

Abstract
Subchondral bone remodelling is an integral part of osteoarthritis and involves the development of subchondral sclerosis seen on plain imaging, along with osteophyte formation.
The development of these changes is due to persistent abnormal mechanical stresses which create a cellular and biomolecular response to microfractures in the subchondral bone and osteochondral junction.
An early sign is bone marrow lesions seen on MRI scanning. Healing can occur at this stage by correcting the abnormal loads. Persistence leads to what is thought to be a delayed union or nonunion response by the bone.
Microfractures of the osteochondral junction, coupled with articular cartilage fissuring and loss, allows synovial fluid to penetrate the subchondral bone along with cytokines and other molecules reacting with the bone cells to increase the pathological effects.
This review gives an overview of the current thoughts on subchondral bone remodelling in osteoarthritis that is aimed at orthopaedic surgeons to help in the understanding of the pathogenesis of osteoarthritis and the role of surgical management.
Cite this article: EFORT Open Rev 2019;4 DOI: 10.1302/2058-5241.4.180102
Keywords: subchondral, bone remodelling, osteoarthritis
Introduction
Subchondral bone is the bedrock of a joint on which sits the articular cartilage. Traditionally, osteoarthritis (OA) has been considered to be wear and tear of articular cartilage, but more recent evidence has shown that subchondral bone disturbance and synovial inflammation can initiate and lead to disease progression.1 OA is characterised as ‘a multi-disease with inflammation, immune and central nervous system dysfunction playing central roles in whole joint damage, injury progression, pain and disability’.2 It is distinguished from rheumatoid arthritis by the hypertrophic changes in the subchondral bone seen on plain radiographs. The pathognomonic signs of OA on plain radiographs are joint space narrowing, osteophytes, subchondral sclerosis and subchondral cysts. The subchondral sclerosis is due to the thickening of the subchondral bone. The osteochondral junction is the transition between soft and hard tissues and so is critical in absorbing the stresses during joint loading. Abnormal loading leads to microfractures within the osteochondral junction and within the subchondral bone. The advent of MRI scanning has shown bone marrow lesions within the subchondral bone, associated with degenerative changes in the joint. The subchondral signals are now termed ‘bone marrow lesions’ (previously bone marrow oedema) and do not just reflect increased vascularity. The histological features change over the course of OA progression; increased vascularity occurs early. Increased radiological bone density (sclerosis) is thought to be a later finding when changes have become permanent and drug therapies aimed at reversing the disorder are not possible.3 Besides the altered vascularity, the bone marrow lesions also show subchondral microdamage.4 With increased subchondral sclerosis, there is a reduction in the flexibility of the osteochondral junction and a reduction in the density of the underlying subchondral bone.5
Articular cartilage degeneration itself comes from a number of different molecular pathways6 that include matrix metalloproteinases (MMP-1, MMP-13),7 complement component-5,8 hypoxia-inducible factor 2α (HIF-2α)9,10 and inhibition of TGF-β signalling.11 ‘Studies aimed at elucidating the pathophysiological roles of these enzymes in cartilage will contribute to our understanding of OA pathogenesis and enable design of targeted inhibitors that effectively target metalloproteinase-mediated cartilage degradation while sparing cartilage repair pathways’.12 These enzymes are produced by the cells of the tissues that make up the joint, in particular the synovial membrane. As OA progresses, the abnormal signals from one cell can lead to responses from other cell types and drive the pathological process. The osteochondral junction (‘tidemark’) is normally a barrier to this type of ‘cross-talk’. Loss of its integrity therefore is critical in the progression of OA.
This review explores our current understanding of subchondral bone in OA. It cannot be treated in isolation as all tissues that make up a joint are altered and interplay in the process. It should also be remembered that OA in humans is a clinical diagnosis and much of the biochemical pathways, especially early in the disorder, come from animal studies and may not translate into humans. The pathognomonic feature of OA in humans is pain. This cannot be seen on imaging and is not reflected in biochemical pathways.
