

Abstract
Regulation of replication and expression of mitochondrial DNA (mtDNA) is essential for cellular energy conversion via oxidative phosphorylation. The mitochondrial transcription elongation factor (TEFM) has been proposed to regulate the switch between transcription termination for replication primer formation and processive, near genome‐length transcription for mtDNA gene expression. Here, we report that Tefm is essential for mouse embryogenesis and that levels of promoter‐distal mitochondrial transcripts are drastically reduced in conditional Tefm‐knockout hearts. In contrast, the promoter‐proximal transcripts are much increased in Tefm knockout mice, but they mostly terminate before the region where the switch from transcription to replication occurs, and consequently, de novo mtDNA replication is profoundly reduced. Unexpectedly, deep sequencing of RNA from Tefm knockouts revealed accumulation of unprocessed transcripts in addition to defective transcription elongation. Furthermore, a proximity‐labeling (BioID) assay showed that TEFM interacts with multiple RNA processing factors. Our data demonstrate that TEFM acts as a general transcription elongation factor, necessary for both gene transcription and replication primer formation, and loss of TEFM affects RNA processing in mammalian mitochondria.
Keywords: mtDNA replication, RNA processing, transcription elongation
Subject Categories: RNA Biology, Transcription
Introduction
Mitochondria harbor the oxidative phosphorylation (OXPHOS) system, which performs cellular energy conversion resulting in the production of adenosine triphosphate (ATP) 1, 2. Deficient OXPHOS is a well‐established primary cause of mitochondrial disease and is also secondarily implicated in a variety of pathophysiological conditions, such as neurological disease, age‐related diseases, and aging 3. The regulation of OXPHOS is complex because subunits of the respiratory chain and the ATP synthase are encoded by both the nuclear genome and mtDNA 4, 5.
Mammalian mtDNA is a gene‐dense, circular double‐stranded DNA molecule, and the two strands are defined as the heavy (H) and the light (L) strand according to their base composition 6. The mtDNA only encodes 13 of the ∼90 proteins present in the OXPHOS system, but all of these are essential subunits 4, 5. In addition, mtDNA encodes 2 ribosomal RNAs (mt‐rRNAs) and 22 transfer RNAs (mt‐tRNAs), which are necessary for translation of mitochondrial mRNAs (mt‐mRNAs). The compact mtDNA only contains one longer noncoding region named the displacement loop (D‐loop), where the origin of replication of the H strand (OH) and the promoters for transcription of H and L strands (HSP and LSP) are located 7, 8.
Transcription of mtDNA initiates from HSP and LSP and generates two, near genomic‐length polycistronic precursor RNAs, which sequentially go through processing to yield individual mitochondrial RNAs (mtRNAs) 5. Transcription initiation has been extensively studied using in vitro and in vivo models, showing that the mitochondrial RNA polymerase (POLRMT) and mitochondrial transcription factors A (TFAM) and B2 (TFB2M) are core components in this process 9, 10, 11. In brief, TFAM unwinds the promoter region of double‐stranded DNA (dsDNA) and introduces a transcription bubble covering the initiation site. This loose structure is essential for the recruitment of POLRMT and enables TFB2M binding, which completes the assembly of the initiation complex 12, 13. However, once POLRMT successfully transcribes the promoter region, TFB2M is released from the initiation complex 14, 15. Thus, TFB2M is necessary for transcription initiation, but not for elongation. Recent studies have also identified and characterized TEFM as another component of the mitochondrial transcription machinery that interacts with POLRMT and promotes transcription processivity to enable near genome‐length transcription 16, 17. The structures of the transcription initiation 18 and elongation complexes 19 have recently been determined. Binding of TEFM to POLRMT allows the complex to form a “sliding clamp” around the DNA, which facilitates high processivity of transcription 19. TEFM also enhances POLRMT transcription by reducing the duration and frequencies of long‐lived transcription pauses 20.
Transcription initiated from LSP also supplies primers for the replication of mtDNA. Using a recombinant in vitro transcription system, previous studies have suggested that a large proportion of transcription events initiated at LSP are prematurely terminated at the conserved sequence block II (CSBII) within the D‐loop region 21. This termination occurs because of the formation of a G‐quadruplex structure between nascent RNA and the non‐template strand of mtDNA and has been proposed to be linked to primer formation for initiation of H‐strand DNA replication 22, 23. The newly transcribed RNA remains associated with the CSB region, where it forms an R‐loop that is resistant to treatment by RNase A and RNase T1 24, 25, 26. Interestingly, TEFM has been reported to regulate the generation of replication primers in human mitochondria 27 by helping POLRMT to bypass the highly structured CSBII region 19, 20 and abolish R‐loop formation 17. Results from an in vitro study have been interpreted to support a model where TEFM serves as a molecular switch that coordinates the balance between mtDNA transcription for replication primer formation and gene expression 27, whereas another in vitro study argues that TEFM is a general unspecific transcription elongator needed for mtDNA gene expression 17.
Processing of the newly synthesized polycistronic precursor RNAs to release mature mtRNAs is thought to occur co‐transcriptionally and is mainly performed in distinct foci, named mitochondrial RNA granules 28, 29, 30. The RNA granules provide an organizational platform for spatiotemporal regulation of mitochondrial RNA processing and maturation 31. The majority of mt‐rRNAs and mt‐mRNAs are flanked by mt‐tRNAs and processing starts with the excision of these flanking mt‐tRNAs, according to the widely accepted tRNA punctuation model 32. The endonucleolytic cleavage of the 5′‐ and 3′‐ends of the tRNAs is performed by the mitochondrial RNase P complex, which consists of MRPP1, MRPP2, and MRPP3 33, 34 and the mitochondrial RNase Z (ELAC2) 35, 36, respectively, situated in close proximity to the RNA granules 29, 37. The G‐rich RNA sequence binding factor 1 (GRSF1) can melt dsRNA 38 and is localized in RNA granules where it interacts with RNase P 29, 30. Other processing factors are also present in the RNA granules, such as the Fas‐activated serine/threonine kinase (FASTK) protein family members FASTK, FASTK2 and FASTKD5 39, 40, the mitochondrial poly(A)‐polymerase (mtPAP) 41, methyltransferases 42, RNA helicases, and the degradosome (SUPV3L1‐PNPase) complex 43.
In this study, we have established the in vivo function of TEFM by generating and characterizing conditional Tefm‐knockout mice. We report that TEFM is essential for embryonic survival and that loss of TEFM in the mouse heart causes mitochondrial cardiomyopathy with severe OXPHOS deficiency. Depletion of TEFM drastically reduces the levels of promoter‐distal transcripts encoded by both mtDNA strands. In contrast, there is a marked increase in the steady‐state levels of promoter‐proximal transcripts at both LSP and HSP in the absence of TEFM. At LSP, these short transcripts are prematurely terminated and shorter than the RNA primers needed for initiation of mtDNA replication. Consistently, de novo mtDNA replication is drastically decreased in isolated mitochondria lacking TEFM. Unexpectedly, RNA sequencing (RNA‐Seq) and northern blot analyses show that there is an increase of unprocessed mitochondrial transcripts in the absence of TEFM. TEFM proximity‐labeling (BioID) assays show that TEFM interacts with diverse RNA processing factors, including the RNase P complex (MRPP1‐3), ELAC2, GRSF1, and SUPV3L1‐PNPase, which may explain why transcription elongation affects RNA processing. Our results thus show that TEFM is necessary for transcription of mtDNA to generate both short replication primers and near genome‐length transcripts for gene expression. Furthermore, we show that loss of TEFM affects both transcription elongation and RNA processing in vivo.
Results
TEFM is essential for embryonic survival
To determine the in vivo function of TEFM, we generated a conditional knockout allele by flanking exon 2 of Tefm with loxP sites (Fig 1A). The targeted allele was transmitted through the germline, and heterozygous mice (Tefm +/loxP‐puro) were mated to mice expressing flp‐recombinase to excise the puromycin (puro) cassette to obtain mice that were heterozygous for the loxP‐flanked Tefm allele (Tefm +/loxP). Heterozygous knockout mice (Tefm +/−) were obtained by breeding Tefm +/loxP mice to mice ubiquitously expressing cre‐recombinase (+/β‐actin‐cre) (Fig 1A). Intercrossing of Tefm +/− mice produced no viable homozygous knockout (Tefm −/−) mice (analyzed offspring, n = 94; Tefm +/+, n = 34, Tefm +/−, n = 60 and Tefm −/−, n = 0), demonstrating that loss of Tefm results in embryonic lethality. Next, we performed an intercross of Tefm +/− mice and analyzed staged embryos at embryonic day 8.5 (E8.5; analyzed embryos, n = 43). All embryos with the Tefm −/− genotype (n = 11) were small and lacked heart structure, while embryos with the Tefm +/+ (n = 10) or Tefm +/− (n = 22) genotype were well developed with normal appearance typical of E8.5 embryos (Fig 1B). Thus, loss of TEFM causes severe developmental defects and embryonic lethality at E8.5, consistent with the phenotype of other knockout mice with homozygous disruption of genes critical for mtDNA expression and maintenance 11, 44, 45, 46.
Figure 1. Disruption of Tefm in the germline and heart.
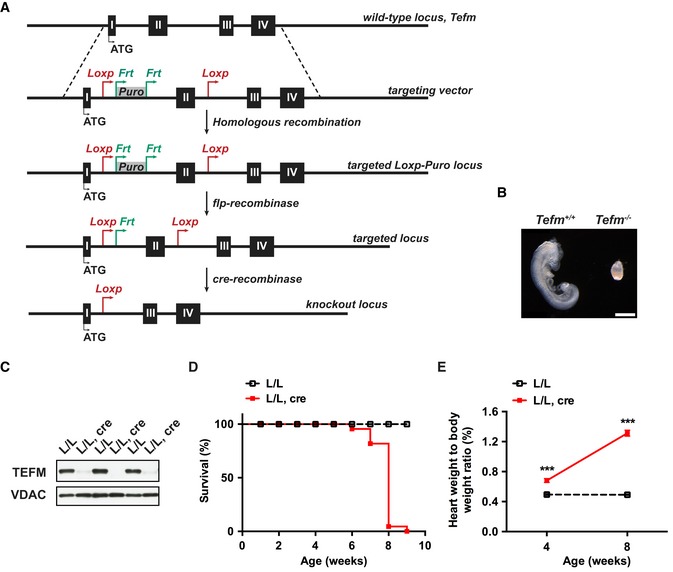
- Targeting strategy for disruption of the Tefm gene. LoxP sites flanking exon 2 of the Tefm gene together with puromycin selection marker (PuroR) were inserted into the mouse genome by homologous recombination. The PuroR cassette was excised by mating with flp‐recombinase mice to obtain heterozygous floxed Tefm mice (Tefm +/loxP). Whole‐body and tissue‐specific Tefm knockout mice were obtained by crossing Tefm +/loxP with different cre‐recombinase mice.
- Morphology of wild‐type (Tefm +/+) and Tefm homozygous knockout (Tefm −/−) embryos at embryonic day 8.5 (analyzed embryos, n = 43). Scale bar, 500 μm.
- Western blot analyses of the TEFM protein level in heart mitochondrial extracts from 8‐week‐old control (L/L) and tissue‐specific Tefm knockout (L/L, cre) mice (n = 12 mice for each group). VDAC was used as loading control.
- Survival curve for control (L/L; n = 60) and Tefm knockout (L/L, cre; n = 22) mice.
- Heart weight to body weight ratio in 4‐ and 8‐week‐old control (L/L) and tissue‐specific Tefm knockout mice (L/L, cre). At 4 weeks: L/L n = 22, L/L, cre n = 15; 8 weeks: L/L n = 41, L/L, cre n = 39.
Disruption of Tefm in heart leads to cardiomyopathy
Next, we disrupted Tefm in heart and skeletal muscle by breeding Tefm +/loxP mice with transgenic mice expressing cre‐recombinase from the muscle creatinine kinase promoter (Ckmm‐cre). The tissue‐specific Tefm knockout mice (Tefm loxP/loxP, +/Ckmm‐cre), hereafter denoted Tefm knockout mice (L/L, cre), were born at expected Mendelian ratios. The depletion of TEFM in heart was verified at the age of 8 weeks by western blot (Fig 1C) and reverse transcriptase quantitative PCR (RT–qPCR) analyses (Fig EV1A). The Tefm knockout mice had a drastically shortened life span with a maximal longevity of 9 weeks (Fig 1D). We also observed a progressively decreased body weight (Fig EV1B) and enlargement of the heart (Fig EV1C–E) in the tissue‐specific Tefm knockout mice, resulting in a significant increase in the heart weight to body weight ratio (Fig 1E). TEFM is thus essential for normal heart function.
