

Abstract
Type I interferon (IFN)‐induced Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling drives the expression of IFN‐stimulated genes (ISGs) to mediate antiviral response. The strength and duration of JAK‐STAT signaling are tightly regulated to ensure effective antiviral defense while avoiding pathological inflammation and autoimmunity. Here, we report that cTAZ, an isoform of the Hippo pathway effector TAZ, is transcribed by an alternative promoter. Although majority of C‐terminal sequences of TAZ is retained, cTAZ is not regulated by the Hippo signaling and does not mediate its growth‐inhibitory functions. Instead, cTAZ negatively regulates JAK‐STAT signaling by inhibiting STAT1/2 nuclear localization and ISG expression, and its expression is induced by type I IFN. Thus, cTAZ functions as a modulator of JAK‐STAT signaling and may play a role in fine‐tuning cellular antiviral response.
Keywords: alternative transcript, Hippo pathway, JAK‐STAT pathway, viral infection
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Introduction
Type I interferons (IFNs), consisting mainly of IFN‐α and IFN‐β, are cytokines produced upon microbial infections to activate both innate immunity and adaptive immunity 1. Mutations in components of the type I IFN signaling pathway frequently lead to immunodeficiency and susceptibility to infection 2. Type I IFNs have been developed as therapeutic agents to treat viral infections and various cancers 3, 4. However, prolonged type I IFNs’ exposure can lead to autoimmune diseases, such as systemic lupus erythematosus 5. Thus, the activity of type I IFN signaling should be tightly controlled.
Type I IFNs exert their biological functions through the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling pathway. Briefly, upon type I IFNs binding to their transmembrane receptors, type I IFNs trigger the activation of receptor‐associated JAKs, which in turn phosphorylate STAT1 and STAT2. Phosphorylated STAT1 and STAT2 dimerize and translocate into nucleus and form a trimeric complex with IFN‐regulatory factor 9 (IRF9). This complex specifically binds to the promoters containing IFN‐sensitive response elements (ISREs) to drive the transcription of IFN‐stimulated genes (ISGs) 6, 7, 8. The strength of type I IFN response is modulated at multiple levels. The expression of type I IFNs is mainly regulated by IRFs, such as IRF3 and IRF7 9, 10, 11. IRF3/7 integrates upstream signaling from membrane‐associated Toll‐like receptors and cytosolic nucleic acid sensors, such as AIM2‐like receptors and RIG‐I‐like receptors, to induce transcription of type I IFNs 10, 12, 13. Meanwhile, IRF7 is a direct target gene of STAT proteins, which constituents a positive feedback mechanism of JAK‐STAT signaling by enhancing type I IFN production 9, 14. Moreover, JAK‐STAT signaling can be modulated by ISGs directly. For instance, ISGs such as STAT1, STAT2, and IRF9, positively regulate JAK‐STAT signaling. On the other hand, ISGs, including suppressor of cytokine signaling 1/3 (SOCS1/3) and ubiquitin‐specific peptidase 18 (USP18), negatively regulate JAK‐STAT signaling 13, 15. Hence, JAK‐STAT signaling is regulated by both positive and negative feedback mechanisms to meet the requirement of host tissues.
The Hippo signaling pathway plays a critical role in early development, tissue regeneration, and tumorigenesis 16, 17, 18, 19. The physiological and pathological functions of the Hippo pathway are largely mediated by two homologous transcriptional co‐activators, YAP and TAZ. When the Hippo pathway is on, YAP and TAZ are phosphorylated by upstream kinases LATS1/2, leading to their cytoplasmic retention. Conversely, when Hippo signaling is off, dephosphorylated YAP and TAZ translocate into nucleus, interact with TEAD family transcription factors, and induce the expression of target genes. YAP and TAZ target genes promote cell proliferation, survival, migration, and epithelial–mesenchymal transition (EMT), which together leads to organ growth and tumorigenesis.
Recently, novel functions of YAP/TAZ in immune system have been uncovered. In the Drosophila immune organ fat bodies, Yorkie (Yki, a YAP ortholog) activity is repressed by Gram‐positive bacteria, which leads to lower production of antimicrobial peptides 20. In mammals, by interacting with TBK1 or IRF3, YAP/TAZ inhibits production of type I IFNs 21, 22. Moreover, it has been revealed recently that TAZ is required for the differentiation of pro‐inflammatory TH17 cells, whereas YAP is involved in maintaining immunosuppressive regulatory T (Treg) cells 23, 24 . Together, these evidences demonstrate that YAP/TAZ activity regulates both innate immunity and adaptive immunity.
In this study, we identified a novel TAZ isoform called cTAZ that was transcribed by an alternative promoter. cTAZ contains the majority of the C‐terminus sequence of TAZ, but not the TEAD‐binding domain (TBD) and WW domain, and thus lacks canonical Hippo pathway functions. Type I IFN‐triggered JAK‐STAT signaling directly induces the expression of cTAZ, and cTAZ in turn inhibits JAK‐STAT signaling by disrupting the dimerization and nuclear translocation of STAT1 and STAT2, thereby down‐regulating the expression of ISGs and cellular antiviral response. Thus, cTAZ serves as a modulator to restrain type I IFN responses following viral infections.
Results and Discussion
A short TAZ isoform, cTAZ, is transcribed by an alternative promoter
We used anti‐YAP/TAZ and anti‐TAZ (CST) antibodies targeting the C‐terminus of TAZ to determine the expression of YAP/TAZ across different cell lines (Fig 1A). In immunoblotting (IB), these C‐terminus‐specific antibodies detected YAP (~70 kDa), TAZ (~55 kDa), and unexpectedly a smaller protein (~37 kDa, as indicated by an asterisk; Fig 1B). The small protein was encoded by the TAZ gene as its expression was reduced by treating cells with shRNA targeting TAZ but not YAP (Fig 1C). In an immunoprecipitation (IP) assay, this smaller protein was immunoprecipitated and recognized by anti‐TAZ (CST) or anti‐YAP/TAZ antibody, but failed to be immunoprecipitated or react with anti‐TAZ (SA), an antibody targeting the N‐terminus of TAZ (aa36–175; Figs 1A and D, and EV1A and B). These results suggested that the ~37‐kDa protein is a shorter TAZ isoform comprising mainly the C‐terminal TAZ sequence; thus, it was dubbed as cTAZ (C‐terminus of TAZ). cTAZ protein was detected in about 30% of the cell lines tested in this study (Appendix Table S1), and cTAZ mRNA was detected in most human tissues, albeit at low levels (Appendix Table S2). In mouse tissues, we failed to detect cTAZ protein expression in organs like liver and heart, whereas a band at the molecular weight of cTAZ was detected in lymph nodes and thymus (Fig EV1C). Moreover, mRNA and protein expression of cTAZ was detected in surgically removed lymph nodes of ~50% thyroid cancer patients (Fig EV1D).
Figure 1. Identification of a short TAZ isoform transcribed by an alternative promoter.
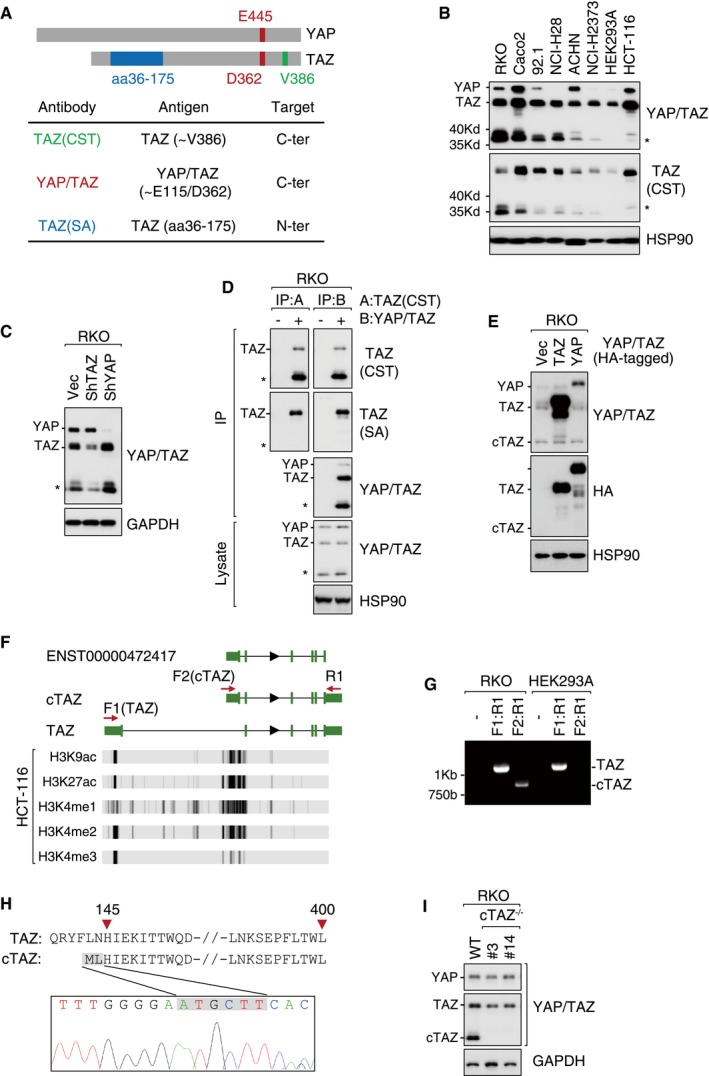
- YAP and/or TAZ antibodies and their target regions.
- Expression of YAP/TAZ and a smaller protein (asterisk) in different cell lines, protein expression was determined by immunoblotting (IB).
- The shRNA targeting TAZ, but not YAP, knocked down the expression of the smaller protein (asterisk).
- Antibodies targeting C‐terminus YAP/TAZ, such as TAZ (CST) and YAP/TAZ, effectively pulled down the smaller protein (asterisk, dubbed as cTAZ hereafter) in RKO cells in an immunoprecipitation (IP) assay. cTAZ was not recognized by TAZ (SA), an antibody targeting N‐terminus of TAZ.
- Exogenous TAZ/YAP was not processed proteolytically to cTAZ. RKO cells were transfected with the indicated plasmids expressing C‐terminal HA‐tagged TAZ or YAP.
- UCSC Genome Browser view of TAZ isoforms. Displayed tracks include a short (cTAZ?) and the full‐length (TAZ) transcript of TAZ assembled using RNA‐seq data of HCT‐116 cells form SRA. The short TAZ isoform was similar to transcript ENST00000472417 annotated in Ensembl database. Below: the H3K9ac, H3K27ac, H3K4me1, H3K4me2, and H3K4me3 histone‐modification signal peaks across TAZ gene in HCT‐116 cells (data from ENCODE database). The red arrows indicate the primers (F: forward; R: reverse) used in (G). UTR and exons are shown in green blocks.