The normal osteochondral junction
Anatomy of the osteochondral junction
The osteochondral junction is the tissue layer between the deep layers of the articular cartilage and the underlying subchondral bone (Fig. 1). It comprises the non-calcified deeper articular cartilage, the tidemark, the calcified layer, the cement line and the subchondral bone plate.5 On histological sections, the layer between the deep articular cartilage and the calcified layer appears as a basophilic line and is called the ‘tidemark’. The cement line is the junction between bone and the calcified zone of the cartilage. The epiphyseal bone contains sensory nerves and blood vessels. In the normal adult, the osteochondral junction seals these off from the deep layer of articular cartilage. The absence of sensory nerve endings in articular cartilage means that, when loaded, there is no experience of pain. However, it is thought by some that nutrition of the deep articular cartilage comes from the subchondral bone, rather than the synovial fluid, which nourishes the superficial layers.
Fig. 1.

Diagram showing the structure of the osteochondral junction
Function of subchondral bone and the osteochondral junction
Subchondral bone acts as the mechanical support of the joint linking to the diaphyseal bone. The length of the diaphyseal bone allows an increase in the lever arm of appropriate muscles to improve the power of the joint’s action. The bone contains cells, haemopoietic tissue, and a nerve and blood supply.
The osteochondral junction has three functions:
1) It is the transitional zone between the soft articular cartilage and the hard subchondral bone and therefore has mechanical properties that are important;1
2) The vertical portions of the arcades of collagen type II pass through it, anchoring the articular cartilage to the subchondral bone (Fig. 1);
3) Cytokines pass across it at a molecular level, creating ‘cross-talk’ between the chondrocytes and bone cells.
Changes at the osteochondral junction associated with OA
There is a direct correlation between the loss of articular cartilage and the expansion of the subchondral bone. Structural, biochemical and biomechanical changes occur at the osteochondral junction in association with OA. These changes can be viewed in two ways:5
compromise of the barrier between the synovium and the subchondral bone; and
osteochondral plasticity with responses from the adjacent structures, i.e. the synovial membrane, cartilage and bone, forming new tissue with different morphological and functional characteristics.
Bone marrow lesions and subchondral bone remodelling
The earliest signs of OA in the subchondral bone are bone marrow lesions (BML; excessive water signals in bone on MRI) seen on MRI scanning.13 They are thought to result from remodelling of the subchondral bone due to mechanical overload. If the abnormal loading continues, then the BMLs persist; there is an associated loss of articular cartilage and symptomatic OA develops (Fig. 2). In the subchondral tissue, the amount of bone increases but the mineral density is reduced. This is seen radiographically as osteosclerosis where there are fewer, but thicker trabeculae14 (Fig. 3). The loss of mineralisation and increased bone volume lowers the tissue stiffness (Young’s modulus) but increases the structural stiffness.
Fig. 2.

Proposed scheme for the development of bone marrow lesions and progression of osteoarthritis (based on data from Hügel and Guerts13)
Fig. 3.

Matched Magnetic Resonance (MR) and histology images at (top row) medial and (bottom row) lateral tibial plateau. Note the area of homogeneous low signal on MR (white arrowheads) corresponds to an area of trabecular thickening on histology (black arrowheads). (reproduced from Mackay et al,14 with permission from Elsevier)
Histology of bone marrow lesions
In a systematic review of the histology of bone marrow lesions, Loef et al3 found 13 studies of OA, of which eight used pre-operative MRI scans and three correlated these with tissue samples. Three investigated bone remodelling using micro-computed tomography.
Signs of bone remodelling were demonstrated by an increase in bone density, thickening of the subchondral plate and an increase in trabecular number, volume and thickness when compared with non-BML samples. A ‘structural model index’ that reflected the trabecular shape was found to be increased in BML samples in one article but decreased in two others.