Figure EV1. Disruption of Tefm in the germline and heart.
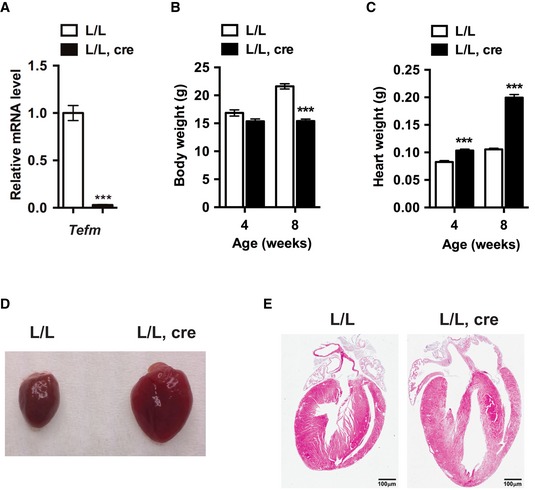
- RT–qPCR analyses of the RNA level of Tefm in 8‐week‐old control (L/L) and Tefm knockout (L/L, cre) mice (n = 10 mice for each group).
- Body weight of 4‐ and 8‐week‐old control and Tefm knockout mice. At 4 weeks: L/L n = 22, L/L, cre n = 15; 8 weeks: L/L n = 41, L/L, cre n = 39.
- Heart weight of 4‐ and 8‐week‐old control and Tefm knockout mice. At 4 weeks: L/L n = 22, L/L, cre n = 15; 8 weeks: L/L n = 41, L/L, cre n = 39.
- Representative images of hearts of 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group).
- Representative images of hematoxylin and eosin staining showing heart structure and morphology in 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group). Scale bar, 100 μm.
Loss of TEFM causes severe mitochondrial dysfunction
Transmission electron microscopy analysis of heart tissue from end‐stage knockout mice showed disrupted mitochondrial alignment and disorganized cristae, consistent with severe respiratory chain dysfunction (Fig 2A). Quantification of mitochondrial density revealed a 1.5‐fold increase of relative mitochondrial mass in Tefm knockout hearts in comparison with controls (Fig EV2A). We also observed an increased ratio of levels of mtDNA relative to nuclear DNA (18S rDNA) as determined by Southern blot (Fig 2B) and quantitative PCR (qPCR) (Fig EV2B) analyses. Further supporting an activation of mitochondrial biogenesis, we also found an increased ratio of VDAC to histone H3 protein levels as determined by western blots (Fig 2B). This increase of mitochondrial biogenesis typically occurs in severely respiratory chain deficient tissues, as exemplified by analysis of tissue‐specific Lrpprc knockout and Mterf4 knockout hearts (Fig 2B) 45, 47. The increase of mitochondrial mass likely represents a compensatory biogenesis response typically seen in human patients and mice with severe mitochondrial dysfunction 48, 49, 50. Next, we measured the activities of the individual complexes in the respiratory chain 51. We found a drastic decrease in the activity for complexes I, III, and IV in the Tefm knockout mice (Fig 2C). A moderate decrease in complex II activity was also observed in the absence of TEFM (Fig 2C), similar to what has been observed in other models with severe reduction of mtDNA expression 52. Consistently, mitochondrial oxygen consumption assays showed decreased OXPHOS capacity after depletion of TEFM (Fig EV2C and D). Additionally, the expression and the assembly of the respiratory chain complexes were negatively affected as determined by western blot analyses (Fig EV2E) and blue native polyacrylamide gel electrophoresis (BN‐PAGE), followed by western blotting (Fig EV2F). Thus, loss of TEFM profoundly impairs OXPHOS, demonstrating that mtDNA transcription elongation is essential for maintaining mitochondrial function.
Figure 2. Knockout of Tefm in heart causes mitochondrial dysfunction.
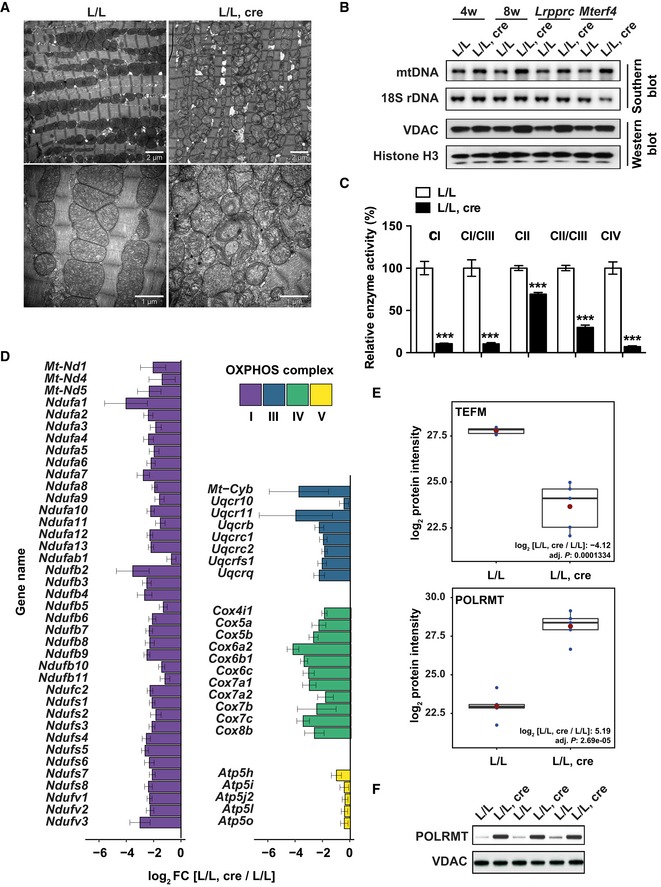
- Transmission electron micrographs of heart tissue in 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group). Scale bar, 2 μm (upper panel) and 1 μm (lower panel).
- Southern blot analyses of mtDNA levels in control and Tefm knockout hearts at the age of 4 and 8 weeks (upper 2 blots; n = 6 mice at each time‐point for each group). 18S rDNA was used as loading control. DNA isolated from tissue‐specific Lrpprc and Mterf4 knockout hearts used as controls. Western blot analyses of VDAC and histone H3 protein levels in control and Tefm knockout hearts at the age of 4 and 8 weeks (bottom 2 blots; n = 6 mice at each time‐point for each group). VDAC represents mitochondria loading, and histone H3 was used as a loading control for nucleus DNA.
- Respiratory chain complex activities were measured spectrophotometrically and normalized to citrate synthase activity in heart mitochondria from 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group). The analyzed enzyme activities are NADH coenzyme Q reductase (complex I), NADH cytochrome c reductase (complex I/III), succinate dehydrogenase (complex II), and cytochrome c oxidase (complex IV).
- Barplot of the significantly changed proteins (Benjamini‐Hochberg adjusted P < 0.05) in Tefm knockout mice compared to control mice (n = 5 mice for each group), organized by individual OXPHOS complexes. Proteins are organized in alphabetical order based on gene name.
- Boxplots of the protein intensity (LFQ) of TEFM (top) and POLRMT (bottom). Red dot: mean; blue dots: intensity value for each biological replicate (n = 5 mice for each group). Horizontal lines: median; box represents the interquartile range (the first quartile (bottom) and third quartile (top) of the intensity value of 5 replicates). The whiskers represent the maximum and minimum values excluding outliers that represent values extend beyond 1.5 times of the interquartile range.
- Western blot analyses of POLRMT protein levels in control and Tefm knockout heart mitochondria at the age of 8 weeks (n = 12 mice for each group). VDAC was used as loading control.
Figure EV2. Depletion of TEFM in heart causes mitochondrial dysfunction.
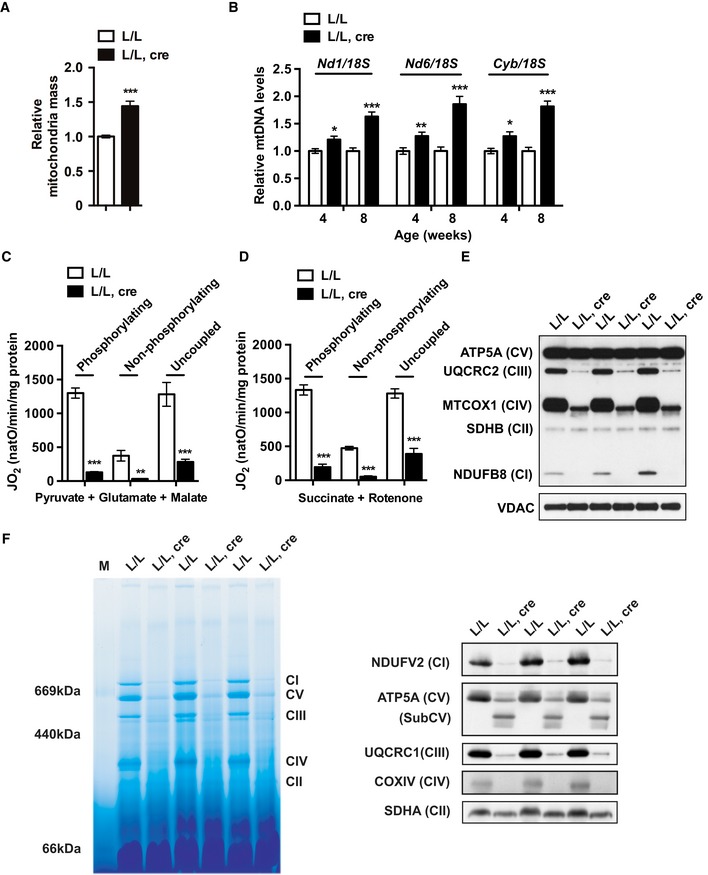
-
AQuantification of the relative mitochondrial mass by electron microscopy analysis of the heart of 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group).
-
BQuantitative PCR analyses of the relative mtDNA levels in 4‐ and 8‐week‐old control and Tefm knockout mice (n = 8 mice at each time‐point for each group).
-
C, DOxygen consumption rates measured using an Oroboros oxygen electrode in heart mitochondria isolated from 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group). Phosphorylating, non‐phosphorylating, and uncoupled respiration under carbonyl cyanide 3‐chlorophenylhydrazone states were measured using pyruvate, glutamate, and malate or succinate and rotenone as substrates.
-
EWestern blot analyses of subunits of OXPHOS at the age of 8 weeks (n = 15 mice for each group). Nucleus‐encoded subunits of complex I (NDUFB8), complex II (SDHB), complex III (UQCRC2), complex IV (MTCOX1), and complex V (ATP5A) were analyzed. VDAC was used as loading control.
-
FAnalysis of the assembly of the OXPHOS complexes by BN‐PAGE in heart mitochondria of 8‐week‐old control Tefm knockout mice (n = 6 mice for each group). OXPHOS complexes were detected by western blot following the BN‐PAGE, including complex I (NDUFV2), complex II (SDHA), complex III (UQCRC1), complex IV (COXIV), and complex V (ATP5A).
Massive changes of the mitochondrial proteome in Tefm knockout hearts
To better understand how Tefm disruption affects mitochondrial function, purified mitochondria from hearts were subjected to label‐free quantitative proteomics. We identified a total of 813 proteins classified as mitochondrial according to MitoCarta 2.0 53. Of these, 62.5% (508/813) were significantly changed and showed either increased (338/508) or decreased (170/508) protein levels in mitochondria lacking TEFM. Manual classification of the differentially expressed proteins revealed that several mitochondrial processes, including oxidative phosphorylation, fatty acid and amino acid metabolic pathways, protein translation (ribosome assembly), mitochondrial structure/morphology, apoptosis, and RNA metabolism, were massively affected by depletion of TEFM (Fig EV3A–C). A closer inspection of the mitochondrial proteome showed a significant decrease of the subunits of OXPHOS complexes I, III, IV, and V (Fig 2D) and increase of respiratory chain complex assembly factors (Fig EV3B). As expected, TEFM protein levels were significantly decreased (Fig 2E). Interestingly, protein levels of nearly all of the small ribosomal subunits (28S) were increased while the majority of the large ribosomal (39S) subunits were decreased (Fig EV3C). These data show that loss of TEFM alters mitochondrial ribosome composition, which may impair mitochondrial protein translation. Furthermore, we observed a remarkable increase of POLRMT protein levels in TEFM‐depleted mitochondria (Fig 2E and F), consistent with our previous results showing that knockout of a variety of genes needed for mtDNA expression leads to increased POLRMT levels 54.
Figure EV3. Proteome of Tefm knockout heart mitochondria.