- RKO, but not HEK293A cells, expressed both full‐length TAZ and cTAZ transcripts. RT–PCR primers targeting different regions are shown in (F).
- Alignment of TAZ and cTAZ protein sequences. cTAZ protein sequence was derived from the Sanger sequencing results of the short PCR product (RKO, F2:R1) in (G).
- Knockout of cTAZ in RKO cells using CRISPR/Cas9 technology. Monoclones (#3 and #14) were selected.
Source data are available online for this figure.
Figure EV1. Identification of a short TAZ isoform transcribed by an alternative promoter.
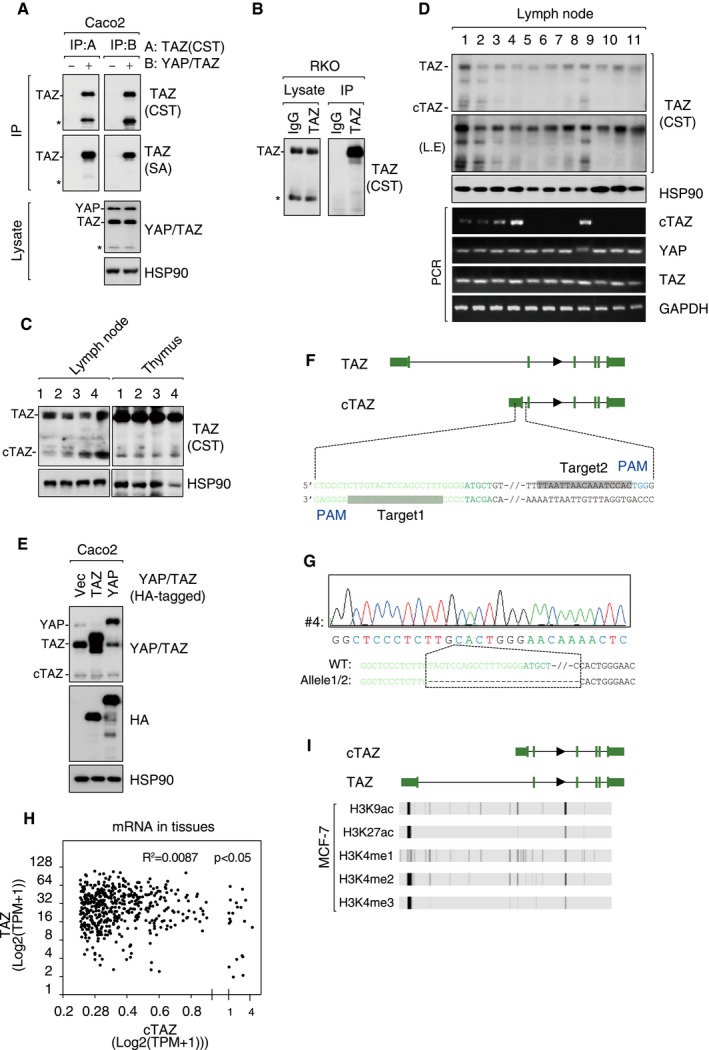
- Antibodies targeting C‐terminus YAP/TAZ, such as TAZ (CST) and YAP/TAZ, effectively pulled down the smaller protein (asterisk) in Caco2 cells in an IP assay.
- The smaller protein (asterisk) was not pulled down using anti‐TAZ (SA) recognizing N‐terminus of TAZ.
- cTAZ is expressed in mouse lymph nodes and thymus.
- cTAZ is expressed in human lymph nodes. Protein and mRNA expression of cTAZ in lymph nodes of thyroid cancer patients were analyzed.
- Exogenous YAP/TAZ was not processed to cTAZ. Caco2 cells were transfected with C‐terminal HA‐tagged TAZ or YAP, and protein expression was determined.
- Information about CRISPR/Cas9 guide RNAs targeting cTAZ. UTR and exons were in green.
- The deletion of the alternative exon of cTAZ gene. Genomic DNA of cTAZ KO cells was subjected to Sanger sequencing.
- No correlation between cTAZ and full‐length TAZ expression at mRNA level in human tissues. 492 tissue samples were used in this analysis. Pearson's correlation coefficient R 2 was calculated.
- Low H3K9ac, H3K27ac, H3K4me1, H3K4me2 and H3K4me3 signals around cTAZ promoter of MCF‐7 cells.
Source data are available online for this figure.
To determine whether cTAZ was generated by proteolytic cleavage of full‐length TAZ or encoded by an alternative transcript, we overexpressed a C‐terminal HA‐tagged YAP or TAZ in RKO or Caco2 cell. A band corresponding to cTAZ was not detected following overexpression (Figs 1E and EV1E), indicating that cTAZ was unlikely to be generated through proteolysis. We then analyzed RNA‐seq data of HCT‐116 cells (which also expresses cTAZ, Appendix Table S1) from Sequence Read Archive (SRA) database and spotted a short TAZ transcript (ENST00000472417) comprising a novel 5′UTR, an alternative exon, and exons encoding the C‐terminal sequence of conventional TAZ (Fig 1F). PCR with primers targeting this novel 5′UTR from cDNA of RKO cells expressing cTAZ, but not HEK293A cells that did not express cTAZ, yielded products of the expected band size (Fig 1F and G). The PCR product was identical in sequence to the variant available in the SRA database, it included amino acids 145–400 of full‐length TAZ and two novel amino acids (extreme N‐terminal) encoded by the alternative exon (Fig 1H). These results suggested that cTAZ was encoded by a transcription variant. Using CRISPR/Cas9 technology, we were able to ablate cTAZ from RKO cells by targeting cTAZ‐specific 5′UTR region (Figs 1I, and EV1F and G).
We further sought to ascertain whether the cTAZ mRNA was generated by alternative splicing or transcribed by an alternative promoter. By comparing the mRNA levels of cTAZ and TAZ in various human tissues using RNA‐seq data from Genotype‐Tissue Expression database, we found no association (Fig EV1H). At the protein level, expression levels of cTAZ and TAZ were also not correlated in diverse cell lines (Fig 1B). Moreover, the region around cTAZ 5′UTR harbors a typical promoter sequence with a predicted TATA box and was intensively marked by active epigenetic modifications such as H3K9ac, H3K27ac, H3K4me1, H3K4me2, and H3K4me3 (Fig 1F, and Appendix Fig S1). On the other hand, active epigenetic modifications were not present near cTAZ 5′UTR in MCF‐7 cells, a cell line without cTAZ expression (Fig EV1I, Appendix Table S1). These evidences collectively suggest that cTAZ was transcribed by an alternative promoter.
cTAZ is not regulated by Hippo signaling
The interaction between YAP/TAZ and their upstream kinases LATS1/2 is mediated by WW domains in YAP/TAZ and PPxY motifs in LATS1/2 25. cTAZ lacks an intact WW domain (Fig EV2A), suggesting that it cannot interact with LATS1/2 or respond to upstream signals. In a co‐immunoprecipitation (Co‐IP) assay, wild‐type YAP and TAZ efficiently pulled down endogenous LATS2, whereas cTAZ or WW‐mutated YAP/TAZ (YAP WW1/2 or TAZ WW) failed to precipitate LATS2 (Fig EV2B). Thus, cTAZ was unlikely affected by LATS1/2 kinase activity.
Figure EV2. cTAZ was not regulated by Hippo signaling.
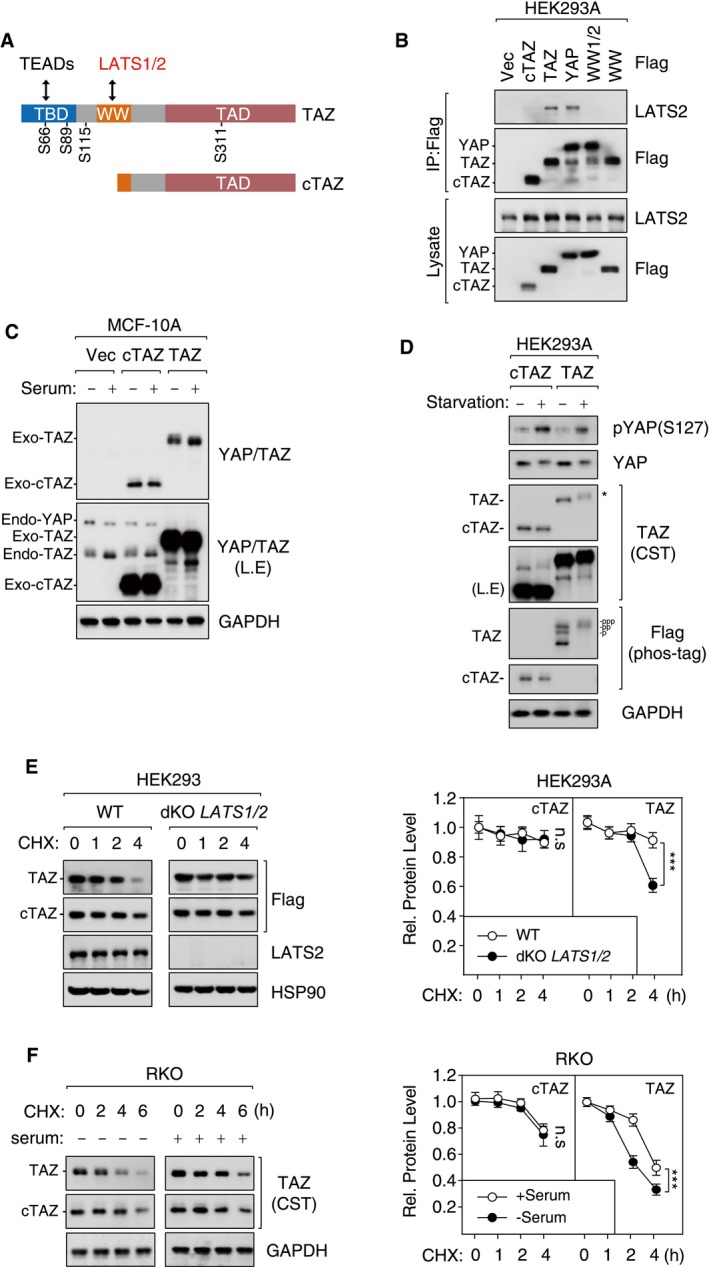
- Protein domains and interacting proteins of TAZ. Phosphorylation sites of LATS kinases are shown.
- cTAZ did not interact with LATS2. HEK293A cells were transfected with Flag‐tagged cTAZ, TAZ, YAP, WW‐mutated TAZ (WW), and WW1/2‐mutated YAP (WW1/2), and cell lysates were prepared and used for IP.
