
- Krieter P, Chiang N, Gyaw S, Skolnick P, Crystal R, Keegan F, Aker J, Beck M, Harris J. (2016) Pharmacokinetic properties and human use characteristics of an FDA-approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol 56:1243–1253. [DOI] [PubMed] [Google Scholar]
- Krieter P, Gyaw S, Chiang CN, Crystal R, Skolnick P. (2019) Enhanced intranasal absorption of naltrexone by dodecyl maltopyranoside: implications for the treatment of opioid overdose. J Clin Pharmacol DOI: 10.1002/jcph.1384 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Wagner HN, Jr, Tanada S, Frost JJ, Bice AN, Dannals RF. (1988) Duration of occupancy of opiate receptors by naltrexone. J Nucl Med 29:1207–1211. [PubMed] [Google Scholar]
- Li K, Armenian P, Mason J, Grock A. (2018) Narcan or Nar-can’t: tips and tricks to safely reversing opioid toxicity. Ann Emerg Med 72:9–11. [DOI] [PubMed] [Google Scholar]
- Maggio ET, Pillion DJ. (2013) High efficiency intranasal drug delivery using Intravail® alkylsaccharide absorption enhancers. Drug Deliv Transl Res 3:16–25. [DOI] [PubMed] [Google Scholar]
- Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. (2002) Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther 303:1029–1037. [DOI] [PubMed] [Google Scholar]
- Meyer MC, Straughn AB, Lo MW, Schary WL, Whitney CC. (1984) Bioequivalence, dose-proportionality, and pharmacokinetics of naltrexone after oral administration. J Clin Psychiatry 45:15–19. [PubMed] [Google Scholar]
- Rabiner EA, Beaver J, Makwana A, Searle G, Long C, Nathan PJ, Newbould RD, Howard J, Miller SR, Bush MA, et al. (2011) Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward-related brain activation in humans. Mol Psychiatry 16:826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SA, Dunne RB. (2018) Pharmacokinetic properties of intranasal and injectable formulations of naloxone for community use: a systematic review. Pain Manag 8:231–245. [DOI] [PubMed] [Google Scholar]
- Sakane T, Akizuki M, Yamashita S, Sezaki H, Nadai T. (1994) Direct drug transport from the rat nasal cavity to the cerebrospinal fluid: the relation to the dissociation of the drug. J Pharm Pharmacol 46:378–379. [DOI] [PubMed] [Google Scholar]
- Seki T, Sato N, Hasegawa T, Kawaguchi T, Juni K. (1994) Nasal absorption of zidovudine and its transport to cerebrospinal fluid in rats. Biol Pharm Bull 17:1135–1137. [DOI] [PubMed] [Google Scholar]
- Setnik B, Bramson C, Bass A, Levy-Cooperman N, Malhotra B, Matschke K, Sommerville KW, Wolfram G, Geoffroy P. (2015) Intranasal administration of crushed ALO-02 (extended-release oxycodone with sequestered naltrexone): a randomized, controlled abuse-potential study in nondependent recreational opioid users. J Clin Pharmacol 55:1351–1361. [DOI] [PubMed] [Google Scholar]
- Shao D, Massoud E, Anand U, Parikh A, Cowley E, Clarke D, Agu RU. (2013) Organic cation transporters in human nasal primary culture: expression and functional activity. Ther Deliv 4:439–451. [DOI] [PubMed] [Google Scholar]
- Vanky E, Hellmundt L, Bondesson U, Eksborg S, Lundeberg S. (2017) Pharmacokinetics after a single dose of naloxone administered as a nasal spray in healthy volunteers. Acta Anaesthesiol Scand 61:636–640. [DOI] [PubMed] [Google Scholar]
- Wermeling DP. (2013) A response to the opioid overdose epidemic: naloxone nasal spray. Drug Deliv Transl Res 3:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey-Wrona J, Dallaire B, Bilsky E, Bath B, Burkart J, Webster L, Magiera D, Yang X, Phelps, Sadee W. (2011) 6β-Naltrexol, a peripherally selective opioid antagonist that inhibits morphine-induced slowing of gastrointestinal transit: an exploratory study. Pain Med 12:1727–1737. [DOI] [PubMed] [Google Scholar]
- Yuen KH, Peh KK, Billa N. (1999) Comparative bioavailability study of a generic naltrexone tablet preparation. Drug Dev Ind Pharm 25:353–356. [DOI] [PubMed] [Google Scholar]