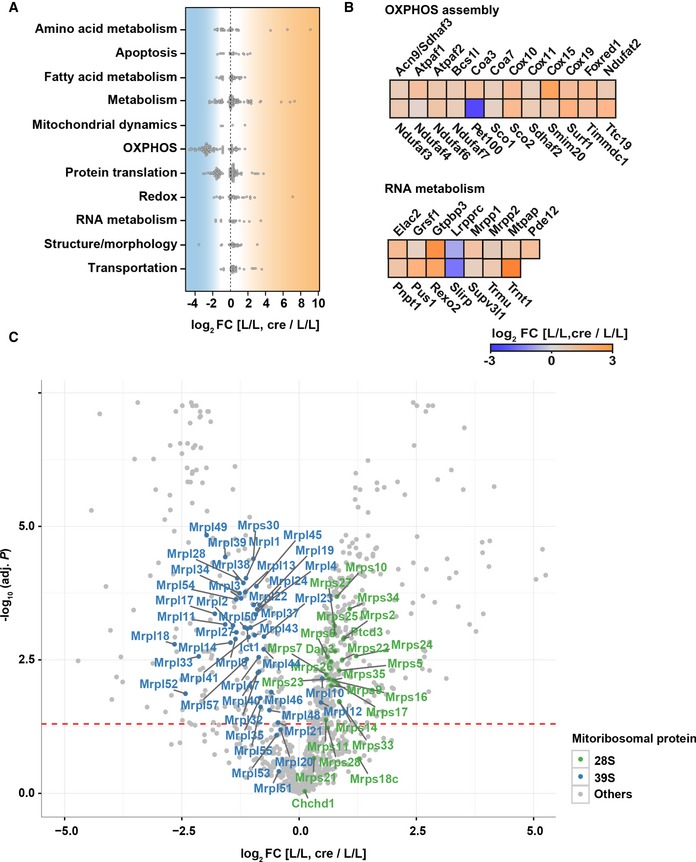
- Beeswarm plot presenting the distribution of differentially expressed proteins (Benjamini–Hochberg adj. P < 0.05) in mitochondrial pathways after knockout of Tefm at 8 weeks of age (n = 5 mice for each group).
- Heatmap showing differentially expressed respiratory chain complexes assembly factors (upper panel) and RNA metabolism‐related proteins (lower panel) in Tefm knockout mice in comparison to controls [L/L, cre/L/L].
- Volcano scatterplots depicting the expression levels of the subunits of the small (28S) and large (39S) ribosomes in green and blue, respectively. Red dotted line, Benjamini–Hochberg adj. P = 0.05.
Tefm and Polrmt double‐heterozygous deletion does not affect mitochondrial function or mtDNA transcription
We proceeded to investigate how a moderate reduction of TEFM and/or POLRMT levels affects mitochondrial function and mtDNA transcription. Tefm +/− mice were crossed with Polrmt heterozygous knockout (Polrmt +/−) mice 11 to generate double‐heterozygous knockout mice (Tefm +/−, Polrmt +/−). Western blot analyses showed decreased protein levels of TEFM and POLRMT in double‐heterozygous knockouts, in accordance with the 50% reduction in Tefm and Polrmt gene dosage (Fig EV4A). The whole‐body reduction of TEFM and POLRMT levels did not lead to any obvious phenotype, and all mice were viable and healthy at the age of 52 weeks. The mtDNA levels were assessed by qPCR at 16 weeks of age and showed no difference between double‐heterozygous knockout and control mice (Fig EV4B). Additionally, western blot analyses showed normal levels of the OXPHOS subunits in heart mitochondria from double‐heterozygous mice (Fig EV4C), showing that OXPHOS is not affected by a combined moderate decrease of TEFM and POLRMT levels. Furthermore, northern blot analyses of the steady‐state levels of individual mtRNA transcripts, including mt‐rRNAs, mt‐mRNAs (Fig EV4D and Appendix Fig S1A), and mt‐tRNAs (Fig EV4E and Appendix Fig S1B and C), showed no change in double‐heterozygous mice in comparison with controls. These data show that a mild reduction in TEFM and POLRMT protein levels does not affect mtDNA expression.
Figure EV4. Characterization of whole‐body Tefm and Polrmt double‐heterozygous knockout mice and the effects of TEFM depletion on mitochondrial transcripts and the mtDNA replication machinery.
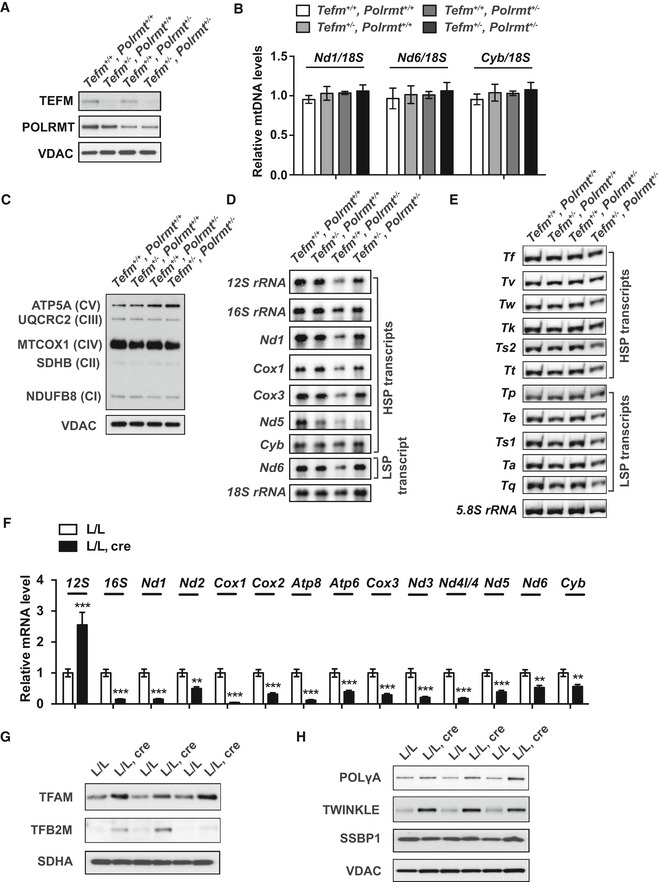
- Western blot analyses of POLRMT and TEFM protein levels in heart mitochondrial extracts from 16‐week‐old Tefm and Polrmt double‐heterozygous knockout mice (n = 6 mice for each group). VDAC was used as loading control.
- Quantitative PCR analyses of the relative mtDNA levels in 16‐week‐old Tefm and Polrmt double‐heterozygous knockout mice (n = 6 mice for each group). 18S was used to normalize for nuclear input.
- Western blot analyses of subunits from individual OXPHOS complexes at the age of 16 weeks (n = 6 mice for each group). Nucleus‐encoded subunits of complex I (NDUFB8), complex II (SDHB), complex III (UQCRC2), complex IV (MTCOX1), and complex V (ATP5A) were analyzed. VDAC was used as loading control.
- Northern blot analyses of mitochondrial transcripts of mt‐rRNAs and mt‐mRNAs in 16‐week‐old Tefm and Polrmt double‐heterozygous knockout mouse hearts (n = 8 mice for each group). 18S rRNA was used as loading control.
- Northern blot analyses of mitochondrial mt‐tRNA transcripts in 16‐week‐old Tefm and Polrmt double‐heterozygous knockout mouse hearts (n = 8 mice for each group). 5.8S rRNA was used as loading control.
- RT–qPCR analyses of the mitochondrial transcripts in 8‐week‐old control and Tefm knockout mice (n = 12 mice for each group).
- Western blot analyses of TFAM and TFB2M in the 8‐week‐old control and Tefm knockout mice (n = 12 mice for each group). SDHA was used as loading control.
- Western blot analyses of the subunit A of the DNA polymerase γ (POLγA), DNA helicase TWINKLE, and ssDNA‐binding protein (SSBP1) in 8‐week‐old control and Tefm knockout mice (n = 12 mice for each group). VDAC was used as loading control.
Loss of TEFM impairs mtDNA transcription elongation and drastically decreases steady‐state levels of promotor‐distal transcripts
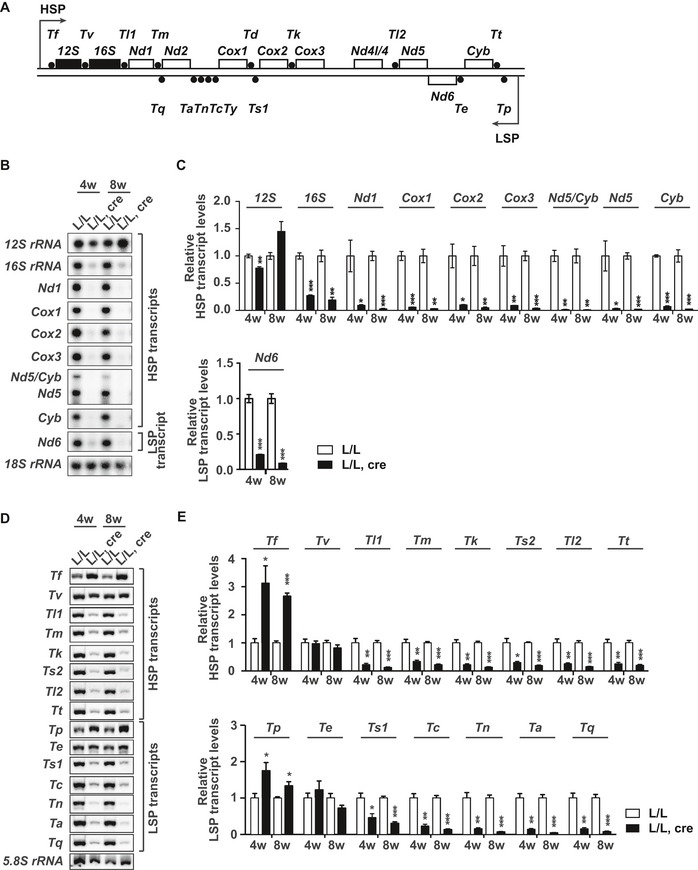
Since TEFM has previously been identified as a component of the mitochondrial transcription machinery in vitro 16, 17, 19, we proceeded to analyze mtRNA transcript levels in hearts from 4‐ and 8‐week‐old Tefm knockout mice. Northern blot analyses showed a drastic reduction in the steady‐state levels of promoter‐distal transcripts (mt‐rRNAs, mt‐mRNAs, and mt‐tRNAs) encoded on both strands in the absence of TEFM (Fig 3A–E). In contrast, the levels of promoter‐proximal transcripts were either increased, such as mt‐tRNAF and mt‐tRNAP, or remained near normal, such as 12S rRNA, mt‐tRNAV, and mt‐tRNAE (Fig 3A–E). The alterations in levels of mt‐rRNAs and mt‐mRNAs were independently confirmed by RT–qPCR analyses (Fig EV4F). Moreover, the near normal 12S rRNA levels and decreased 16S rRNA levels explain the alteration of steady‐state levels of mitoribosomal proteins found in the mitoproteomics data set (Fig EV3C) and are also consistent with a previous report showing that normal levels of the 12S rRNA and low levels of 16S rRNA decrease the levels of large ribosomal subunit proteins, whereas the levels of the small ribosomal subunit proteins remain unaffected 34. Furthermore, we investigated de novo mtRNA synthesis by in organello labeling with 32P‐labeled uridine triphosphate (32P‐UTP) and observed that loss of TEFM led to a dramatically reduced mtRNA levels, predominantly affecting the production of longer transcripts (Fig 4A). Therefore, loss of TEFM impairs mtDNA transcription, particularly transcription elongation, resulting in a severe reduction of promoter‐distal transcripts from both strands. The defect in transcription elongation is accompanied by an increase of transcription initiation events, which may also be supported by the increased levels of TFAM and TFB2M (Fig EV4G) and POLRMT (Fig 2E and F), thus leading to drastically increased levels of promoter‐proximal transcripts.
Figure 3. Loss of TEFM impairs mtDNA transcription elongation and decreases steady‐state levels of promotor‐distal transcripts.
-
ASchematic linear map of mouse mtDNA indicating the relative position of mtRNA transcripts analyzed by northern blot.
-
B–ENorthern blot analyses (B and D) and relative quantification (C and E) of mtRNA transcripts in control and Tefm knockout mouse hearts at the age of 4 and 8 weeks (n = 12 mice at each time‐point for each group). 18S rRNA and 5.8S rRNA were used as loading controls, respectively.
Figure 4. Accumulated shorter LSP transcripts in TEFM‐depleted mitochondria do not contribute to mtDNA replication.