- cTAZ was not regulated by serum stimulation. cTAZ‐ or TAZ‐overexpressing MCF‐10A cells were stimulated by 10% FBS for 1 h, and protein expression was determined. Ectopic and endogenous TAZ were down‐shifted upon serum stimulation, whereas no change for cTAZ was detected.
- cTAZ was not regulated by serum starvation. cTAZ‐ or TAZ‐overexpressing HEK293A cells were serum‐starved for 8 h and harvested for IB. The phosphorylation of full‐length TAZ was also determined using phos‐tag gel. The asterisk indicates a shift of TAZ on regular gel.
- Deletion of LATS kinases stabilized TAZ, but not cTAZ. WT or LATS1/2‐double‐knockout (dKO) HEK293A cells were transfected with Flag‐tagged cTAZ or TAZ. Cells were treated with CHX (50 μg/ml) in a time course (0–4 h), and protein levels were determined. Error bars indicate SD, n = 3. ***P < 0.001; two‐way ANOVA test was used for statistical analysis.
- Serum treatment stabilized TAZ, but not cTAZ. RKO cells cultured in the presence or absence of 10% serum were treated with CHX (50 μg/ml) in a time course (0–6 h), and protein levels were determined. Error bars indicate SD, n = 3. ***P < 0.001; two‐way ANOVA test was used for statistical analysis.
Source data are available online for this figure.
The Hippo pathway senses many environmental cues, such as cell–cell contact, matrix stiffness, and diffusible ligands for G‐protein‐coupled receptors (GPCR) 25, 26, 27. YAP/TAZ phosphorylation by LATS1/2 leads to subsequent ubiquitination and degradation of YAP/TAZ 28. Notably, serum starvation controls the phosphorylation and protein levels of YAP/TAZ (S61, S109, S127, S164, and S381 of YAP, and S66, S89, S117, and S311 of TAZ) in a LATS1/2‐dependent manner 28, 29. Both endogenous and exogenous full‐length TAZ exhibited a significant increase in phosphorylation (as indicated by an upshift in regular or Phostag gel electrophoresis, or blotting using a YAP S127 phosphorylation‐specific antibody), whereas cTAZ was not regulated by serum starvation (Fig EV2C and D). Indeed, when cells were treated with cycloheximide (CHX), an inhibitor of protein synthesis, the half‐life of cTAZ protein was longer than that of full‐length TAZ (Fig EV2E and F). Moreover, half‐life of full‐length TAZ but not cTAZ was stabilized in LATS1/2‐double‐knockout cells or under serum‐rich conditions (Fig EV2E and F). Taken together, these results suggest that cTAZ was not regulated by LATS1/2 or upstream signals.
cTAZ lacks TEAD‐mediated transcriptional activity and canonical TAZ functions
The biological functions of YAP/TAZ are mainly mediated by TEAD family transcription factors, and TEAD‐binding domains (TBD) are mapped to the N‐terminus of YAP/TAZ (Fig EV2A). The entire TBD is absent in cTAZ; thus, cTAZ is not expected to induce TEAD‐mediated transcription and canonical Hippo pathway functions. Indeed, cTAZ could not interact with TEAD proteins as TAZ did (Fig EV3A). In a luciferase assay, the activity of an 8×GTIIC reporter, which reflects TEAD‐mediated transcriptional activity, was up‐regulated by full‐length TAZ, but unaffected by cTAZ (Fig EV3B). Moreover, expression of CTGF and CYR61, two TEAD‐mediated YAP/TAZ target genes, was significantly induced by full‐length TAZ, but not by cTAZ (Fig EV3B–D). Together, these results indicate that cTAZ lacks TEAD‐mediated transcriptional activity.
Figure EV3. cTAZ had no TEAD‐dependent transcriptional activity.
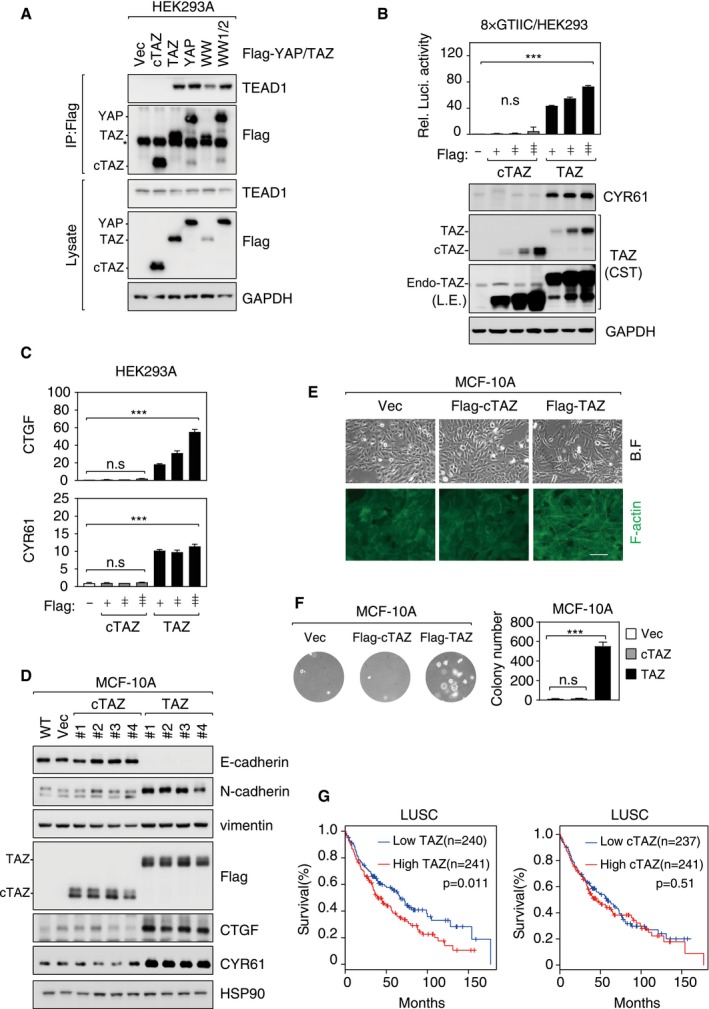
- cTAZ did not interact with TEADs. HEK293A cells were transfected with Flag‐tagged cTAZ, TAZ, YAP, WW‐mutated TAZ (WW), or WW1/2‐mutated YAP (WW1/2), and cells were lysed 24 h later for IP using Flag antibody. The asterisk indicates position of IgG.
- cTAZ could not activate TEAD reporter. HEK293A cells were transfected with 8×GTIIC‐luciferase reporter along with cTAZ or TAZ plasmid. Luciferase activity was measured. The expression levels of TAZ, cTAZ, and CYR61 (a TAZ targeting gene) were determined by IB. Error bars indicate SD, n = 3. ***P < 0.001; Student's t‐test.
- cTAZ could not induce expression of YAP/TAZ target genes. Cells were transfected as in (B). Endogenous CTGF and CYR61 mRNA levels were determined by qPCR. Error bars indicate SD, n = 3. ***P < 0.001; Student's t‐test.
- Overexpression of cTAZ could not induce EMT. cTAZ‐ or TAZ‐overexpressing MCF‐10A cell lines were established. Expression of TAZ target genes (CYR61 and CTGF) and EMT markers (epithelial marker: E‐cadherin; mesenchymal markers: N‐cadherin and vimentin) was assessed by IB.
- cTAZ could not induce EMT phenotype. MCF‐10A cells (also used in D) expressing cTAZ remained in an organized monolayer, and actin cytoskeleton was similar to that of wild‐type cells. On the other hand, full‐length TAZ induced cell morphological change and formation of strong actin fibers. Filamentous actin (F‐actin) was visualized by FITC‐conjugated phalloidin. Scale bar, 100 μm.
- cTAZ could not induce anchorage‐independent growth. MCF10A cells (also used in D) were seeded into soft agar; after 3 weeks, colony formation was determined. Error bars indicate SD, n = 3. ***P < 0.001; Student's t‐test.
- The expression of TAZ, but not cTAZ, served as a prognostic marker. Lung squamous cell carcinoma (LUSC) mRNA expression data were collected from TCGA database, and the relationship between TAZ (or cTAZ) expression and patient overall survival was analyzed using GEPIA application. The P‐values are derived by using the Mantel–Cox test.
Source data are available online for this figure.
As oncoproteins, YAP/TAZ can promote cell proliferation, migration, EMT, and transformation 29, 30, 31. However, overexpression of cTAZ failed to down‐regulate E‐cadherin or up‐regulate N‐cadherin and vimentin, which are hallmarks of EMT (Fig EV3D). In addition, overexpression of full‐length TAZ but not cTAZ induced cellular morphological changes, F‐actin reorganization, and anchorage‐independent growth of MCF‐10A cells (Fig EV3E and F). Moreover, in lung squamous cell carcinoma (LUSC), high expression of TAZ could predict a poor patient survival, whereas cTAZ expression was not associated with prognosis (Fig EV3G). These evidences suggest that cTAZ does not possess the canonical growth‐promoting and oncogenic activities of TAZ.
cTAZ directly suppresses JAK‐STAT signaling
To further characterize the function of cTAZ, we performed RNA‐seq in wild‐type, cTAZ‐knockout (cTAZ −/−), and cTAZ‐complemented (cTAZ −/− GFP‐cTAZ) cells. We identified 103 differentially expressed genes, which were affected by cTAZ deficiency and rescued by GFP‐cTAZ complementation (Figs 2A and EV4A, Dataset EV1). Gene ontology (GO) analysis indicated that these differentially expressed genes were mainly enriched in type I IFN signaling, defense response to virus, and immune system process (Fig 2B), with many being well‐defined ISGs 32, 33. Some cTAZ‐targeted genes, such as DDX58, IRF7, IFIH1, STAT1, OAS1, MX1, and IRF9, were assessed by qPCR in multiple cTAZ −/− clones (Fig 2C). The protein levels of RIG‐I (encoded by DDX58), IRF9, and MX1 in cTAZ‐knockout cells were also elevated, whereas IRF7 and STAT1 protein levels were not significantly changed upon cTAZ ablation (Fig EV4B). Thus, expression of an array of ISGs was repressed by cTAZ.
Figure 2. cTAZ directly represses JAK‐STAT signaling.