The histological findings were:
1) Cell death
a) Bone necrosis (empty lacunae suggesting lack of osteocytes) (one study)
b) Swollen or disintegrated adipose tissue cells (three studies), fat necrosis affecting 5% to 60% of marrow fat (one study) and necrosis significantly greater in BMLs than in areas without BMLs (one study)
2) Fibrosis
a) Adipose tissue partially replaced with fibrous and fibrovascular tissue (ten studies)
b) Fibrosis found in 85% of patients amounting to 20% of the tissue volume (one study)
3) Cellular infiltration
a) Increased cellular infiltration was shown in the BML group compared to the non-BML group (one study)
b) No study described the cellular infiltration
4) Vascularity
a) Increased bone vascularity in BMLs compared to controls (three studies) being four times that of controls in BMLs
They concluded that more studies of better quality are needed.
Subchondral bone circulation in OA
Subchondral bone perfusion kinetics in human OA has been investigated by using dynamic contrast-enhanced (DCE) MRI. Aaron et al15 compared 15 patients with severe OA of the knee with eight normal controls. They investigated the lateral tibial plateau to avoid the multiple bone changes seen in the affected medial compartment. They found that OA bone has venous outflow stasis and a decrease in arterial inflow when compared to normal bone. They commented that this venous outlet syndrome has known effects on intraosseous hypertension, reductions in perfusion and hypoxia. Since osteoblasts are affected by hypoxia changes in their cytokine profiles in OA suggest that there might be a relationship between tissue perfusion and osteoblast function.
Cellular and biomolecular response of osteochondral tissues
The principal cells involved in subchondral bone remodelling are osteoblasts, osteoclasts and macrophages (Fig. 4). In humans with OA, microfractures and neovascularisation are seen in the subchondral bone and at the osteochondral junction. This triggers a reparative response with new blood vessel formation and inflammation with the typical radiological findings in subchondral bone of osteophyte formation, cysts and subchondral sclerosis (Fig. 5).1 The effect of age on the osteochondral tissue in humans is poorly understood; as bone demineralisation occurs there is a corresponding increase in organic content. In OA, the organic content does not increase with demineralisation implying that there is an increase in osteoid tissue. Hypo-mineralisation is associated with an altered alpha-1 to alpha-2 chain ratio in the composition of collagen type I in the subchondral zone of osteoarthritic samples.16 The normal homeostasis between osteoblasts and osteoclasts is disrupted and osteoclast activity increases. In normal bone, fractures heal back to bone, but in OA the microfractures heal with fibrous vascular tissue and under-mineralised sclerotic bone, implying a similar process to delayed fracture healing.17
Fig. 4.

Diagram showing the cells involved in bone remodelling. Note the perforation of the osteochondral junction by an osteoclast taking blood vessel and sensory nerve supply into the deep layer of articular cartilage
Fig. 5.
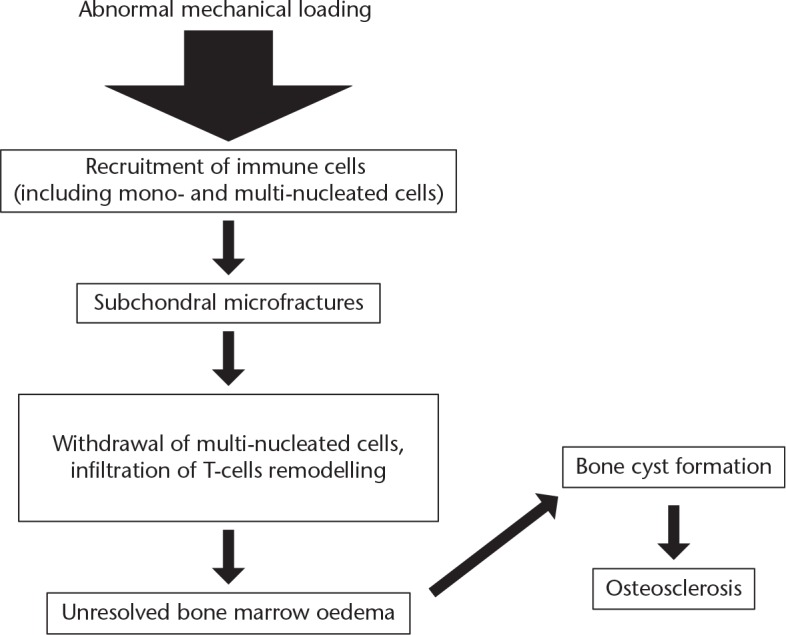
Proposed scheme for the development of osteoarthritis in a post-traumatic model (based on data from Weber et al1)