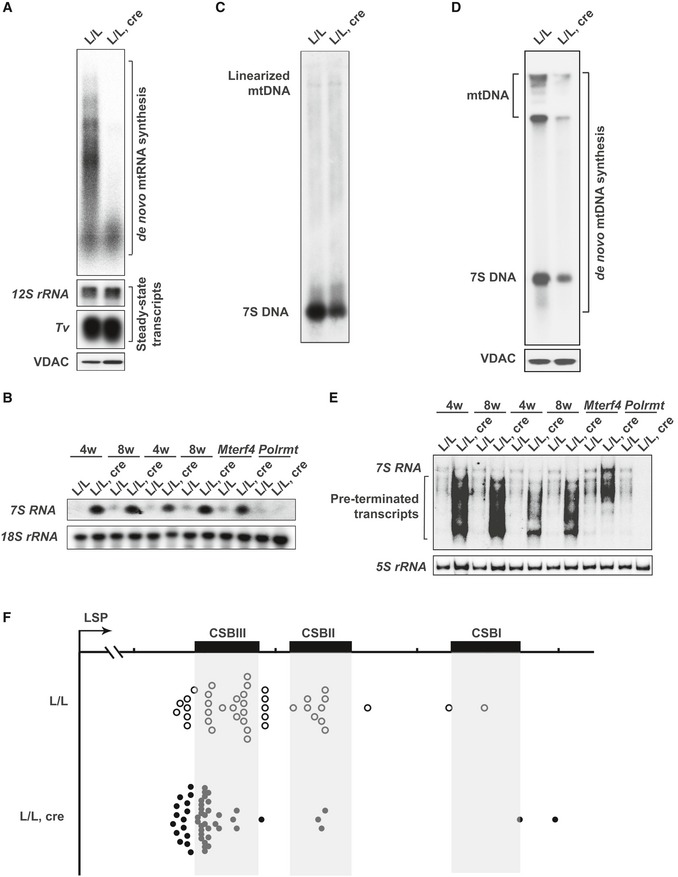
- In organello transcription analyses using heart mitochondria isolated from 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group). The steady‐state levels of 12S rRNA and mt‐tRNAV were used as endogenous controls, due to their unchanged levels. VDAC was used as loading control for the equal input of mitochondrial extracts. This experiment has performed five times with either two or three replicates.
- Northern blot analyses of 7S RNA level in 4‐ and 8‐week‐old control and Tefm knockout mouse hearts (n = 12 mice at each time‐point for each group). 18S rRNA was used as loading control. RNA from tissue‐specific Mterf4 knockout mouse hearts and tissue‐specific Polrmt knockout mouse hearts was loaded as positive and negative control, respectively.
- Southern blot analyses of mtDNA to obtain 7S DNA in 8‐week‐old control and Tefm knockout mice. Heart mitochondria from three control or knockout mice were pooled for preparation of mtDNA. Equal amounts of mtDNA shown on the top of the gel were loaded as the endogenous control. This experiment has performed three times.
- In organello replication assays were performed on heart mitochondria isolated from 8‐week‐old control and Tefm knockout mice (n = 5 mice for each group). VDAC was used as loading control.
- Northern blot analyses of the 7S RNA levels in 4‐ and 8‐week‐old control and Tefm knockout mouse hearts (n = 12 mice for each group). 5S rRNA was used as loading control. RNA from tissue‐specific Mterf4 knockout mouse hearts and tissue‐specific Polrmt knockout mouse hearts was loaded as positive and negative control, respectively.
- Mapping 3′ termini of mitochondrial 7S RNA/early LSP transcripts in control (L/L) and Tefm knockout (L/L, cre) mouse hearts at the age of 8 weeks. A 3′ RACE was performed using linker‐ligated RNA and a forward primer corresponding to the first 45 bp from the transcription initiation site at LSP. CSB, conserved sequence block.
Accumulation of short LSP transcripts in TEFM‐deficient mitochondria does not contribute to mtDNA replication
Transcripts initiated from LSP frequently terminate at the conserved sequence blocks (CSBs) resulting in the formation of 7S RNA, which is the most promoter‐proximal L strand transcript 5. To measure 7S RNA levels in Tefm knockout mice, a 32P‐radiolabeled RNA probe, corresponding to the first 45 base pairs (bp) of the RNA produced by transcription initiation at LSP, was used for northern blot analyses (Fig 4B). Samples from tissue‐specific Mterf4 knockouts, which are known to have increased 7S RNA levels, and tissue‐specific Polrmt knockouts, which are known to lack 7S RNA 11, 47, were included as positive and negative controls, respectively. The levels of 7S RNA were markedly increased in Tefm knockout mice in comparison with controls (Fig 4B). The drastically increased abundance of 7S RNA prompted us to evaluate mtDNA replication, because LSP transcripts are used to prime initiation of mtDNA replication in the CSB region 7, 21, 55. Surprisingly, when loaded with equal amounts of mtDNA, the steady‐state levels of 7S DNA within the D‐loop were markedly reduced in Tefm knockout heart mitochondria (Fig 4C). Furthermore, analysis of de novo mtDNA synthesis revealed a drastic reduction in the synthesis of 7S DNA and full‐length mtDNA in the absence of TEFM (Fig 4D). These results show that loss of TEFM impairs mtDNA replication. To address the question why the high levels of 7S RNA are not associated with increased mtDNA replication, we performed further analysis of the promoter‐proximal transcripts using high‐resolution polyacrylamide gels (Fig 4E). A range of prematurely terminated RNA species shorter than full‐length 7S RNA were observed in the Tefm knockout mice (Fig 4E). The tissue‐specific Mterf4 knockout mice also contain high levels of 7S RNA reflecting the increase in the rate of transcription in these mice, but in this case the size distribution of 7S RNA transcripts is the same as in controls without evidence for premature termination. Further analysis with the rapid amplification 3′ cDNA ends method (3′RACE) confirmed the findings from the northern blots and showed premature termination of 7S RNAs (Fig 4F and Table EV1). Promoter‐proximal transcription termination for RNA primer formation has been attributed to the G‐quadruplex structure of CSBII 22, and it has been reported that TEFM is necessary for reading through this sequence 27. However, loss of TEFM does not generate a distinct termination product at CSBII, but rather a range of short, prematurely terminated transcripts (Fig 4E and F).
We also investigated the expression of core factors involved in mtDNA replication, including the subunit A of the DNA polymerase γ (POLγA), the DNA helicase TWINKLE, and the ssDNA‐binding protein (SSBP1) by western blot analyses. We found that POLγA and TWINKLE were significantly increased at the protein level, while SSBP1 expression remained unchanged in TEFM‐depleted mitochondria (Fig EV4H). Thus, the defective mtDNA replication capacity does not result from a lack of replication‐related proteins. Therefore, our results show that the absence of TEFM causes accumulation of very short, promoter‐proximal transcripts initiated from LSP, which prevent proper RNA primer formation to promote mtDNA replication.
Transcriptome‐wide analysis of RNA from Tefm knockout hearts suggests a link between transcription elongation and RNA processing
We further analyzed the mitochondrial transcriptome in total heart tissue and purified mitochondria from Tefm knockout and control mice by RNA sequencing (RNA‐Seq). We performed TruSeq RNA‐Seq, to capture long transcripts, such as mt‐mRNAs and mt‐rRNAs (Fig 5A and Appendix Fig S2A), and small RNA sequencing to selectively capture mt‐tRNAs and the 7S RNA (Appendix Fig S2B and C and Fig EV5A). Analyses of both RNA‐Seq datasets showed a drastic increase in reads corresponding to promoter‐proximal transcripts from both LSP and HSP (e.g., mt‐tRNAF and mt‐tRNAP), whereas reads corresponding to promoter‐distal transcripts from both strands (16S rRNA, all mt‐mRNAs and most mt‐tRNAs) were decreased (Fig 5A and Appendix Fig S2). These data are consistent with the results from our northern blot and RT–qPCR analyses (Figs 3 and EV4F). The small RNA‐Seq dataset revealed the presence of shorter, prematurely terminated transcripts in Tefm knockout heart mitochondria (Fig EV5A), which provides independent confirmation of the results shown in Fig 4E and F.
Figure 5. Transcriptome‐wide analysis reveals that mtDNA transcription elongation is linked to RNA processing.
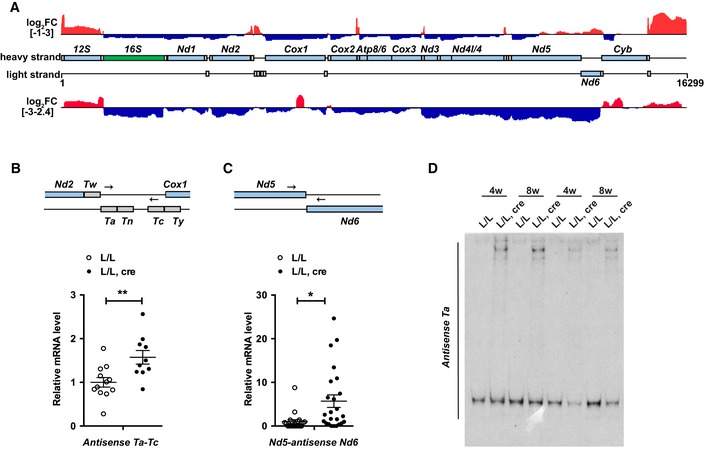
- Transcriptome‐wide analysis of the effects of TEFM loss on mtRNA transcripts by TruSeq RNA sequencing. A complete map of mtDNA showing the change in sequence read coverage (log2 fold change [L/L, cre/L/L]) between Tefm knockout (L/L, cre; n = 4) and control (L/L; n = 3) mice on both strands. Increased reads are shown in red and decreased reads in blue. The mtDNA is displayed in the central track, with mt‐rRNAs in green, mt‐mRNAs in light blue and mt‐tRNAs in gray.
- Strand‐specific RT–qPCR detection of the mitochondrial precursor transcripts containing the antisense of mt‐tRNAA and antisense of mt‐tRNAC in control and Tefm knockout mice (n = 12 mice for each group). The mt‐mRNAs are shown in light blue and mt‐tRNAs in gray. Arrows show the binding position of qPCR primers. Actin transcript was used as endogenous control.
- Strand‐specific RT–qPCR detection of the mitochondrial precursor transcripts spanning the junction of Nd5 and antisense of Nd6 in control and Tefm knockout mice (n = 25 mice for each group). The mt‐mRNAs are shown in light blue. Arrows show the binding position of qPCR primers. Actin transcript was used as endogenous control.
- Northern blot analyses of the precursor transcripts containing the antisense of mt‐tRNAA in 4‐ and 8‐week‐old control and Tefm knockout mouse hearts (n = 6 mice for each group).
Figure EV5. Transcriptome‐wide analysis of Tefm knockout hearts and TEFM interacts with mtRNA processing factors.
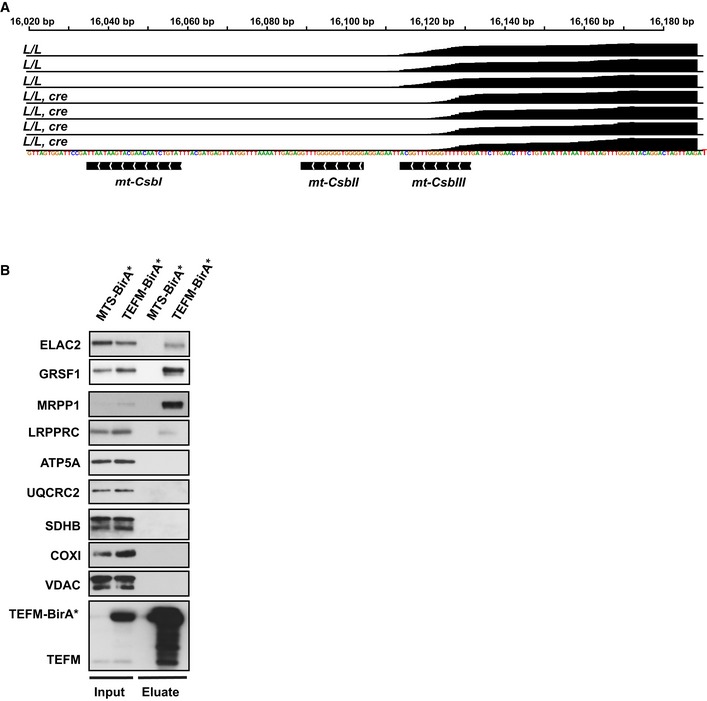
- Normalized read counts corresponding to the light‐promoter region from small RNA sequencing of mitochondrial transcripts showing the 7S RNA read counts (L/L, n = 4; L/L, cre n = 3).