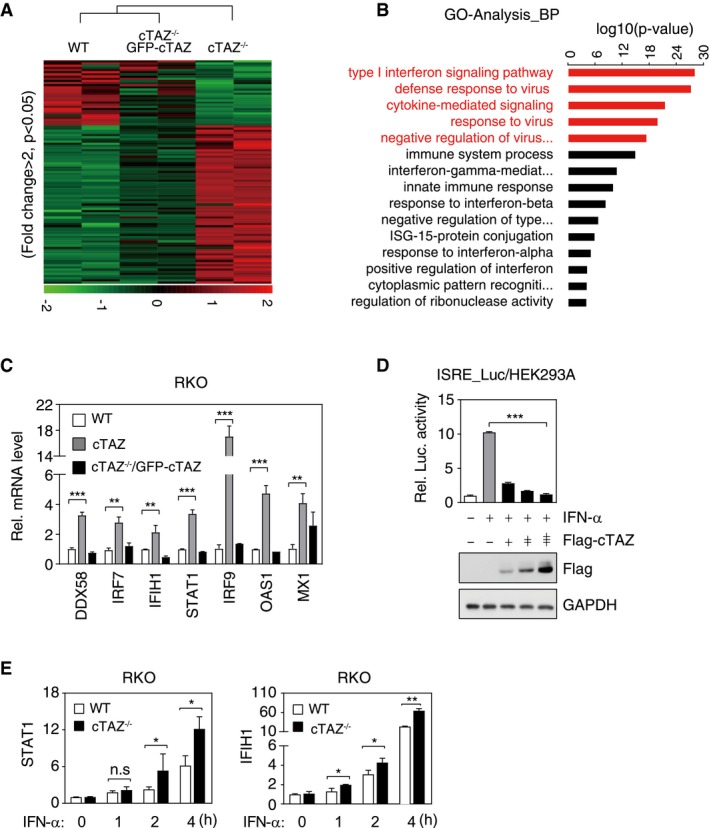
- Cluster analysis of RNA expression of WT, cTAZ−/−, and cTAZ−/−/GFP‐cTAZ put‐back RKO cells. Genes down‐ and up‐regulated more than twofold (P < 0.05) in cTAZ KO cells were included in cluster analysis. Green and red indicate down‐ and up‐regulated genes, respectively.
- Gene ontology (GO) analysis identified differentially regulated pathway by cTAZ.
- cTAZ suppressed expression of ISGs. RNA levels of selected ISGs in WT, cTAZ−/−, and cTAZ−/−/GFP‐cTAZ RKO cells were quantified by qPCR. Error bars indicate SD, n = 3. **P < 0.01; ***P < 0.001; Student's t‐test.
- cTAZ repressed 5×ISRE‐luciferase activity induced by IFN‐α. HEK293A cells were transfected with 5×ISRE‐luciferase reporter along with the indicated plasmids, treated with or without IFN‐α (50 ng/ml) for 12 h. The expression of ectopic gene was determined by IB. Error bars indicate SD, n = 3. ***P < 0.001; one‐way ANOVA test was used for statistical analysis.
- Knockout of cTAZ promoted ISG expression in the presence of IFN‐α. WT and cTAZ −/− RKO cells were treated with or without of IFN‐α (50 ng/ml) for indicated time, and expression of selected genes was quantified by qPCR. WT or cTAZ −/− group was normalized by basal level, respectively. Error bars indicate SD, n = 3. *P < 0.05; **P < 0.01; Student's t‐test.
Source data are available online for this figure.
Figure EV4. cTAZ directly repressed JAK‐STAT signaling.
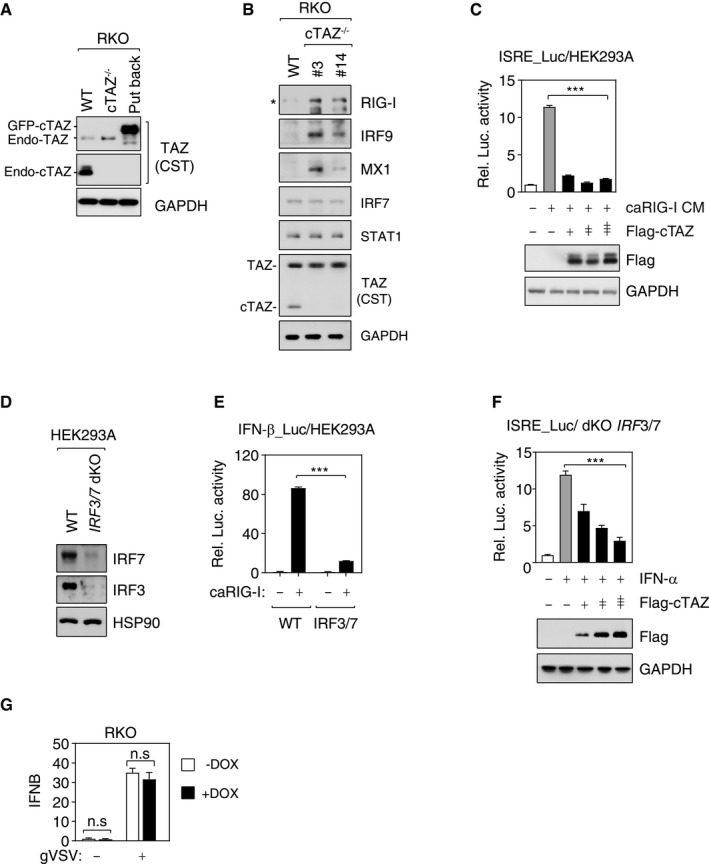
- cTAZ protein levels in cells used for RNA‐seq (Fig 2A).
- Depletion of cTAZ induced protein expression of several ISGs. WT RKO cells and cTAZ−/− clones (#3, #14) were used. The asterisk indicates position of RIG‐I.
- cTAZ repressed 5×ISRE‐luciferase activity. HEK293A cells were transfected with 5×ISRE‐luciferase reporter along with indicated plasmids, treated with or without conditional medium (CM) from caRIG‐I expressing cells for 12 h, and luciferase activity was measured. Protein levels were determined by IB. Error bars indicate SD, n = 3. ***P < 0.001; Student's t‐test.
- Knockout (pooled) of IRF3 and IRF7 (dKO) using CRISPR/Cas9 technology. The expression of IRF3/7 was assessed by IB.
- Knockout of IRF3/7 significantly downregulated IFN‐β‐promoter activity induced by caRIG‐1. Error bars indicate SD, n = 3. ***P < 0.001; Student's t‐test.
- cTAZ repressed 5×ISRE‐reporter activity induced by IFN‐α in IRF3/7 dKO cells. Error bars indicate SD, n = 3. ***P < 0.001; one‐way ANOVA test was used for statistical analysis.
- cTAZ did not affect IFNB mRNA expression. Control or cTAZ‐expressing (DOX induced) RKO cells were infected by gVSV for 12 h. IFNB expression was determined using qPCR. Error bars indicate SD, n = 3. Student's t‐test.
Source data are available online for this figure.
Type I IFNs activate JAK‐STAT signaling and promote ISG expression 34, 35. We then tested whether cTAZ could regulate the expression of ISGs induced by JAK‐STAT signaling. cTAZ expression dramatically repressed the activity of an ISRE reporter induced by IFN‐α (Fig 2D). cTAZ‐mediated repression of the ISRE reporter was similarly observed in cells treated with condition medium containing type I IFNs from cells expressing constitutively active RIG‐I (caRIG‐I, aa1–284 of RIG‐I 22, 36; Fig EV4C).
YAP/TAZ can block IRF3 activation and inhibit type I IFNs production 21, 22. IRF7 is a target gene of STAT1/2 and can also induce IFN production; thus, IRF3 and IRF7 may interfere the assay by regulating IFN levels. We generated IRF3/7‐double‐knockout (dKO) cells, which were significantly less competent to caRIG‐I in activating the ISRE reporter (Fig EV4D and E). However, the response of IRF3/7 dKO cells to IFN‐α was identical to that of wild‐type cells (Figs 2D and EV4F). More importantly, under IFN‐α stimulation, the expression of ISGs, such as IFIH1 and STAT1, was super‐induced when cTAZ was ablated (Fig 2E). In addition, the mRNA levels of IFNB in RKO cells were not significantly repressed when cTAZ was overexpressed (Fig EV4G). Taken together, these results suggest that, rather than modulating the production of IFNs, cTAZ can regulate the robustness of JAK‐STAT signaling directly.
cTAZ inhibits dimerization and nuclear translocation of phosphorylated STAT1/2
We next investigated the molecular mechanism by which cTAZ regulates JAK‐STAT signaling. Upon IFN stimulation, phosphorylation of STAT proteins (such as tyrosine 701 (Y701) of STAT1) by JAK kinases serves as a molecular switch to activate the pathway 37, 38. In wild‐type and cTAZ −/− RKO cells, no difference in Y701 phosphorylation of STAT1 protein was observed following IFN‐α treatment (Fig EV5A). Moreover, ectopic cTAZ failed to block the phosphorylation of other sites of STAT proteins (such as Y705 of STAT1 and Y694 of STAT5) induced by IFN‐α in HEK293A cells (Fig EV5B). Thus, cTAZ does not regulate STAT phosphorylation by their upstream kinases.
Figure EV5. cTAZ attenuated dimerization of STAT1/2.
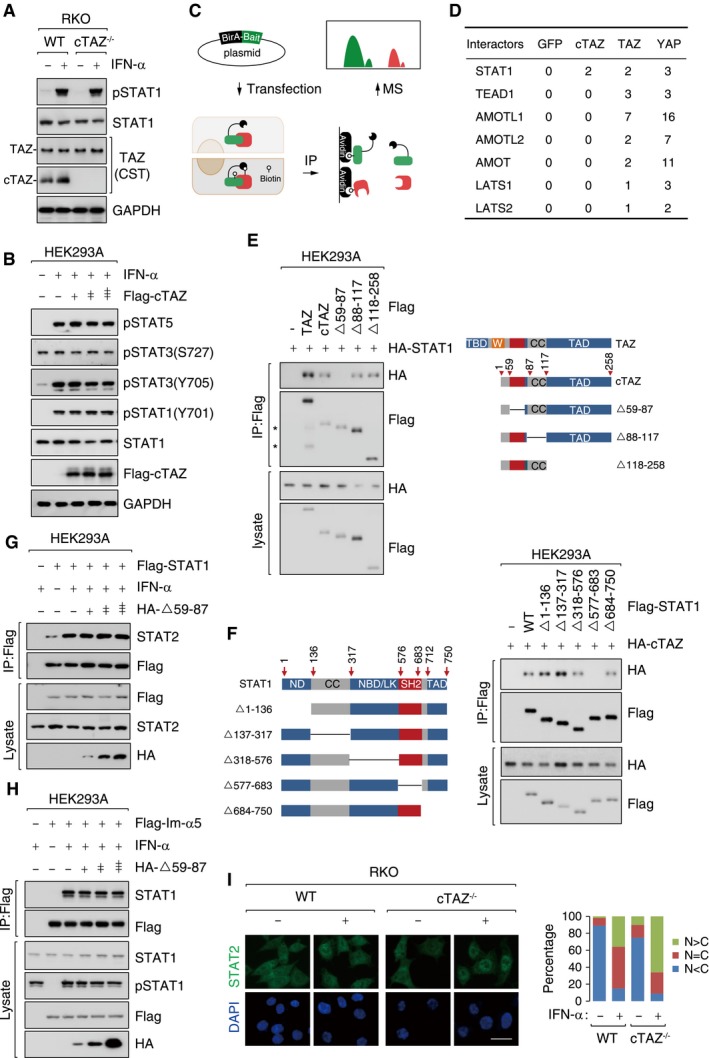
- Deletion of cTAZ had no effect on the phosphorylation of STAT1 induced by IFN‐α. WT and cTAZ−/− RKO cells were treated with IFN‐α (50 ng/ml) for 1 h.