Both osteoblast and osteoclast activity increase in OA by 96% and 190%, respectively, but since bone loss overall is not observed, this indicates that the osteoblast-mediated bone formation has a distinct role in OA.1 Bone remodelling involves the interplay between the osteoblasts, osteoclasts and bone marrow mesenchymal stem cells (MSCs). Rather than mature osteoblasts lining the resorption pits in damaged subchondral bone, the osteoblasts come from bone marrow-derived MSCs migrating and transforming into osteoblasts.18 MSCs derived from bone marrow lesions in OA have lower mineralisation capacity and slower proliferation. It is thought that this may be an effect of the inflammatory process.19
Osteoblasts from the subchondral bone can induce chondrocytes to increase MMP-13 expression and decrease aggrecan production, a pro-catabolic state. OA chondrocytes can affect osteoblast function by causing the latter to increase MMP-1 expression; although these data are from end-stage, rather than early-stage OA.1 OA osteoblasts also express high levels of inflammatory cytokines such as transforming growth factor b1 (TGFb1) and prostaglandin E2 (PGE2). Overexpression of inflammatory cytokines is thought to contribute to subchondral bone disturbance. Figure 6 shows a proposed biomolecular interaction at the osteochondral junction.
Fig. 6.

Diagram of the biomolecular interactions in osteoarthritis (original drawing, adapted from Suri and Walsh5)
In addition, the bone marrow osteocytes send apoptotic signals in response to microfractures; these increase osteoclastogenesis20 and also decreased sclerostin production.21 Sclerostin inhibits osteoblasts. When the subchondral osteoclasts become activated, they form cutting cones that breech the subchondral plate and enter the non-calcified cartilage layer (Fig. 7). This may be associated with fissuring of the articular cartilage down to the tidemark and into the subchondral plate. Separation between the chondral layer and the subchondral plate is seen macroscopically as a chondral defect. Breeching of the subchondral plate coupled with the chondral damage allows the movement of fluid, cells and molecules between the bone and the joint cavity. Mechanical forces can also affect and worsen the destruction.
Fig. 7.

Diagram of the interaction of synovial inflammation and mechanical subchondral damage in osteoarthritis (original drawing, adapted from Hügel and Guerts13)
The tissues may respond by duplicating the tidemark within the non-calcified layer with a ‘second front’ of perivascular ossification and subchondral thickening. New blood vessel formation may occur with ingrowth into the cartilage accompanied by sensory nerves. The formation of osteophytes, an attempt to decrease the load across the joint by increasing its surface area, depends on neovascularisation. Normal chondrocytes are resistant to vascular invasion secreting anti-angiogenic factors, such as the regulatory peptides troponin I and thrombospondin-1,22,23 and inhibitors of matrix degrading enzymes, including tissue inhibitors of metalloproteinases (TIMPs), secretory leukocyte proteinase inhibitor and plasminogen activator inhibitor-1.24 The fissures in the articular cartilage may be filled with fibrocartilage as an attempt at repair, with invasion of fibroblasts via the new blood vessels.