- Western blot analyses of RNA processing‐related proteins in isolated mitochondria (input) and eluates when TEFM‐BirA* was used as a bait. Eluate (100%), input (1%).
The TruSeq technique only captures mt‐tRNAs (or other short RNAs) if they are part of longer, unprocessed transcripts and does not detect free mt‐tRNAs 34. This analysis revealed a marked increase of mt‐tRNA reads in the Tefm knockout datasets, indicating an accumulation of RNA processing intermediates (Fig 5A). We found that the enrichment of unprocessed transcripts was particularly prominent in regions containing mt‐tRNA clusters or regions spanning tRNA‐mRNA junctions (Fig 5A). To further confirm the increase of unprocessed transcripts in Tefm knockout mice, we performed strand‐specific RT–qPCR (Fig 5B and C). H‐strand cDNAs were generated using the anti‐mt‐tRNAC‐R primer, which is complementary to the antisense strand relative to mt‐tRNAC, and the anti‐Nd6‐R primer, which is complementary to the antisense strand of Nd6. Synthesis of cDNA with these primers thus generates ssDNA that corresponds to RNAs that are antisense to mt‐tRNAC and Nd6, respectively. In the next step, pairs of primers were used to amplify the cDNA to get dsDNA for analysis by qPCR. As shown in Fig 5B and C, these unprocessed transcripts were upregulated in the absence of TEFM. Moreover, northern blot analyses with a probe corresponding to antisense mt‐tRNAA confirmed the presence of long unprocessed transcripts in TEFM knockout mice identified by TruSeq (Fig 5D). We thus conclude that absence of TEFM leads to impaired RNA processing.
TEFM interacts with RNA processing factors
To further investigate the role of TEFM in RNA processing, we searched for TEFM interacting partners using a proximity‐labeling (BioID) assay 56 followed by affinity purification and mass spectrometry of biotinylated proteins. Using Flp‐In T‐REx 293 cells, we generated a stable cell line that constitutively expressed human TEFM as a fusion protein containing a carboxyterminal BirA*‐HA tag (TEFM‐BirA*). As expected, our results confirmed that TEFM interacts with POLRMT (Fig 6), as previously reported 16, 17, 19. In addition, we identified a number of RNA processing‐related proteins that interacted with TEFM, including the RNase P complex (MRPP1‐3), GRSF1, FASTKD5, ELAC2, mtPAP, and the components of mitochondrial RNA degradosome (SUPV3L1‐PNPase) complex (Fig 6), which are all reported to be localized in or adjacent to RNA granules 29, 30, 37, 40, 41, 57. Western blot analyses of the eluates confirmed the association between RNA processing factors (ELAC2, GRSF1, and MRPP1) and TEFM (Fig EV5B). As expected, naturally biotinylated proteins, such as carboxylases, were also identified. Furthermore, some very abundant mitochondrial proteins, including respiratory chain subunits, ribosomal proteins, and proteases, were also identified and likely represent the commonly found contaminants, as previously reported in a similar study 58.
Figure 6. TEFM interacts with RNA processing factors.
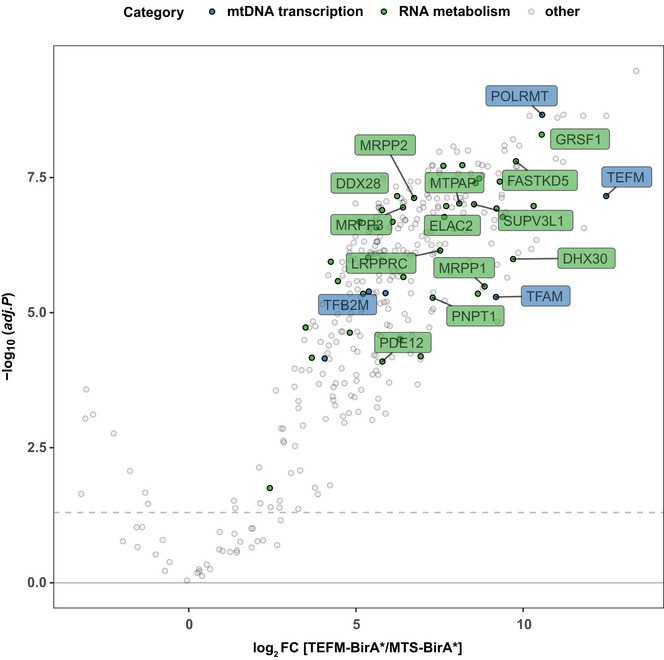
Volcano plot showing TEFM‐BioID. TEFM‐BirA*‐HA was stably expressed in HEK 293 cells, followed by streptavidin affinity purification and mass spectrometry analysis to detect the associated proteins. Mitochondrial targeted BirA* (MTS‐BirA*) was used as a control. The significantly enriched proteins are presented above the horizontal dashed line with 5% false discovery rate (FDR) significance. The blue dots highlight proteins involved in mtDNA transcription, and the green dots represent proteins involved in RNA metabolism. All other proteins are in gray.
Discussion
We report here that TEFM acts as a general elongation factor for mtDNA transcription in mammalian mitochondria. Without TEFM, the vast majority of transcripts initiated at LSP does not reach very far and is mostly terminated near CSBIII after initiation. We also report that some regions of mtDNA, such as those with clusters of mt‐tRNA genes, seem to be particularly difficult sequences to bypass in the absence of TEFM, likely because of strong secondary structures. Recent computational analyses of the human mtDNA sequence suggest that there are ∼200 putative G‐quadruplex forming sequences 59, 60, whose presence has been confirmed in live cells 61. The existence of G‐quadruplexes in the non‐transcribed DNA strand constitutes a very strong transcription barrier for a progressing RNA polymerase 62, and a G‐quadruplex in nascent RNA strongly stimulates transcription termination at CSBII 22, 23. It should be noted that the region just upstream of CSBIII is predicted to have the potential to form G‐quadruplex by the QGRS mapper, although it has only a modest effect on transcription termination in vitro 22. Clearly, transcription in the absence of TEFM in vivo leads to an accumulation of very short transcripts that terminate prematurely and fail to pass through the predicted G‐quadruplex at CSBIII.
Unexpectedly, the in vivo data we present here also show that loss of TEFM affects RNA processing. By using several independent methods, i.e., RNA‐Seq, northern blots, and RT–qPCR, we found abundant, unprocessed transcripts in the absence of TEFM, particularly in regions with strong secondary structures, such as mt‐tRNA gene clusters. Label‐free quantitative proteomics showed increased levels of many proteins involved in RNA processing in the absence of TEFM. By performing BioID assays, we found interactions between TEFM and a range of proteins indispensable for endonucleolytic processing of mtRNAs, e.g., the RNase P complex, ELAC2, and GRSF1. Our results thus suggest that TEFM may interact with the RNA processing machinery in mammalian mitochondria, although further studies are needed to reveal possible molecular mechanisms. A possible coupling between transcription elongation and RNA processing could increase the accuracy and efficiency of pre‐RNA maturation and may also provide new regulatory opportunities to control mtDNA gene expression. It should be pointed out that transcription and pre‐mRNA processing are coupled processes also in the nucleus, with important consequences for transcript maturation and gene regulation. In the nucleus, the carboxyl‐terminal domain (CTD) of the largest RNA polymerase II subunit is phosphorylated during transcription initiation and this leads to recruitment of proteins involved in RNA processing, including factors required for 5′‐capping, splicing, and polyadenylation. By acting as a landing pad that brings processing factors in close proximity to the actively transcribing RNA polymerase II, the nascent pre‐mRNA can be efficiently processed during active transcription elongation. For instance, the nascent pre‐mRNA is capped (attachment of 7‐methylguanosine by a 5′‐5′ triphosphate linkage) already after transcription of the first 20–30 nucleotides, just as the 5′ end of the nascent transcript emerges from the RNA exit channel 63. Co‐transcriptional processing also helps to explain how chromatin modification, such as histone methylation (H3K36), can mark expressed exons 64. Future work is needed to reveal additional factors required for co‐transcriptional processing in mitochondria and to define their regulatory potential.
Based on results from in vitro transcription experiments, previous work suggested that TEFM regulates the formation of the RNA primers needed for initiation of mtDNA replication 27. This study led to the postulation of a model whereby regulation of TEFM controls a switch between mtDNA gene expression and replication. On the one hand, high levels of TEFM will allow transcription to proceed past CSBII to promote near genome‐length transcription for expression of mtDNA‐encoded genes. On the other hand, low levels of TEFM will lead to transcription arrest at CSBII and formation of RNA primers needed for mtDNA replication. Data presented here and elsewhere suggest that the mechanisms of primer formation are more complicated than described in the replication–transcription switch model. First, as we show here, loss of TEFM does not lead to an upregulation of mtDNA replication initiation as predicted by the model. Instead, low levels of TEFM cause a drastic decrease in de novo mtDNA replication and a depletion of nascent D‐loop strands (7S DNA). Second, transcripts formed by premature termination at CSBII cannot be directly used as primers for initiation of mtDNA replication, but must first be processed to shorter transcripts by RNase HI 65. A role for RNase H in primer maturation has also been reported in other systems. For instance, RNase H is required for R‐loop processing and replication primer formation during ColE1 plasmid replication in bacteria 66.
Another interesting finding is that loss of TEFM leads to a dramatic increase of POLRMT levels (Fig 2E and F) and a mild increase of MRPL12 levels (Fig EV3C). One may argue that MRPL12 binds to and stabilizes POLRMT 67, 68, but we have failed to confirm this proposed interaction in previous studies 11, 54. In density gradients with wild‐type mitochondrial extracts, POLRMT is found in fractions containing mtDNA, whereas MRPL12 is present in mtDNA‐free fractions containing other ribosomal proteins 11, arguing against a significant interaction between MRPL12 and POLRMT. Recently, we analyzed the proteome of 5 tissue‐specific knockout mouse mutants with disruption of mtDNA gene expression at different levels and found that the expression of 12S and 16S rRNA was critical for the stability of most mitoribosomal proteins 54. In all of the 5 knockouts, the POLRMT levels were substantially increased (except in the Polrmt knockout mice), but the overall levels of mitoribosomal proteins followed the levels of 12S and 16S rRNA. It should be noted that MRPL12 levels were near normal in the absence of 12S and 16S rRNA, but were increased in the two knockouts that can form mitochondrial ribosomes (Fig 5 in 54). MRPL12 thus belongs to a small subset of mitoribosomal proteins that remains stable in the absence of 12S and 16S rRNA, and the POLRMT protein levels are upregulated independent of changes in MRPL12 levels.
In summary, the in vivo results we report here establish TEFM as a crucial component of the mitochondrial transcription machinery. In the absence of TEFM, transcription elongation is severely impaired, resulting in reduced mtDNA expression and a bioenergetics crisis. In contrast to a recently presented model, we find that reduced levels of TEFM do not increase the formation of RNA primers needed for DNA replication. Instead, we show that TEFM is necessary both for replication primer formation and for near genome‐length transcription for mtDNA expression. Our in vivo results are thus inconsistent with a model where TEFM acts as a switch that regulates the balance between replication and gene expression of mtDNA, and instead, we report a possible connection between transcription elongation to RNA processing that needs to be explained in future molecular studies.
Materials and Methods
Experimental model and subject details
Mouse models
Generation of conditional Tefm‐knockout mice. The targeting vector for disruption of Tefm in embryonic stem cells (derived from C57BL/6N) was generated by Taconic Artemis using BAC clones from the C57BL/6J RPCIB‐731 BAC library. To generate the conditional Tefm‐knockout allele, exon 2 of the Tefm locus was flanked by loxP sites and a puromycin resistance cassette (PuroR) flanked by Frt sites was introduced in intron 1 as a positive selection marker. The PuroR was removed by the mating of Tefm +/loxP‐Puro mice with transgenic mice with ubiquitously expressing flp‐recombinase. The resulting Tefm +/loxP mice were subsequently crossed with mice ubiquitously expressing cre‐recombinase under the control of (i) the β‐actin promoter (+/β‐actin) to generate heterozygous Tefm knockout mice (Tefm +/−) or (ii) the muscle creatinine kinase promoter (+/Ckmm‐cre) to generate heart and skeletal muscle tissue‐specific Tefm knockout mice (Tefm loxP/loxP, +/Ckmm‐cre; L/L, cre). The Tefm and Polrmt double‐heterozygous knockout mice (Tefm +/−, Polrmt +/−) were obtained by intercrossing Tefm +/− with heterozygous Polrmt knockout mice (Polrmt +/−) 11. All the mice strains used in this study are listed in Table EV2.