- Ectopic cTAZ did not modulate the phosphorylation of STAT proteins induced by IFN‐α. HEK293A was transfected with Flag‐tagged cTAZ, and treated with or without IFN‐α (50 ng/ml) for 1 h.
- The flowchart of BioID assay. BirA*‐fused GFP, cTAZ, TAZ, or YAP was stably overexpressed in HEK293A cells and subjected to BioID assays.
- Potential interacting proteins of TAZ and/or cTAZ identified in BioID assay. The values were normalized PSM (peptide–spectrum match).
- The amino acids 59–87 of cTAZ were required for interaction with STAT1. A schematic representation of cTAZ deletion mutants used is shown on the left, CC: coiled‐coil domain; TAD: transcriptional activation domain. Transfection and IP assays were performed as in Fig 3A. Asterisks indicate degraded TAZ in lane 2.
- The CC domain of STAT1 was required for interaction with cTAZ. A schematic representation of STAT1 deletion mutants used is shown on the left, ND: N‐terminal Domain; DBD/LK: DNA binding domain/linker domain; SH2: SH2 domain; CC: coiled‐coil domain; TAD: transcriptional activation domain. Transfection and IP assays were performed as in Fig 3A.
- Amino acids 59–87‐deleted cTAZ failed to attenuate STAT1/2 dimerization induced by IFN‐α. HEK293A cells were transfected and stimulated by IFN‐α (50 ng/ml) for 1 h, and cells lysates were prepared for IP Flag antibody.
- Amino acids 59–87 deleted cTAZ failed to attenuate interaction between STAT1 and importin‐α5.
- Deletion of cTAZ promoted nuclear accumulation of STAT2 under IFN‐α stimulation. Same cells and treatment were used as in Fig 3E. Scale bar, 50 μm.
Source data are available online for this figure.
We then used the proximity‐dependent biotin identification (BioID) to interrogate the potential interactions between cTAZ and components of the JAK‐STAT pathway (Fig EV5C). The only hit of this screen was STAT1 (Fig EV5D and Dataset EV2). In a Co‐IP assay, ectopic cTAZ interacted with STAT1, and the interaction is increased upon IFN‐α treatment (Fig 3A). Following extensive mapping experiments, we found that amino acids 59–87 of cTAZ and the SH2 domain of STAT1 were essential for cTAZ–STAT1 interaction (Fig EV5E and F). The SH2 domain of STAT1/2 was critical for their homo‐ or heterodimerization 39. We speculated that cTAZ may perturb the interaction between STAT1 and STAT2. Indeed, ectopic cTAZ, but not amino acids 59–87‐deleted cTAZ (cTAZ▵59–87), abrogated STAT1/2 dimerization induced by IFN‐α (Figs 3B and EV5G). Moreover, knockout of endogenous cTAZ could promote STAT1/2 interaction stimulated by IFN‐α in RKO (Fig 3C).
Figure 3. cTAZ attenuates dimerization and nuclear translocation of STAT1/2.
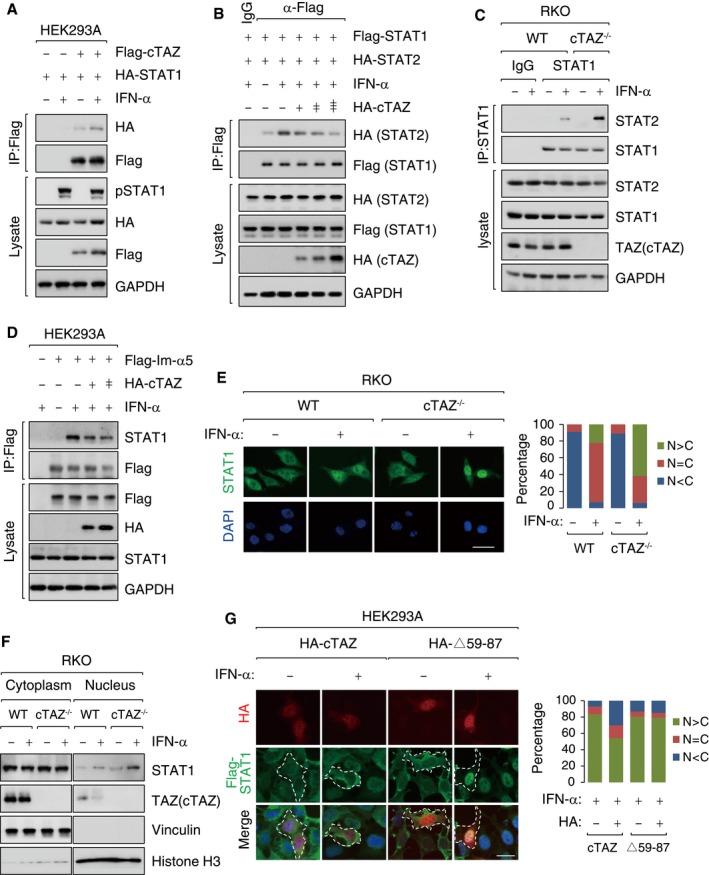
- STAT1 interacted with cTAZ, and the interaction was enhanced by IFN‐α. HEK293A cells were transfected with indicated plasmids, treated with or without IFN‐α (50 ng/ml) for 1 h. IP were performed using Flag antibody.
- cTAZ attenuated heterodimerization between STAT1 and STAT2 induced by IFN‐α. Following transfection, cells were treated with or without IFN‐α (50 ng/ml) for 1 h and subjected to IP assays using Flag antibody.
- Depletion of cTAZ enhanced the interaction between STAT1 and STAT2. RKO cell lines (WT and #14) were treated with IFN‐α as in (B). IP assays were performed using STAT1 antibody.
- cTAZ attenuated interaction between STAT1 and importin‐α5 (Im‐α5). Following transfection, HEK293A cells were stimulated using IFN‐α as in Fig 2B. IP assays were performed using Flag antibody.
- Deletion of cTAZ promoted nuclear accumulation of STAT1 upon IFN‐α stimulation. RKO (WT and cTAZ−/−) cells treated with or without IFN‐α (50 ng/ml) for 1 h, fixed, and subjected to IF using STAT1 antibody (green). DNA was labeled by DAPI (blue). Scale bar, 50 μm. Quantification was performed using ImageJ software (right). N>C: nuclear; N = C: nuclear plus cytoplasmic; N<C: cytoplasmic localization.
- Deletion of cTAZ promoted nuclear accumulation of STAT1 upon IFN‐α stimulation. Cells were treated as in (E) and subjected to subcellular fractionation.
- cTAZ defective in STAT1‐binding failed to block nuclear translocation of STAT1. HEK293A stably overexpressing STAT1 were transfected with the indicated plasmids, treated with or without IFN‐α (50 ng/ml) for 1 h, fixed, and subjected to IF as in (E). Scale bar, 25 μm.
Source data are available online for this figure.
The dimerization of phosphorylated STAT1/2 is essential for interaction with importin‐α5 and nuclear translocation 37, 38, 40, 41. We found that ectopic cTAZ, but not cTAZ▵59–87, attenuated the interaction between STAT1 and importin‐α5 in a dose‐dependent manner (Figs 3D and EV5H). In an IF assay, about 30% of WT cells and 50–60% of cTAZ–/– cells, respectively, showed strong nuclear STAT1/2 localization following IFN‐α stimulation (Figs 3E and EV5I). Nuclear enrichment of STAT1 in cTAZ‐knockout cells was also observed in a cell fractionation assay (Fig 3F). Moreover, overexpression of cTAZ, but not cTAZ▵59–87, blocked STAT1 nuclear entry upon IFN‐α treatment (Fig 3G). Taken together, cTAZ attenuates JAK‐STAT signaling by disrupting the dimerization of phosphorylated STAT1/2 and blocking their nuclear translocation.
cTAZ negatively regulates cellular antiviral response
A major function of type I IFN‐induced JAK‐STAT signaling is to limit viral infection 15. We subsequently tested whether cTAZ could regulate antiviral response. In HEK293A or HCT‐116 cells, overexpression of STAT1/2 efficiently blocked the replication of GFP‐labeled vesicular stomatitis virus (gVSV, an RNA virus), and cTAZ overexpression abolished the antiviral effect of STAT1/2 without affecting STAT1/2 expression (Fig 4A and B, and Appendix Fig S2A and B). Similar results were obtained using GFP‐labeled herpes simplex virus (gHSV, a DNA virus; Appendix Fig S2C). In contrast, cTAZ▵59–87, which was defective in binding to STAT1/2, failed to repress the antiviral effect of STAT1/2 (Appendix Fig S2D). We also found that ectopic expression of cTAZ could neutralize the antiviral activity of IFN‐α in a dose‐dependent manner (Fig 4C). To prove JAK‐STAT signaling pathway is required for the inhibitory function of cTAZ in cellular antiviral activity, we generated JAK1‐knockout cells (Appendix Fig S2E), and in these cells, ectopic cTAZ could not repress viral replication (Appendix Fig S2F).
Figure 4. cTAZ negatively regulates cellular antiviral response.