Immune cells in OA
Immune cells are known to be active in fracture healing. It is postulated that the persistent bone marrow lesions have similarities with the pathogenesis of delayed fracture healing.1 Macrophages, as well as T and B cells participate actively in the healing process by migrating into the fracture site to serve various functions.25 T and B cells are involved in osteoclastogenesis and bone catabolism. The osteoprotegerin/receptor activator of the nuclear factor kappa-B ligand (OPG/RANKL) system allows cross-talk between immune cells and bone cells. RANKL is secreted by osteocytes and can bind to RANK on the surface of osteoclast precursors. This initiates osteoclastogenesis and bone resorption during the remodelling process.26 However, immune cells such as B lymphocytes could be another source of RANKL for osteoclastogenesis.27
In late-stage OA, the number of CD68+ macrophages and CD20+ B lymphocytes are significantly higher in the sclerotic region of subchondral bone than in non-sclerotic areas.28 B cells and their secretion of IL-10 are associated with delayed bone fracture healing.29 Therefore, B cells are also suspected to have a hand in the defective repair of subchondral bone in OA. However, more work is needed to support the notion that persistent BMLs have the same pathogenesis as delayed fracture healing.
TGF-β1 is activated in the subchondral bone in response to altered mechanical loading.11 TGF-β1 concentration also increases in the subchondral bone of human osteoarthritis. High concentrations of TGF-β1 induce formation of nestin + MSC clusters, leading to aberrant bone formation accompanied by increased angiogenesis. Transgenic expression of active TGF-β1 in osteoblastic cells induces OA. Inhibition of TGF-β activity in subchondral bone attenuates degeneration of OA articular cartilage.
Effects of inflammation on bone remodelling in OA
Inflammation is a phase of bone fracture healing, with inflammatory cells regulating the process by secreting inflammatory cytokines and chemotactic mediators,30 as well as growth factors, to recruit and stimulate both immune cells and progenitor cells for remodelling of the fracture site. Monocytes and macrophages are capable of releasing cytokines such as BMP-2, BMP-4 and TGF-b1, which can stimulate osteoblast differentiation and proliferation.31 Weber et al1 propose that changes in TGFb1 level after subchondral bone micro-fractures might lead to activation of the adaptive immune system. Imbalance in inflammatory and anti-inflammatory cells might contribute to delayed union or nonunion of micro-fractures, contributing to subchondral bone sclerosis formation and the persistence of the disorder. Identifying a unique pattern of adaptive immune system activation in OA would throw light on the development of new diagnostic and therapeutic strategies for OA.
Although synovial inflammation secondary to cartilage wear-debris is well-known, OA may be a systemically induced inflammatory disease. In the peripheral blood of OA patients, a significant aberrant ratio of CD4+/CD8+ cells has been found.32,33 The highly vascularised synovium may be a gateway for systemic inflammatory cytokines to affect the joint (Fig. 6).13 Synovial membrane is a source of inflammatory cytokines such as IL-1, IL-6 and TNF,34 which can be released by mast cells, which are present in great abundance in OA synovium.35 Mast cells can also recruit innate immune cells.36 Mast cells can therefore contribute to inflammation as well as subchondral bone pathology. It is important to emphasise that there is no direct evidence that inflammation causes osteosclerosis in subchondral bone, but the presence of high numbers of macrophages and B lymphocytes suggests there might be.28
Conclusions
Subchondral bone remodelling is an integral part of the pathology of OA. However, the response of subchondral bone is not independent of the rest of the joint. Alterations from normal in the articular cartilage results in changes in subchondral bone and vice versa. The usual initiator of subchondral remodelling is abnormal mechanical loads. These lead to microfractures and what may be a fracture healing response. This response involves macrophages, MSCs forming osteoblasts and osteoclastogenesis through chemical signals. The effect is to increase bone tissue coupled with demineralisation which results in fewer, but thicker, trabeculae. This is seen as osteosclerosis on imaging. Increased vascularisation can be seen on MRI early in the evolution of OA. Osteoclasts penetrate the osteochondral junction allowing new blood vessels and sensory nerves to infiltrate the deep layer of the articular cartilage. Coupled with articular cartilage fissuring and loss, the synovial fluid and its associated cytokines can directly infiltrate the subchondral bone and affect all the cells present. The progression of OA occurs when the abnormal mechanical environment continues to the point where recovery of the tissue is not possible. It is thought that this is when subchondral sclerosis has occurred. Drug therapies to alter the chemical pathways are not expected to work from this point and, apart from pain relief, surgical therapies such as osteotomy are more likely to be limited to reducing the progression of the disorder.