All mice in this study had an inbred C57BL/6N background. Animal studies were approved by the animal welfare ethics committee and performed in compliance with National and European law.
Method details
Morphological analysis
Hearts collected from 8‐week‐old mice were fixed in 4% paraformaldehyde (PFA) at 4°C for 24 h. Then, the hearts were processed routinely and embedded in paraffin and sectioned to 5 μm thickness. Hematoxylin and eosin was performed for structural analysis.
Transmission electron microscopy
Left myocardium was cut into small pieces and fixed in 2% glutaraldehyde, 0.5% paraformaldehyde, 0.1 M sodium cacodylate, 0.1 M sucrose, and 3 mM CaCl2, pH 7.4 at room temperature for 30 min, followed by 24 h at 4°C. Specimens were rinsed in a buffer containing 0.15 M sodium cacodylate and 3 mM CaCl2, pH 7.4; postfixed in 2% osmium tetroxide, 0.07 M sodium cacodylate, 1.5 mM CaCl2, pH 7.4 at 4°C for 2 h; dehydrated in ethanol followed by acetone; and embedded in LX‐112 (Ladd Research Industries). Ultra‐thin sections (40–50 nm) from longitudinal parts were cut and examined in a transmission electron microscope (Tecnai 10; FEI) at 80 kV. Digital images at a final magnification of 8,200× were randomly taken on myofibrils from sections of the myocardium.
Crude mitochondrial isolation
Mitochondria from mouse heart were isolated as described previously 49, using mitochondrial isolation buffer (320 mM sucrose, 10 mM Tris–HCl, pH 7.4, and 1 mM EDTA), supplemented with protease inhibitor (Roche) by differential centrifugation. Briefly, hearts were homogenized using Potter‐Homogenizer on ice (20 strokes, 500 rpm). Nuclei and cell debris were pelleted at 1,000 g for 10 min at 4°C. Next, the supernatant was centrifuged at 10,000 g for 10 min at 4°C. The differential centrifugation was repeated twice to obtain the crude mitochondria.
Western blot analysis
50–100 μg of crude mitochondria were resuspended in 1× NuPAGE™ LDS Sample Buffer. Mitochondrial proteins were then separated by SDS–PAGE (4–12% Bis‐Tris Protein Gels; Invitrogen) and transferred onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore). Immunoblotting was performed using standard procedures and developed using ECL reagent. The antibodies used in this study are listed in the Table EV2.
RT–qPCR and northern blot analysis
For RT–qPCR, northern blotting, and 3′ RACE, total RNA was extracted from heart using Trizol reagent (Invitrogen) according to the manufacturer's instructions and then treated with TURBO DNA‐free™ DNase (Invitrogen). For RT–qPCR expression analysis, cDNA was reversed transcribed from 1 μg total RNA using High‐Capacity cDNA Reverse Transcription Kit (Invitrogen). The qPCR was performed in a QuantStudio 6 Flex Real‐Time PCR System (Life Technologies), using TaqMan™ Universal Master Mix II, with UNG (Applied Biosystems) to quantify mitochondrial transcripts (mt‐rRNAs and mt‐mRNAs), Tefm, Actin, and 18S.
For RT–qPCR detection of transcripts across the tRNA‐mRNA junctions, H‐strand cDNA was generated by reverse transcriptase PCR using High‐Capacity cDNA Reverse Transcription Kit; however, synthesized reverse primers, including Anti‐mt‐tRNAC‐R or Anti‐Nd6‐R and Actin‐R, were used instead of commercial random primers. Then, qPCR was performed using Platinum SYBR Green qPCR supermix‐UDG (Invitrogen) to quantify the transcripts containing tRNA‐mRNA junctions. Primer pair 1 (mt‐tRNAA‐F and Anti‐mt‐tRNAC‐R) was used to detect the antisense transcript of mt‐tRNAs cluster on the H strand, shown in Fig 5B. Primer pair 2 (Nd5‐F and Anti‐Nd6‐R) was used to detect the Nd5‐antisense Nd6 junction on the H strand, shown in Fig 5C. Actin was used as endogenous control.
For northern blotting, 3 μg of total RNA was separated in either 1% MOPS–formaldehyde agarose gels to detect mt‐mRNAs and mt‐rRNAs or neutral 10% polyacrylamide gel to detect mt‐tRNAs, 7S RNA, and unprocessed mtRNA intermediates. Separated RNAs were then transferred to Hybond‐N+ membranes (GE Healthcare) and hybridized with (i) randomly [32P] dCTP‐labeled dsDNA probes, following the Prime‐It II Random Primer Labeling Kit (Agilent), to detect mt‐mRNAs (except Nd6), mt‐rRNAs, 18S rRNA, 5S rRNA, and 5.8S rRNA; (ii) in vitro transcribed single‐stranded [32P] UTP‐labeled RNA probe for Nd6, using the Riboprobe Combination System‐SP6/T7 RNA Polymerase (Promega); (iii) strand‐specific oligonucleotide probes labeled with [32P] dATP, using T4 Polynucleotide Kinase (New England Biolabs), to detect mt‐tRNAs, 7S RNA, and unprocessed mtRNA intermediates. Radioactive signals were captured in a PhosphorImager screen and was quantified using a Typhoon 7000 FLA and the ImageQuant TL 8. 1 software (GE Healthcare). All the Taqman probes and the primer sequences are listed in the Table EV2.
3′ RACE
For rapid amplification of 3′ cDNA ends (3′ RACE), a phosphorylated oligonucleotide linker was first ligated to 2.5 μg of a total RNA using T4 RNA ligase 1 (New England Biolabs). RNA was precipitated and cDNA synthesized using the GeneAmp RNA PCR kit and a primer complementary to the linker sequence (anti‐linker). The cDNA was then amplified by PCR using the anti‐linker primer and a forward primer corresponding to the first 45 nucleotides from the transcription initiation site at LSP. PCR products were cloned into pCRII‐TOPO using TOPO TA Cloning Kit (Invitrogen), following manufacturer's instructions, and direct sequencing of the plasmid from single colonies was performed using M13 forward and M13 reverse primers. Sequence details are listed in Table EV1.
DNA isolation and mtDNA quantification
Genomic DNA from mouse heart was isolated with the DNeasy Blood and Tissue Kit (Qiagen) following manufacturer's instructions, followed by treatment with RNase A. mtDNA was measured by quantitative PCR using 5 ng of DNA in a QuantStudio 6 Flex Real‐Time PCR System using TaqMan™ Universal Master Mix II, with UNG. The TaqMan assays were used for the detection of mtDNA, which were Nd1, Nd6, and Cyb. 18S was used to normalize for nuclear input. Three technical replicates were performed.
Southern blot analysis and D‐loop Southern
For Southern blot analysis, 3 μg of DNA samples was digested with SacI to linearize mtDNA molecules, followed by separation on a 0.8% agarose gel and blotted to Hybond‐N+ membranes. The D‐loop was assessed as described previously 11, and mtDNA was isolated by phenol/chloroform extraction from purified mitochondria. Samples were heated for 3 min at 100°C before loading. Membranes were hybridized with [32P] dCTP‐labeled dsDNA probes following the Prime‐It II Random Primer Labeling Kit to detect total mtDNA (pAM1), the D‐loop, or nuclear DNA (18S rDNA) as loading control and exposed to PhosphorImager screens.
In organello transcription assay
In organello transcription assay was performed on crude mitochondria isolated from mouse hearts. 750 μg of mitochondria was washed three times with 500 μl of cold incubation buffer (25 mM sucrose, 75 mM sorbitol, 100 mM KCl, 10 mM K2HPO4, 50 μM EDTA, 5 mM MgCl2, 1 mM ADP, 10 mM glutamate, 2.5 mM malate, 10 mM Tris–HCl, pH 7.4, and 1 mg/ml BSA). Mitochondria were pelleted by centrifugation, resuspended in 500 μl of transcription labeling buffer supplemented with 40 μCi of [32P] UTP, and gently rotated for 1 h at 37°C. After incubation, mitochondria were washed with 500 μl of washing buffer (10% glycerol, 10 mM Tris–HCl, pH 6.8, and 0.15 mM MgCl2) and RNA was isolated with Trizol reagent (Invitrogen), following the manufacturer's instructions. An aliquot of the labeled mitochondria was used for western blot analysis to ensure equal input of mitochondrial extracts. The isolated RNA was analyzed by northern blotting, as described before.
In organello replication assay
In organello replication assay was performed on crude mitochondria isolated from mouse hearts. 750 μg of isolated mitochondria was washed three times with 500 μl of incubation buffer and resuspended in 500 μl of incubation buffer supplemented with 50 μM dCTP, 50 μM dTTP, 50 μM dGTP, and 20 μCi of [32P] dATP, and incubated for 2 h at 37°C. After incubation, mitochondria were washed three times in washing buffer. An aliquot of the labeled mitochondria was used for western blot analysis to ensure equal input of mitochondrial extracts. Then, mtDNA was isolated by phenol/chloroform extraction using standard procedures, and radiolabeled replicated DNA was analyzed by Southern blotting, as described previously.
Measurement of mitochondrial respiration
To obtain high‐quality crude mitochondria for respiration analysis, heart tissue was finely cut and digested with 0.2% trypsin in 2 ml mitochondrial isolation buffer (310 mM sucrose, 20 mM Tris–HCl, and 1 mM EGTA, pH 7.2) for 10 min at 4°C, with constant rotation. Heart tissue was gently hand‐homogenized with 5 ml ice‐cold mitochondrial isolation buffer. Mitochondria were purified by differential centrifugation, as previously described. Mitochondrial oxygen consumption flux was measured at 37°C using 25–50 μg of crude mitochondria diluted in 2.1 ml mitochondrial respiration buffer (120 mM sucrose, 50 mM KCl, 20 mM Tris–HCl, 4 mM KH2PO4, 2 mM MgCl2, and 1 mM EGTA, pH 7) within an Oxygraph‐2k chamber (Oroboros Instruments). The oxygen consumption rate was measured either using 10 mM pyruvate, 10 mM glutamate, and 5 mM malate or 10 mM succinate in the presence of 0.5 μM rotenone. The phosphorylating state was obtained using 2.5 mM ADP and the non‐phosphorylating state using 125 ng/ml oligomycin. Maximal mitochondrial respiration, uncoupling, was reached by successive addition of CCCP up to 0.3 μM.
Blue native polyacrylamide gel electrophoresis
Crude heart mitochondria (65 μg) were solubilized in 16 μl of 1× NativePAGE sample buffer containing 1% n‐dodecyl‐β‐d‐maltoside (DDM) and incubated on ice for 15 min. Samples were then centrifuged at 20,000 g for 30 min at 4°C. Supernatant was collected, and 0.5 μl of NativePage 5% G‐250 sample additive was added to each sample. Next, proteins and high molecular weight native makers were separated on a 3–12% NativePage gel (Invitrogen) at 120V using a dark blue cathode (1× NativePAGE running buffer and 0.5× cathode additive) until the blue‐front migrated 50% of the gel followed by a light blue cathode buffer (1× NativePAGE running buffer and 0.1× cathode additive). The anode buffer consisted of 1× NativePAGE running buffer. For western blot analysis, proteins were then blotted onto a PVDF membrane with transfer buffer (48 mM Tris and 39 mM glycine) for 1.5 h at 400 mA. Membranes were completely destained with methanol before proceeding with standard immunoblotting.
Ultrapure mitochondria isolation
Ultrapure mitochondria were prepared as previously described 54. Crude mitochondrial pellets from mouse hearts were washed in 1xM buffer (220 mM mannitol, 70 mM sucrose, 5 mM HEPES pH 7.4, 1 mM EGTA pH 7.4); pH was adjusted with potassium hydroxide, supplemented with EDTA‐free complete protease inhibitor cocktail and PhosSTOP Tablets (Roche), and purified on a Percoll density gradient (12%:19%:40% prepared with buffer 2xM) via centrifugation in a SW41 rotor at 41,086 g at 4°C for 1 h in a Beckman Coulter Optima L‐100 XP ultracentrifuge using 14 × 89 mm Ultra‐Clear Centrifuge Tubes (Beckman Instruments Inc.). Mitochondria were harvested at the interphase between 19 and 40%, and washed three times with buffer 1xM, and mitochondrial pellets were frozen at −80°C. Purified frozen mitochondria pellets were suspended in lysis buffer (6 M guanidinium chloride, 10 mM Tris(2‐carboxyethyl)phosphine hydrochloride, 40 mM chloroacetamide, and 100 mM Tris–HCl). After complete lysis, samples were diluted 1:10 in 20 mM Tris–HCL pH 8.0 and 80 μg of protein was mixed with 3 μg of Trypsin gold (Promega) and incubated overnight at 37°C to achieve complete digestion. Peptides were cleaned with home‐made STAGEtips 69 (Empore Octadecyl C18; 3M) and eluted in 60% acetonitrile/0.1% formic acid buffer. Samples were dried in a SpeedVac apparatus (Eppendorf concentrator plus 5305) at 45°C, and the peptides were suspended with 0.1% formic acid. Approximately 1.5 μg of peptides was analyzed by LC‐MS/MS.