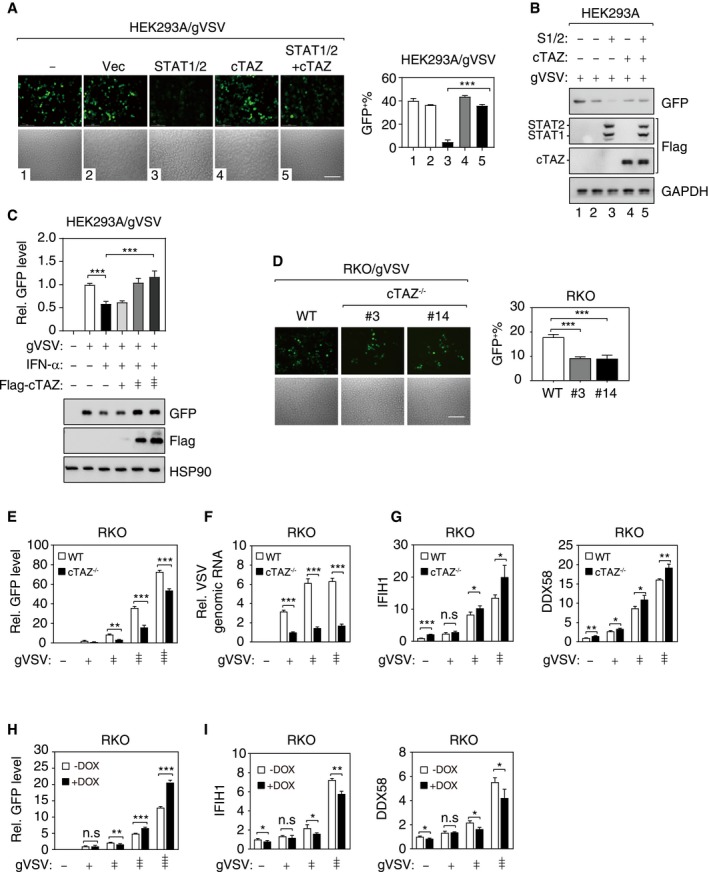
- cTAZ repressed the antiviral activity of ectopic STAT1/2. HEK293A cells were transfected with indicated plasmids, infected with gVSV (MOI = 0.01) for 12 h, fixed, and subjected to microscopy (Left). Viral infection and replication were marked by green fluorescence. Scale bar, 100 μm. (Right) Quantification of GFP‐positive cells. About 300 cells (from three different fields) were analyzed. Error bars indicate SD. **P < 0.01; ***P < 0.001; Student's t‐test.
- cTAZ and STAT1/2 expression levels of cells used in (A).
- cTAZ effectively repressed antiviral activity of IFN‐α. Transfection and infection were performed as in (A). Top: GFP intensity of cell lysates was measured by a fluorometer. Bottom: Viral GFP and ectopic cTAZ protein levels were detected by IB.
- Depletion of cTAZ repressed viral replication. RKO cell lines (WT and two independent cTAZ −/− clones) were infected with gVSV (MOI = 0.1) for 12 h. Scale bar, 100 μm. The ratio of GFP‐positive cells were calculated as in (A). About 300 cells (from three different fields) were analyzed. Error bars indicate SD. ***P < 0.001; Student's t‐test.
- Depletion of cTAZ repressed viral replication. Cells were treated as in (D), and GFP intensity of cell lysates was measured. Error bars indicate SD, n = 3. **P < 0.01; ***P < 0.001; Student's t‐test.
- Depletion of cTAZ repressed viral replication. Cells were treated as in (D), and the viral genomic RNA was quantified by qPCR (normalized to human GAPDH). Error bars indicate SD, n = 3. ***P < 0.001; Student's t‐test.
- cTAZ deficiency enhanced ISG expression. Cells were treated as in (E), and expression of IFIH1 and DDX58 was assessed by qPCR. Error bars indicate SD, n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; Student's t‐test.
- Overexpression of cTAZ promotes virus infection. DOX‐inducible cTAZ‐overexpressing RKO cell lines were pre‐treated with DOX for 48 h (1 μg/ml). gVSV infection and GFP fluorescence intensity quantification were performed as in (D). Error bars indicate SD, n = 3. **P < 0.01; ***P < 0.001; Student's t‐test.
- Overexpression of cTAZ repressed ISG expression. Cells were treated as in (H), and expression of IFIH1 and DDX58 was assessed by qPCR. Error bars indicate SD, n = 3. *P < 0.05; **P < 0.01; Student's t‐test.
Source data are available online for this figure.
We further investigated the inhibitory function of endogenous cTAZ in cellular antiviral activity. In cTAZ −/− RKO cells, the replication of gVSV and gHSV was significantly inhibited, as indicated by fewer GFP‐positive cells, lower intensity of GFP fluorescence, and/or lower viral genome copy number (Fig 4D–F, and Appendix Fig S2G and H). Meanwhile, the expression of ISGs was significantly increased in cTAZ −/− RKO following viral infection (gHSV and gVSV) (Fig 4G and Appendix Fig S2I).
We also established doxycycline (DOX)‐inducible cTAZ‐expressing cells, and observed that inducible cTAZ expression enhanced the replication of virus (gVSV and gHSV) and attenuated ISG expression (Fig 4H and I, and Appendix Fig S2J–L). Together, cTAZ negatively regulates cellular antiviral response by inhibiting JAK‐STAT signaling and expression of ISGs.
cTAZ regulates antiviral response in a Hippo signaling‐independent manner
The major difference between cTAZ and full‐length TAZ and YAP was their response to Hippo pathway kinases and upstream signals. Full‐length TAZ and YAP also interacted with STAT1 as shown in the BioID experiment (Fig EV5D), and they also had the motif (aa59–87 in cTAZ) responsible for STAT1 binding. We therefore tested the effect of full‐length TAZ and YAP on JAK‐STAT signaling and cellular antiviral response. In a Co‐IP assay, full‐length TAZ and YAP could interact with STAT1 (Appendix Fig S3A), indicating that full‐length TAZ and YAP could negatively regulate JAK‐STAT signaling, in a similar fashion as cTAZ did.
We also tested the effect of cTAZ and full‐length TAZ and YAP on the expression of ISGs and ISRE promoter activities under different cell densities and serum concentrations, two robust upstream signals of the Hippo pathway 25, 27. Interestingly, under low cell density or serum‐rich conditions, the effect of cTAZ and full‐length TAZ and YAP on the expression of ISGs and ISRE promoter activity was indistinguishable; however, under high cell density and serum starvation conditions, cTAZ remained effective whereas full‐length TAZ and YAP were unable to repress ISG expression or ISRE promoter activity (Appendix Fig S3B and C). Moreover, the interaction between full‐length TAZ and STAT1 was reduced under serum starvation, whereas cTAZ and STAT1 interaction was not sensitive to serum (Appendix Fig S3D). The effect of full‐length YAP toward ISG expression in a Hippo signaling‐dependent manner was consistent with a previous report 22. These results suggested that, compared to full‐length TAZ and YAP, cTAZ could regulate antiviral response under diverse conditions in a Hippo signaling‐independent manner.
In 2017, two groups reported a role of YAP (and TAZ) in antiviral response 21, 22. Zhang et al 22 demonstrated that YAP prevented TBK1 binding to STING/MAVS and blocked virus‐induced TBK1 activation. Wang et al 21 reported that YAP, independent of its transcriptional activity, blocked the dimerization of the transcription factor IRF3 to impede nuclear translocation following viral infection. Although different mechanisms were proposed, both studies reached a same conclusion that YAP blocked virus‐induced production of type I IFNs and impaired host antiviral response. Our results indicate that cTAZ also represses antiviral response, but not by limiting type I IFN production and instead by inhibiting JAK‐STAT signaling. Furthermore, both full‐length TAZ and YAP could interact with STAT1 and limit antiviral response in sparse culture and serum‐rich conditions; on the other hand, the effect of cTAZ was independent on Hippo signaling (Appendix Fig S3). We propose that, cTAZ and full‐length TAZ and YAP may limit antiviral response via multiple mechanisms, including the production and downstream signaling of type I IFNs. Moreover, compare to full‐length TAZ and YAP, cTAZ may regulate antiviral response under broader situations.
As oncoproteins, YAP/TAZ can profoundly promote cell proliferation and inhibit cell differentiation. Should the expression and activity of YAP/TAZ be regulated by type I IFNs, the fate of the responding cells may undergo dramatic change upon infection. Conceivably, cTAZ does not participate in the canonical Hippo pathway, and can function as a specialized regulator for the JAK‐STAT signaling pathway during antiviral response.
cTAZ expression is induced by JAK‐STAT signaling
The IFN response mediated by the JAK‐STAT signaling pathway is tightly controlled by negative regulators, such as SOCS1 and USP18 5, 42, 43. We noticed that gVSV infection induced expression of cTAZ protein (Fig 5A), indicating cTAZ as a target gene of JAK‐STAT signaling. When cells were treated with IFN‐α, both cTAZ transcript and protein were induced in a dose‐dependent manner; in contrast, TAZ was not regulated by IFN‐α (Fig 5B and C, and Appendix Fig S4A and B). Moreover, a luciferase reporter of cTAZ promoter activity (~2,000 bp upstream of the translation starting site) was activated by ectopic caRIG‐I or STAT1/2 (Fig 5D and Appendix Fig S4C), and a shorter promoter (~1,000 bp upstream of the translation starting site) similarly responded to overexpression of caRIG‐I or STAT proteins (Appendix Fig S4C and D). Using the JASPAR software, we identified three STAT1 (−999, −515, and −75) and three STAT1/2 (−724, −666, and −648) binding sites in the cTAZ promoter (Fig 5E). Subsequent mutagenesis experiments revealed that three STAT1/2‐binding sites, especially the one near −634 position, were required for STAT‐induced promoter activation (Fig 5E). In a chromatin immunoprecipitation (ChIP) assay using anti‐STAT1 antibody, promoters of cTAZ and ISG15 were pulled down in an IFN‐α‐sensitive manner, indicating that STAT1 was recruited to the cTAZ and ISG15 promoters upon IFN‐α stimulation (Fig 5F). The levels of acetylated lysine 27 of histone H3 (H3K27ac), an epigenetic mark of active promoter, around cTAZ and ISG15 promoters were not modulated by IFN‐α (Appendix Fig S4E). Although H3K27ac levels were not modulated by IFN stimulation, it might facilitate the recruitment of STAT proteins to target promoters. Taken together, the expression of cTAZ is directly induced by JAK‐STAT signaling to negatively regulate JAK‐STAT signaling.
Figure 5. cTAZ expression was induced by JAK‐STAT signaling.