Label‐free quantitative proteomics
Peptides were separated on a 25 cm, 75 μm internal diameter PicoFrit analytical column (New Objective) packed with 1.9 μm ReproSil‐Pur media (Dr. Maisch) using an EASY‐nLC 1200 (Thermo Fisher Scientific). The column was maintained at 50°C. Buffer A was 0.1% formic acid in water; buffer B was 80% acetonitrile, 0.1% formic acid. Peptides were separated on a segmented gradient from 3 to 6% buffer B for 10 min, from 6 to 25% buffer B for 100 min, from 25 to 31% buffer B for 10 min, and from 31 to 60% buffer B for 10 min, at 200 nl/min. For the BioID experiment, peptides were separated on a segmented gradient from 6 to 31% buffer B for 80 min and from 31 to 50% buffer B for 10 min. Eluting peptides were analyzed on a QExactive HF mass spectrometer (mitoproteome quantification, Thermo Fisher Scientific) or Orbitrap Fusion (BioID experiment, Thermo Fisher Scientific). Peptide precursor mass to charge ratio (m/z) measurements (MS1) was carried out at 60,000× resolution, in the 300–1,800 m/z range. The top ten most intense precursors with charge state from 2 to 7 only were selected for HCD fragmentation using 25% normalized collision energy. The m/z of the peptide fragments (MS2) was measured at 30,000× resolution, using an AGC target of 2e5 and 80‐ms maximum injection time. Upon fragmentation, precursors were put on an exclusion list for 45 s.
BioID
TEFM‐BirA*‐HA and MTS‐BirA*‐HA (mitochondrial targeting sequence from human COX8a) were cloned into pcDNA5/FRT/TO vector. Human Flp‐In T‐REx 293 cells were nucleofected using NucleofectorTM Lonza device. Cells were seeded in DMEM GlutaMax and supplemented with 10% FBS and 1% pen/strep (100 U/ml). The next day, 200,000 cells were collected and resuspended in 20 μl of Nucleofector™ Solution with 900 ng of pOG44 and 100 ng of construct. 24 h after nucleofection, cells were passaged into 10‐cm2 plates and selected by the addition of hygromycin and blasticidin at final concentration of 100 and 10 μg/ml, respectively. This selection medium was changed every 2–3 days until clear visible colonies were present. Several colonies per construct were picked and expanded. Selected clones were scaled up to one 245 × 245 × 25 mm3 square dish for treatment and harvesting. The cells were induced with 1 μg/μl of doxycycline at around 70% of confluency. The day after, cells were treated with 50 μM biotin for protein labeling. Cells were harvested 24 h later and washed three times with PBS by pelleting at 800 g for 5 min at 4 °C. Mitochondria were isolated using mitochondria isolation buffer (20 mM HEPES pH 7.6, 220 mM mannitol, 70 mM sucrose, 1 mM EDTA, 1× protease inhibitor, 2 mg/ml BSA) by differential centrifugation. The cells were homogenized in homogenizer by applying 15–20 strokes. This suspension was then centrifuged at 800 g for 5 min at 4°C. Collected supernatant was centrifuged at 10,000 g for 10 min at 4°C. Pellet (crude mitochondria) was then resuspended in mitochondria isolation buffer without BSA and centrifuged again at 10,000 g for 10 min at 4°C.
Pull‐down experiments were performed according to the published protocol 56 with small modifications. Mitochondrial pellet was lysed in 1 ml of lysis buffer (50 mM Tris–HCl, pH 7.4, 500 mM NaCl, 0.2% SDS, 1× protease inhibitor, 1 mM DTT). Subsequently, 20 μl of 20% Triton X‐100 was added. Samples were incubated for 30 min on ice and then centrifuged at 16,500 g, 4°C for 10 min. Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific) were incubated with the lysates on a rotator at 4°C overnight. There were 9 following washing steps. Samples were eluted in elution buffer (2 M Urea, 5 ng/μl Trypsin, 1 mM TCEP, 50 mM Tris–HCl, pH 7.5) at room temperature. A volume of 5 mM CAA was added to the samples, and reaction was incubated at 37°C overnight. The samples were further analyzed by LC‐MS/MS mass spectrometry. The proteins of TEFM‐BioID are listed in Dataset EV1.
LC‐MS/MS data analysis
The raw data were analyzed with MaxQuant 70 version 1.5.2.8 using the integrated Andromeda search engine 71. Peptide fragmentation spectra were searched against the canonical and isoform sequences of the mouse reference proteome (proteome ID UP000000589, downloaded August 2015) or the human reference proteome (BioID experiment, proteome ID UP000005640, downloaded September 2018) from UniProt. The database was automatically complemented with sequences of contaminating proteins by MaxQuant. Methionine oxidation and protein N‐terminal acetylation were set as variable modifications. Digestion parameters were set to “specific” and “Trypsin/P”, allowing for cleavage after lysine and arginine, also when followed by proline. The minimum number of peptides and razor peptides for protein identification was 1; the minimum number of unique peptides was 0. Protein identification was performed at a peptide spectrum matches and protein false discovery rate of 0.01. The “second peptide” option was on to identify co‐fragmented peptides. Successful identifications were transferred between the different raw files using the “Match between runs” option, using a match time window of 0.7 min. Label‐free quantification (LFQ) 72 was performed using an LFQ minimum ratio count of 2.
Analysis of the label‐free quantification results was performed with Perseus computation platform 73, version 1.5.0.0 and R 74. Proteins marked as “Reverse”, “Only identified by site”, and “Potential contaminant” were removed. LFQ intensity values were log2 transformed and proteins that contained at least three valid values in L/L or L/L, cre groups, and five valid values for the BioID experiment were kept for further analysis.
Missing values were imputed with a width of 0.3 and down shift of 1.8. Differential expression analysis was performed using the two‐sided moderated t‐test from the limma package 75. Proteins with a Benjamini‐Hochberg adjusted P < 0.05 were designated as differentially regulated. Differentially expressed proteins were manually classified into selected mitochondrial pathways.
RNA sequencing analyses
RNA was isolated from total mouse heart and crude heart mitochondria using the miRNeasy Mini Kit (Qiagen), according to manufacturer's instructions. RNA quantity, purity, and integrity were verified using a Bioanalyzer.
TruSeq library analysis
Sequenced reads were aligned to the mouse genome reference sequence (mm10), masked for NUMTs regions, with bowtie2 v2.2.9 76, and the subsequent alignments filtered with SAMtools v.1.3.1 77 (‐q 2 ‐f 2 ‐F 256) to retain mapped proper pairs and exclude secondary alignments and multireads (read alignments with equal primary and secondary alignment scores). Mitochondrial alignments were extracted, split by template strand, and converted to full‐length RNA fragment BED files, before coverage was calculated with BEDtools 78 genomecov (‐d ‐scale [1e+06/total mapped reads]), normalized to the total number of fragments mapped to the mouse whole genome (reads per million; RPM), and converted to wig format for visualization. Gene‐level counts for the nuclear genome were calculated with featureCounts 79 (‐s 2) using the GENCODE vM12 annotation (excluding mitochondrial annotations), while counts for mitochondrial genes were calculated with BEDtools coverage (‐counts ‐s ‐F 0.80) using a GENCODE‐based annotation with contiguous mt‐Atp8/6 and mt‐Nd4 l/4 intervals, and an approximate 7S RNA annotation (chrM:16035‐16188). BEDtools coverage was used for mitochondrial genes due to their compact genomic organization and the subsequent overlapping of multiple genes by read alignments with post‐transcriptional 5′ or 3′ extensions, which is addressed by specifying a minimum 80% overlap between the read and the annotation. Count tables were collated in R v3.3.2 74, and differential expression between conditions was analyzed with DESeq2 v1.14.1 80.
Small RNA library analysis
Reads were trimmed of 3′ adapter sequences with cutadapt v1.10 81 (‐m 15 ‐a TGGAATTCTCGGGTGCCAAGG). Trimmed reads were aligned to the mouse genome reference sequence (mm10), masked for NUMTs regions, with bowtie2 76, and the subsequent alignments filtered with SAMtools v1.3.1 77 (‐q 2 ‐F 260) to exclude unmapped reads, secondary alignments, and multireads. Mitochondrial alignments were extracted and split by strand, and coverage was calculated with BEDtools 78 genomecov (‐d ‐scale [1e+06/total mapped reads]), normalized to the total number of mapped reads to the mouse whole genome (reads per million; RPM), and converted to wig format for visualization. Gene‐level counts for the nuclear genome were calculated with featureCounts 79 (‐s 1), while counts for mitochondrial genes were calculated with BEDtools coverage (‐counts ‐s ‐F 0.80). Count tables were collated in R v3.3.2 74, and differential expression between conditions was analyzed with DESeq2 v1.14.1 80.
Statistical analysis
Experiments were performed at least three times, and results are representative of n > 5 independent biological replicates, unless indicated otherwise. All values are expressed as means ± SEM. Statistical analysis of proteomics and RNA sequencing are described above. Other statistical analyses were conducted using GraphPad Prism 5 software. Statistical significance between two groups was assessed by two‐tailed unpaired Student's t‐test, and statistical significance between more than two groups was performed using one‐way analysis of variance (ANOVA). Differences were considered statistically significant at a value of P < 0.05.
Author contributions
SJ, CK, and N‐GL conceived the project, designed the experiments, and wrote the manuscript. SJ performed and interpreted the majority of the experiments. SJ, CK, N‐GL, IK, and MM advised on methodology and interpreted the data. JM, SS, IK, ESR, MM, MJ, OL, IA, FAS, VP, RW, and KH performed experiments and analyzed the data. DM, AF, and CMG contributed on experiment design, interpreted the data, and edited the manuscript. N‐GL supervised the project. All the authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Review Process File
Acknowledgements
We thank Maria Falkenberg, Christoph Freyer, and Paula Clemente for technical suggestions and critical input, and Thomas J. Nicholls and Susanne Virding for technical assistance. We are also grateful for the technical support from the Proteomics Core Facility, Max Planck Institute for Biology of Ageing; in particular, we acknowledge Xinping Li. RNA library construction and sequencing were performed at the Cologne Center for Genomics. This work was supported by the Knut och Alice Wallenbergs Stiftelse (Knut and Alice Wallenberg Foundation), Swedish Research Council: 2015‐00418, Novo Nordisk Fonden and the Max Planck Society to N.‐G. Larsson. A. Filipovska receives support from the Australian Research Council (DP170103000) and the National Health and Medical Research Council of Australia (APP1067837 and APP1058442).
EMBO Reports (2019) 20: e48101
Data availability
The RNA sequencing data have uploaded to GEO database, with the access number of GSE102255. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 82 partner repository with the dataset identifier PXD013398.