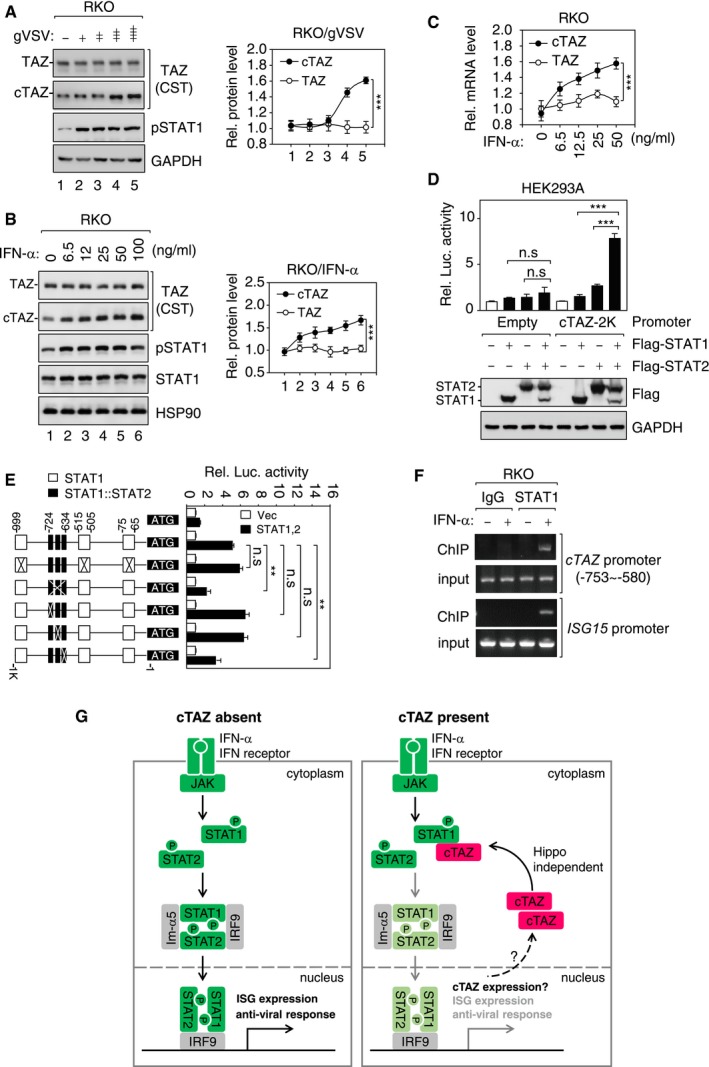
- gVSV induced expression of cTAZ. RKO cells were infected with gVSV (MOI = 0, 0.001, 0.01, 0.01, 1) for 8 h. Protein expression was determined by IB (Left) and quantified (Right). Error bars indicate SD, n = 3. ***P < 0.001; two‐way ANOVA test was used for statistical analysis.
- IFN‐α induced expression of cTAZ. RKO cells were treated with different doses of IFN‐α for 8 h. Protein expression was determined as in (A). Error bars indicate SD, n = 3. ***P < 0.001; two‐way ANOVA test was used for statistical analysis.
- IFN‐α induced mRNA level of cTAZ. RKO cells were treated with different doses of IFN‐α for 8 h. RNA levels were measured by qPCR. Error bars indicate SD, n = 3. ***P < 0.001; two‐way ANOVA test was used for statistical analysis.
- STAT1/2 synergistically activated a cTAZ promoter. HEK293A cells were transfected with cTAZ reporter with or without STAT proteins. Promoter activity was determined using luciferase assay, and protein expression was determined by IB. Error bars indicate SD, n = 3. ***P < 0.001; Student's t‐test.
- STAT1/2 binding sites on cTAZ promoter. HEK293A cells were transfected with cTAZ promoters (wild type or mutants). Luciferase activity and responses to STAT1/2 expression were measured. Error bars indicate SD, n = 3. **P < 0.01; Student's t‐test.
- STAT1 occupied cTAZ promoter in an IFN‐α‐sensitive manner. RKO cells were treated with or without IFN‐α (50 ng/ml) for 1 h and subjected to ChIP assays. cTAZ PCR primers were designed to target STAT1/2 binding site. ISG15 promoter was included as a positive control.
- A negative feedback mechanism regulating JAK‐STAT signaling involving cTAZ. Upon IFN‐α stimulation, STAT1/2 are phosphorylated and undergo dimerization. STAT1/2 dimer interacts with importins (such as Im‐α5), enters nucleus, and activates transcription of target genes including ISGs. The expression of cTAZ is induced by STAT1/2. Elevated cTAZ blocks the dimerization and nuclear translocation of STAT1/2, which in turn leads to repression of the JAK‐STAT signaling. cTAZ represents a critical node of a negative feedback mechanism regulating the JAK‐STAT signaling.
Source data are available online for this figure.
In summary, this study has identified a new transcript variant cTAZ and revealed its function in modulating the JAK‐STAT signaling. In the absence of cTAZ, JAK‐STAT signaling can effectively induce expression of ISGs and boost cellular antiviral response. On the other hand, in the presence of cTAZ, it will repress the nuclear translocation of STAT proteins by inhibiting the dimerization of phosphorylated STAT1/2 and their subsequent interaction with importin‐α5, which in turn lead to reduced expression of ISGs and antiviral response (Fig 5G). The expression of cTAZ is also slightly induced by JAK‐STAT signaling, and elevated cTAZ expression may keep STAT1/2 activity in check and prevents uncontrolled expression of ISGs and subsequent detrimental effect (Fig 5G). In light of the findings in this study, we envisage that the impact of cTAZ‐mediated regulation of JAK‐STAT signaling goes beyond antiviral responses. For instance, cTAZ expression may confer resistance to IFN therapy for certain types of cancer 1, 2, 4. Conversely, defects in cTAZ expression and function may contribute to hypersensitivity to IFNs and autoimmune diseases.
Materials and Methods
Antibodies, plasmids, and other materials
The information about vendor and catalog number of antibodies used in this study is shown in Appendix Table S3.
TAZ, cTAZ, YAP, STAT1/2, and MAVS were cloned into pLVX vector. 8×GTIIC‐luciferase reporter and cTAZ luciferase reporter were in‐house constructed. IFN‐β luciferase reporter, 5×ISRE‐luciferase reporter, and caRIG‐I were described in 22. The shRNA plasmids (YAP, TRCN0000300325; TAZ, TRCN0000370007) were from Sigma‐Aldrich.
IFN‐α was purchased from Sino Biological. Primers and oligos (see Appendix Table S4 for sequence information) were synthesized by Huajin Biotech, China.
C57BL/6 wild‐type male mice (4 weeks old) were dissected to obtain tissues, proteins, and mRNA. The use of mice for this study was approved by Ethic Committee of Institutes of Biomedical Sciences, Fudan University.
Patient specimens
Lymph nodes near thyroid were collected during surgery, from 11 thyroid cancer patients who were treated at Fudan University Shanghai Cancer Center. Tissue samples were immediately frozen and stored at −80°C for protein extraction, or stored in RNAlater at 4°C for 24 h and then transferred to −80°C for RNA extraction. Following pathological examination, lymph nodes without cancer metastasis were used in this study. This study was approved by the Ethical Committee of Fudan University Shanghai Cancer Center (050432‐4‐1212B), and informed consent was obtained from all patients.
Cell culture, transfection, and lentivirus transduction
RKO cells were cultured in RPMI‐1640 (Hyclone) containing 10% FBS (Gibco) and 50 mg/ml penicillin/streptomycin (P/S). MCF‐10A cells were cultured in DMEM/F12 (Hyclone) supplemented with 5% horse serum (Invitrogen), 0.5 mg/ml hydrocortisone, 20 ng/ml EGF, 10 mg/ml insulin, 100 ng/ml cholera toxin, and 50 mg/ml P/S. Other type cell lines, such as HEK293A, Caco2, ACHN, NCI‐H2372, and NCI‐H2452, were cultured in DMEM (Hyclone) containing 10% FBS and P/S. All cells were incubated at 37°C under 5% CO2.
DNA transfection using PolyJet (SignaGen) was performed according to manufacturer's instructions. Lentivirus was produced by co‐transfecting HEK293A cells with viral vectors and packaging plasmids (psPAX.2 and pMD2.G). After 48 h of transfection, lentivirus supernatant was filtered through a 0.45‐μm filter, followed by infecting cells together with 5 mg/ml polybrene. One day after infection, the medium was replaced with fresh culture medium containing 1 mg/ml puromycin (Invivogen).
Immunoblotting
Cells were lysed in 1× SDS loading buffer containing 50 mM Tris pH 6.8, 2% SDS, 0.025% bromophenol blue, 10% glycerol, and 5% BME. Proteins were resolved by 7.5–10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to PVDF membrane. Membranes were blocked with 5% BSA/TBST or skim milk, incubated with different primary antibodies (4°C, overnight) and HRP‐conjugated secondary antibodies. Molecular weight marker, ECL substrates, and image acquisition equipment (5200S) were from Tanon Science & Technology Co., Ltd. If required, ImageJ software was used to facilitate quantification.
Immunofluorescence (IF)
Cells were seeded on fibronectin‐coated coverslips, incubated, and treated with condition medium, IFN‐α, or virus. Cells were washed with cold PBS, fixed with 4% paraformaldehyde for 15 min, and permeabilized with 0.2% Triton X‐100 at room temperature (RT). After blocking in 3% BSA or 3% goat serum, cells were incubated with first antibodies diluted in 3% BSA for 1.5 h at room temperature or overnight at 4°C. Next, cells were incubated with Alexa Fluor 488‐ or Alexa Fluor 594‐conjugated secondary antibodies (1:1,000 diluted) for 1–2 h, washed, mounted, and subjected to microscopy.
Immunoprecipitation (IP)
Cells were lysed using a mild lysis buffer (MLB) containing 20 mM Tris, 2 mM EDTA, 100 mM NaCl, 1% NP‐40, 50 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail (Biotool). Cleared cell lysates were then subjected to IP using various antibodies for 2–4 h. After 3–4 washes with MLB, the proteins were eluted by 1× SDS loading buffer, resolved by SDS–PAGE, and subjected to IB.
GFP fluorescence intensity measurement
Cells were lysed using MLB as mentioned in IP assay, and the fluorescence intensity of cleared lysates was determined by BioTek microplate spectrophotometer at excitation wavelength (485/20) and emission wavelength (528/20).
RNA isolation and qPCR
Total RNA was extracted using the TRIzol reagent (Invitrogen). cDNA was generated by the PrimeScript™ RT Reagent Kit (TaKaRa), and quantitative qPCR was conducted using SYBR Green qPCR Master Mix (TaKaRa) on 7500 Real‐Time PCR systems (Applied Biosystems). Relative abundance of mRNA was calculated by normalization to GAPDH mRNA.
Luciferase reporter assay
HEK293A cells were transfected with the indicated reporters (100 ng), control Renilla luciferase reporter (50 ng), and other expression plasmids. After 12 h of transfection, cells were treated with condition medium or IFN for 24 h. Luciferase activity was determined by using a Dual‐Luciferase Assay Kit (Promega). Firefly luciferase activity was measured and normalized with co‐transfected Renilla luciferase activity.
Soft agar colony formation assay
Agar (1.0%) and 2× DMEM (or RPMI‐1640) media were mixed at > 42°C and placed onto 3.5‐cm dishes to generate 0.5% base agar. MCF‐10A cells (vector, Flag‐cTAZ, Flag‐TAZ) were seeded in 0.35% top agar (1,000 cells per plate for MCF‐10A). Cells were incubated at 37°C with fresh medium replenished regularly for 3 weeks. Cell colonies were counted after staining with crystal violet.