References
- 1. Wallace DC (1999) Mitochondrial diseases in man and mouse. Science 283: 1482–1488 [DOI] [PubMed] [Google Scholar]
- 2. Larsson N‐G (2010) Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79: 683–706 [DOI] [PubMed] [Google Scholar]
- 3. Kauppila TES, Kauppila JHK, Larsson N‐G (2017) Mammalian mitochondria and aging: an update. Cell Metab 25: 57–71 [DOI] [PubMed] [Google Scholar]
- 4. Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88: 611–638 [DOI] [PubMed] [Google Scholar]
- 5. Gustafsson CM, Falkenberg M, Larsson N‐G (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85: 133–160 [DOI] [PubMed] [Google Scholar]
- 6. Clayton DA (1991) Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol 7: 453–478 [DOI] [PubMed] [Google Scholar]
- 7. Chang DD, Clayton DA (1987) A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J 6: 409–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Falkenberg M, Larsson N‐G, Gustafsson CM (2007) DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem 76: 679–699 [DOI] [PubMed] [Google Scholar]
- 9. Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson N‐G, Gustafsson CM (2002) Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet 31: 289–294 [DOI] [PubMed] [Google Scholar]
- 10. Ringel R, Sologub M, Morozov YI, Litonin D, Cramer P, Temiakov D (2011) Structure of human mitochondrial RNA polymerase. Nature 478: 269–273 [DOI] [PubMed] [Google Scholar]
- 11. Kühl I, Miranda M, Posse V, Milenkovic D, Mourier A, Siira SJ, Bonekamp NA, Neumann U, Filipovska A, Polosa PL et al (2016) POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Sci Adv 2: e1600963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Posse V, Hoberg E, Dierckx A, Shahzad S, Koolmeister C, Larsson N‐G, Wilhelmsson LM, Hällberg BM, Gustafsson CM (2014) The amino terminal extension of mammalian mitochondrial RNA polymerase ensures promoter specific transcription initiation. Nucleic Acids Res 42: 3638–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morozov YI, Agaronyan K, Cheung ACM, Anikin M, Cramer P, Temiakov D (2014) A novel intermediate in transcription initiation by human mitochondrial RNA polymerase. Nucleic Acids Res 42: 3884–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mangus DA, Jang SH, Jaehning JA (1994) Release of the yeast mitochondrial RNA polymerase specificity factor from transcription complexes. J Biol Chem 269: 26568–26574 [PubMed] [Google Scholar]
- 15. Sologub M, Litonin D, Anikin M, Mustaev A, Temiakov D (2009) TFB2 is a transient component of the catalytic site of the human mitochondrial RNA polymerase. Cell 139: 934–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minczuk M, He J, Duch AM, Ettema TJ, Chlebowski A, Dzionek K, Nijtmans LGJ, Huynen MA, Holt IJ (2011) TEFM (c17orf42) is necessary for transcription of human mtDNA. Nucleic Acids Res 39: 4284–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Posse V, Shahzad S, Falkenberg M, Hällberg BM, Gustafsson CM (2015) TEFM is a potent stimulator of mitochondrial transcription elongation in vitro . Nucleic Acids Res 43: 2615–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hillen HS, Morozov YI, Sarfallah A, Temiakov D, Cramer P (2017) Structural basis of mitochondrial transcription initiation. Cell 171: 1072–1081.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hillen HS, Parshin AV, Agaronyan K, Morozov YI, Graber JJ, Chernev A, Schwinghammer K, Urlaub H, Anikin M, Cramer P et al (2017) Mechanism of transcription anti‐termination in human mitochondria. Cell 171: 1082–1093.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu H, Xue C, Long M, Jia H, Xue G, Du S, Coello Y, Ishibashi T (2018) TEFM enhances transcription elongation by modifying mtRNAP pausing dynamics. Biophys J 115: 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pham XH, Farge G, Shi Y, Gaspari M, Gustafsson CM, Falkenberg M (2006) Conserved sequence box II directs transcription termination and primer formation in mitochondria. J Biol Chem 281: 24647–24652 [DOI] [PubMed] [Google Scholar]
- 22. Wanrooij PH, Uhler JP, Simonsson T, Falkenberg M, Gustafsson CM (2010) G‐quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. Proc Natl Acad Sci USA 107: 16072–16077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wanrooij PH, Uhler JP, Shi Y, Westerlund F, Falkenberg M, Gustafsson CM (2012) A hybrid G‐quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R‐loop. Nucleic Acids Res 40: 10334–10344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee DY, Clayton DA (1996) Properties of a primer RNA‐DNA hybrid at the mouse mitochondrial DNA leading‐strand origin of replication. J Biol Chem 271: 24262–24269 [DOI] [PubMed] [Google Scholar]
- 25. Xu B, Clayton DA (1995) A persistent RNA‐DNA hybrid is formed during transcription at a phylogenetically conserved mitochondrial DNA sequence. Mol Cell Biol 15: 580–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu B, Clayton DA (1996) RNA‐DNA hybrid formation at the human mitochondrial heavy‐strand origin ceases at replication start sites: an implication for RNA‐DNA hybrids serving as primers. EMBO J 15: 3135–3143 [PMC free article] [PubMed] [Google Scholar]
- 27. Agaronyan K, Morozov YI, Anikin M, Temiakov D (2015) Mitochondrial biology. Replication‐transcription switch in human mitochondria. Science 347: 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reinhard L, Sridhara S, Hällberg BM (2017) The MRPP1/MRPP2 complex is a tRNA‐maturation platform in human mitochondria. Nucleic Acids Res 45: 12469–12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska‐Lightowlers ZM, Martinou J‐C (2013) GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab 17: 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EA (2013) The mitochondrial RNA‐binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab 17: 386–398 [DOI] [PubMed] [Google Scholar]
- 31. Pearce SF, Rebelo‐Guiomar P, D'Souza AR, Powell CA, Van Haute L, Minczuk M (2017) Regulation of mammalian mitochondrial gene expression: recent advances. Trends Biochem Sci 42: 625–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290: 470–474 [DOI] [PubMed] [Google Scholar]
- 33. Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135: 462–474 [DOI] [PubMed] [Google Scholar]
- 34. Rackham O, Busch JD, Matic S, Siira SJ, Kuznetsova I, Atanassov I, Ermer JA, Shearwood A‐MJ, Richman TR, Stewart JB et al (2016) Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep 16: 1874–1890 [DOI] [PubMed] [Google Scholar]
- 35. Brzezniak LK, Bijata M, Szczesny RJ, Stepien PP (2011) Involvement of human ELAC2 gene product in 3′ end processing of mitochondrial tRNAs. RNA Biol 8: 616–626 [DOI] [PubMed] [Google Scholar]
- 36. Siira SJ, Rossetti G, Richman TR, Perks K, Ermer JA, Kuznetsova I, Hughes L, Shearwood A‐MJ, Viola HM, Hool LC et al (2018) Concerted regulation of mitochondrial and nuclear non‐coding RNAs by a dual‐targeted RNase Z. EMBO Rep 19: e46198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bogenhagen DF, Martin DW, Koller A (2014) Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab 19: 618–629 [DOI] [PubMed] [Google Scholar]
- 38. Pietras Z, Wojcik MA, Borowski LS, Szewczyk M, Kulinski TM, Cysewski D, Stepien PP, Dziembowski A, Szczesny RJ (2018) Dedicated surveillance mechanism controls G‐quadruplex forming non‐coding RNAs in human mitochondria. Nat Commun 9: 2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Antonicka H, Shoubridge EA (2015) Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep 10: 920–932 [DOI] [PubMed] [Google Scholar]
- 40. Jourdain AA, Koppen M, Rodley CD, Maundrell K, Gueguen N, Reynier P, Guaras AM, Enriquez JA, Anderson P, Simarro M et al (2015) A mitochondria‐specific isoform of FASTK is present in mitochondrial RNA granules and regulates gene expression and function. Cell Rep 10: 1110–1121 [DOI] [PubMed] [Google Scholar]
- 41. Wilson WC, Hornig‐Do H‐T, Bruni F, Chang JH, Jourdain AA, Martinou J‐C, Falkenberg M, Spåhr H, Larsson N‐G, Lewis RJ et al (2014) A human mitochondrial poly(A) polymerase mutation reveals the complexities of post‐transcriptional mitochondrial gene expression. Hum Mol Genet 23: 6345–6355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee K‐W, Okot‐Kotber C, LaComb JF, Bogenhagen DF (2013) Mitochondrial ribosomal RNA (rRNA) methyltransferase family members are positioned to modify nascent rRNA in foci near the mitochondrial DNA nucleoid. J Biol Chem 288: 31386–31399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ (2013) Human mitochondrial RNA decay mediated by PNPase‐hSuv3 complex takes place in distinct foci. Nucleic Acids Res 41: 1223–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA (1998) Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 18: 231–236 [DOI] [PubMed] [Google Scholar]
- 45. Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Cámara Y, Milenkovic D, Zickermann V, Wibom R, Hultenby K et al (2012) LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J 31: 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Milenkovic D, Matic S, Kühl I, Ruzzenente B, Freyer C, Jemt E, Park CB, Falkenberg M, Larsson N‐G (2013) TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D‐loop strands and complete mtDNA replication. Hum Mol Genet 22: 1983–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cámara Y, Asin‐Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K et al (2011) MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab 13: 527–539 [DOI] [PubMed] [Google Scholar]
- 48. Smeitink J, van den Heuvel L, DiMauro S (2001) The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2: 342–352 [DOI] [PubMed] [Google Scholar]
- 49. Metodiev MD, Lesko N, Park CB, Cámara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson N‐G (2009) Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab 9: 386–397 [DOI] [PubMed] [Google Scholar]
- 50. Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson N‐G (2002) Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci USA 99: 15066–15071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wibom R, Hagenfeldt L, von Döbeln U (2002) Measurement of ATP production and respiratory chain enzyme activities in mitochondria isolated from small muscle biopsy samples. Anal Biochem 311: 139–151 [DOI] [PubMed] [Google Scholar]
- 52. Park CB, Asin‐Cayuela J, Cámara Y, Shi Y, Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument‐Bromage H, Tempst P et al (2007) MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell 130: 273–285 [DOI] [PubMed] [Google Scholar]
- 53. Calvo SE, Clauser KR, Mootha VK (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44: D1251–D1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kühl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A, Larsson N‐G (2017) Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 6: e30952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chang DD, Hauswirth WW, Clayton DA (1985) Replication priming and transcription initiate from precisely the same site in mouse mitochondrial DNA. EMBO J 4: 1559–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roux KJ, Kim DI, Burke B (2013) BioID: a screen for protein‐protein interactions. Curr Protoc Protein Sci 74: Unit19.23.–19.23.14 [DOI] [PubMed] [Google Scholar]
- 57. Jourdain AA, Boehm E, Maundrell K, Martinou J‐C (2016) Mitochondrial RNA granules: compartmentalizing mitochondrial gene expression. J Cell Biol 212: 611–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matic S, Jiang M, Nicholls TJ, Uhler JP, Dirksen‐Schwanenland C, Polosa PL, Simard M‐L, Li X, Atanassov I, Rackham O et al (2018) Mice lacking the mitochondrial exonuclease MGME1 accumulate mtDNA deletions without developing progeria. Nat Commun 9: 1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dong DW, Pereira F, Barrett SP, Kolesar JE, Cao K, Damas J, Yatsunyk LA, Johnson FB, Kaufman BA (2014) Association of G‐quadruplex forming sequences with human mtDNA deletion breakpoints. BMC Genom 15: 677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bharti SK, Sommers JA, Zhou J, Kaplan DL, Spelbrink JN, Mergny J‐L, Brosh RM (2014) DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G‐quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J Biol Chem 289: 29975–29993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang W‐C, Tseng T‐Y, Chen Y‐T, Chang C‐C, Wang Z‐F, Wang C‐L, Hsu T‐N, Li P‐T, Chen C‐T, Lin J‐J et al (2015) Direct evidence of mitochondrial G‐quadruplex DNA by using fluorescent anti‐cancer agents. Nucleic Acids Res 43: 10102–10113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tornaletti S, Park‐Snyder S, Hanawalt PC (2008) G4‐forming sequences in the non‐transcribed DNA strand pose blocks to T7 RNA polymerase and mammalian RNA polymerase II. J Biol Chem 283: 12756–12762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bentley DL (2014) Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kolasinska‐Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J (2009) Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet 41: 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Posse V, Al‐Behadili A, Uhler JP, Clausen AR, Reyes A, Zeviani M, Falkenberg M, Gustafsson CM (2019) RNase H1 directs origin‐specific initiation of DNA replication in human mitochondria. PLoS Genet 15: e1007781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dasgupta S, Masukata H, Tomizawa J (1987) Multiple mechanisms for initiation of ColE1 DNA replication: DNA synthesis in the presence and absence of ribonuclease H. Cell 51: 1113–1122 [DOI] [PubMed] [Google Scholar]
- 67. Surovtseva YV, Shutt TE, Cotney J, Cimen H, Chen SY, Koc EC, Shadel GS (2011) Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc Natl Acad Sci USA 108: 17921–17926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nouws J, Goswami AV, Bestwick M, McCann BJ, Surovtseva YV, Shadel GS (2016) Mitochondrial ribosomal protein L12 is required for POLRMT stability and exists as two forms generated by alternative proteolysis during import. J Biol Chem 291: 989–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- 70. Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- 71. Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- 72. Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- 74. Dalgaard P, R Development Core Team (2010) R: a language and environment for statistical computing. Vienna, Austria: R Development Core Team; Computer programme, Retrieved from http://www.R-project.org/ [Google Scholar]
- 75. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47–e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- 80. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.J 17: 10–12 [Google Scholar]
- 82. Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Review Process File
Data Availability Statement
The RNA sequencing data have uploaded to GEO database, with the access number of GSE102255. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 82 partner repository with the dataset identifier PXD013398.