RNA‐seq data analysis
Total RNA was extracted from RKO cells (WT, cTAZ−/−, and cTAZ−/− GFP‐cTAZ) using RNeasy Mini Kit (Qiagen). The sequencing libraries were constructed using the TruSeq RNA Sample Prep Kit (Illumina) according to the manufacturer's protocol. Sequencing was performed on an Illumina HiSeq 2500 platform. The differentially expressed genes were identified using DESeq2 44. Sequencing reads from RNA‐seq data were aligned using the spliced read aligner HISAT2, which was supplied with the Ensembl human genome assembly (Genome Reference Consortium GRCh38) as the reference genome. Gene expression levels (read counts) were calculated using featureCount. All read count data were provided as input to DESeq2 with default parameters. The differentially expressed genes were extracted with fold change ≥ 2 and P adj < 0.05. The Database for Annotation, Visualization, and Integrated Discovery (DAVID) was employed to explore GO ontologies with the differentially expressed genes. Differentially expressed genes are shown in Dataset EV1.
BioID
MCF‐10A cells stably express promiscuous biotin ligase (BirA*) tagged GFP, cTAZ, TAZ, or YAP were established. BioID was performed following a published protocol 45. Briefly, cells (in a 10‐cm dish) were treated with biotin (50 μM) for 24 h, washed with PBS twice, and lysed in 600 μl of lysis buffer (50 mM Tris, pH 7.4, 500 mM NaCl, 0.2% SDS, and protease inhibitor). Detergent (20% Triton X‐100, 240 μl) was added, and the total lysate was then subjected to sonication to disrupt visible aggregates. The lysate was centrifuged for 10 min at 16,500 × g at 4°C, and the supernatants were incubated with appropriate amount of streptavidin beads (Thermo) overnight at 4°C. Beads were washed six times with wash buffer. 10% of beads was saved for IB, and the rest was resuspended gently in 50 μl of 50 mM ammonium bicarbonate buffer and subjected for mass spectrometry analysis. Interacting proteins identified are shown in Dataset EV2.
Analysis of public datasets
RNA‐seq data were downloaded from the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra), and Galaxy was used for transcript assembly (https://usegalaxy.org/). ENCODE ChIP‐seq data (HCT‐116 and MCF‐7 cells) for H3K9ac, H3K27ac, H3K4me1, H3K4me2, and H3K4me3 were mapped to hg19 and visualized using UCSC Genome Browser. Expression datasets for cTAZ and TAZ transcripts in tissues were downloaded from UCSC xena (https://xenabrowser.net/datapages/), and TOIL RSEM expected_count datasets were used.
ChIP assay
Chromatin immunoprecipitation (ChIP) assays were performed according to the public protocol 46. In brief, RKO cells were fixed with 1% formaldehyde for 15 min at room temperature and quenched by adding 1 M glycine (at final concentration of 125 mM) for 15 min. The fixed cells were washed twice with PBS and then were lysed, and chromatin was sonicated into ~500‐base pair fragments. IP was performed overnight at 4°C with 2 μg anti‐STAT1, anti‐H3K27ac or equal amount of mouse IgG for negative control. The second day, protein‐G agarose beads were added to the lysates and incubated for 2 h at 4°C. Then, the beads were washed sequentially with low‐salt buffer, high‐salt buffer, LiCl buffer, and twice with TE buffer, and DNA was then eluted using 200 μl of fresh elution buffer. The elutes were reverse‐cross‐linked using 5 M NaCl at 65°C for 4 h and followed by treatment with RNase A at 37°C for 30 min and proteinase K at 45°C for 1 h to remove RNA and protein, respectively. DNA was extracted using a PCR Purification Kit (Tiangen).
Statistical analysis
All experiments subjected to statistical test were repeated for three times. Statistical analyses were performed using software GraphPad Prism7. Error bars indicate standard deviation (SD); unless otherwise indicated, three independent experimental data were used for Student's t‐test. P < 0.05 was considered as statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001).
Author contributions
Conceptualization: CF and F‐XY; methodology: CF, JianL, SQ, YL, YZe, PY, ZH, JinL, CD, and SH; formal analysis: CF, SH, CD, KC, and F‐XY; investigation: CF, SH, and F‐XY; resources: SH, CD, YZh, YW, RD, PX, KC, and F‐XY; writing of the original draft: CF; review and editing of the manuscript: CF, KC, and F‐XY; supervision: F‐XY; and funding acquisition: F‐XY.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We would like to thank Drs. Justin Wong and Renhua Song (The University of Sydney) for analyzing public datasets, Licheng Sun for technical assistance, and Dr. Steven Plouffe for proofreading of this manuscript. F.X.Y. would like to gratefully acknowledge financial support from the National Natural Science Foundation of China (81622038, 81772965, and 31571479) and Shanghai Municipal Commission of Health and Family Planning (2017BR018).
EMBO Reports (2019) 20: e47227
See also: S Strano & G Blandino (June 2019)
Data availability
RNA‐seq data are available at GEO database (accession number: GSE128257, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128257). Other datasets are reported in Datasets EV1 and EV2 and the Appendix Tables S1–S4.
References
- 1. Hertzog PJ, Williams BR (2013) Fine tuning type I interferon responses. Cytokine Growth Factor Rev 24: 217–225 [DOI] [PubMed] [Google Scholar]
- 2. Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, McGovern N, Cant AJ, Hackett SJ, Ghazal P et al (2013) STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA 110: 3053–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, Tough DF (2006) Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol 176: 2074–2078 [DOI] [PubMed] [Google Scholar]
- 4. Fuchs SY (2013) Hope and fear for interferon: the receptor‐centric outlook on the future of interferon therapy. J Interferon Cytokine Res 33: 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ronnblom L (2011) The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups J Med Sci 116: 227–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bach EA, Aguet M, Schreiber RD (1997) The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 15: 563–591 [DOI] [PubMed] [Google Scholar]
- 7. Veals SA, Schindler C, Leonard D, Fu XY, Aebersold R, Darnell JE Jr, Levy DE (1992) Subunit of an alpha‐interferon‐responsive transcription factor is related to interferon regulatory factor and Myb families of DNA‐binding proteins. Mol Cell Biol 12: 3315–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schindler C, Fu XY, Improta T, Aebersold R, Darnell JE Jr (1992) Proteins of transcription factor ISGF‐3: one gene encodes the 91‐and 84‐kDa ISGF‐3 proteins that are activated by interferon alpha. Proc Natl Acad Sci USA 89: 7836–7839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Honda K, Takaoka A, Taniguchi T (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25: 349–360 [DOI] [PubMed] [Google Scholar]
- 10. Iwasaki A (2012) A virological view of innate immune recognition. Annu Rev Microbiol 66: 177–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gonzalez‐Navajas JM, Lee J, David M, Raz E (2012) Immunomodulatory functions of type I interferons. Nat Rev Immunol 12: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawai T, Akira S (2011) Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650 [DOI] [PubMed] [Google Scholar]
- 13. Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N et al (2005) IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 434: 772–777 [DOI] [PubMed] [Google Scholar]
- 15. Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14: 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson R, Halder G (2014) The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 13: 63–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pan D (2010) The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piccolo S, Dupont S, Cordenonsi M (2014) The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94: 1287–1312 [DOI] [PubMed] [Google Scholar]
- 19. Yu FX, Zhao B, Guan KL (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu B, Zheng Y, Yin F, Yu J, Silverman N, Pan D (2016) Toll receptor‐mediated hippo signaling controls innate immunity in Drosophila . Cell 164: 406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T, Gao L, Wang L, Ling L, Jia J et al (2017) YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKɛ‐mediated phosphorylation. Nat Immunol 18: 1270 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S, Li X, Zhou R, Wang J, Zhao B et al (2017) Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ‐mediated TBK1 blockade. Nat Cell Biol 19: 362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geng J, Yu S, Zhao H, Sun X, Li X, Wang P, Xiong X, Hong L, Xie C, Gao J et al (2017) The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat Immunol 19: 1036 [DOI] [PubMed] [Google Scholar]
- 24. Ni X, Tao J, Barbi J, Chen Q, Park BV, Li Z, Zhang N, Lebid A, Ramaswamy A, Wei P et al (2018) YAP is essential for Treg‐mediated suppression of antitumor immunity. Cancer Discov 8: 1026–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L et al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183 [DOI] [PubMed] [Google Scholar]
- 27. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H et al (2012) Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell 150: 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta‐TRCP). Genes Dev 24: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL (2008) TAZ promotes cell proliferation and epithelial‐mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 28: 2426–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, Zeng Q, Hong W (2008) A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res 68: 2592–2598 [DOI] [PubMed] [Google Scholar]
- 31. Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, Xiong Y, Lei QY, Guan KL (2009) TEAD transcription factors mediate the function of TAZ in cell growth and epithelial‐mesenchymal transition. J Biol Chem 284: 13355–13362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472: 481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, Jiang Y, Wang C, Cao X (2017) Methyltransferase SETD2‐mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 170: 492–506.e14 [DOI] [PubMed] [Google Scholar]
- 34. Stark GR, Darnell JE Jr (2012) The JAK‐STAT pathway at twenty. Immunity 36: 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A (2015) The JAK‐STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 66: 311–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]
- 37. Reich NC, Liu L (2006) Tracking STAT nuclear traffic. Nat Rev Immunol 6: 602–612 [DOI] [PubMed] [Google Scholar]
- 38. Reich NC (2013) STATs get their move on. JAKSTAT 2: e27080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE Jr, Kuriyan J (1998) Crystal structure of a tyrosine phosphorylated STAT‐1 dimer bound to DNA. Cell 93: 827–839 [DOI] [PubMed] [Google Scholar]
- 40. McBride KM, Banninger G, McDonald C, Reich NC (2002) Regulated nuclear import of the STAT1 transcription factor by direct binding of importin‐alpha. EMBO J 21: 1754–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fagerlund R, Melen K, Kinnunen L, Julkunen I (2002) Arginine/lysine‐rich nuclear localization signals mediate interactions between dimeric STATs and importin alpha 5. J Biol Chem 277: 30072–30078 [DOI] [PubMed] [Google Scholar]
- 42. Hong XX, Carmichael GG (2013) Innate immunity in pluripotent human cells: attenuated response to interferon‐beta. J Biol Chem 288: 16196–16205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou L, Little MT, Malakhova OA, Sipe JC, Orkin SH, Zhang DE (2002) Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev 16: 2207–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roux KJ (2013) Marked by association: techniques for proximity‐dependent labeling of proteins in eukaryotic cells. Cell Mol Life Sci 70: 3657–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li XY, Biggin MD (2012) Genome‐wide in vivo cross‐linking of sequence‐specific transcription factors. Methods Mol Biol 809: 3–26 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
RNA‐seq data are available at GEO database (accession number: GSE128257, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128257). Other datasets are reported in Datasets EV1 and EV2 and the Appendix Tables S1–S4.