Footnotes
ICMJE Conflict of interest statement: The author reports royalties from Corin/Tornier, outside the submitted work.
Funding statement
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
References
- 1. Weber A, Chan PMB, Wen C. Do immune cells lead the way in subchondral bone disturbance in osteoarthritis? Prog Biophys Mol Biol 2017. December 22; 10.1016/j.pbiomolbio.2017.12.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 2. Dobson GP, Letson HL, Grant A, et al. Defining the osteoarthritis patient: back to the future. Osteoarthritis Cartilage 2018;26:1003-1007. [DOI] [PubMed] [Google Scholar]
- 3. Loef M, van Beest S, Kroon FPB, et al. Comparison of histological and morphometrical changes underlying subchondral bone abnormalities in inflammatory and degenerative musculoskeletal disorders: a systematic review. Osteoarthritis Cartilage 2018;26:992-1002. [DOI] [PubMed] [Google Scholar]
- 4. Muratovic D, Findlay DM, Cicuttini FM, et al. Bone matrix microdamage and vascular changes characterize bone marrow lesions in the subchondral bone of knee osteoarthritis. Bone 2018;108:193-201. [DOI] [PubMed] [Google Scholar]
- 5. Suri S, Walsh DA. Osteochondral alterations in osteoarthritis. Bone 2012;51:204-211. [DOI] [PubMed] [Google Scholar]
- 6. Clark I, McGregor A, Donell S. The genetic association between asporin and osteoarthritis. Knee 2008;15:vii-viii. [Google Scholar]
- 7. Kevorkian L, Young DA, Darrah C, et al. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum 2004;50:131-141. [DOI] [PubMed] [Google Scholar]
- 8. Wang Q, Rozelle AL, Lepus CM, et al. Identification of a central role for complement in osteoarthritis. Nat Med 2011;17:1674-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saito T, Fukai A, Mabuchi A, et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med 2010;16:678-686. [DOI] [PubMed] [Google Scholar]
- 10. Yang S, Kim J, Ryu JH, et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med 2010;16:687-693. [DOI] [PubMed] [Google Scholar]
- 11. Zhen G, Wen C, Jia X, et al. Inhibition of TGF–β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med 2013;19:704-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang C-Y, Chanalaris A, Troeberg L. ADAMTS and ADAM metalloproteinases in osteoarthritis - looking beyond the ‘usual suspects’. Osteoarthritis Cartilage 2017;25:1000-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hügle T, Geurts J. What drives osteoarthritis? Synovial versus subchondral bone pathology. Rheumatology (Oxford) 2017;56:1461-1471. [DOI] [PubMed] [Google Scholar]
- 14. MacKay JW, Murray PJ, Kasmai B, et al. Subchondral bone in osteoarthritis: association between MRI texture analysis and histomorphometry. Osteoarthritis Cartilage 2017;25:700-707. [DOI] [PubMed] [Google Scholar]
- 15. Aaron RK, Racine JR, Voisinet A, Evangelista P, Dyke JP. Subchondral bone circulation in osteoarthritis of the human knee. Osteoarthritis Cartilage 2018;26:940-944. [DOI] [PubMed] [Google Scholar]
- 16. Kerns JG, Gikas PD, Buckley K, et al. Evidence from Raman spectroscopy of a putative link between inherent bone matrix chemistry and degenerative joint disease. Arthritis Rheumatol 2014;66:1237-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hunter DJ, Gerstenfeld L, Bishop G, et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res Ther 2009;11:R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park D, Spencer JA, Koh BI, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012;10:259-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Campbell TM, Churchman SM, Gomez A, et al. Mesenchymal stem cell alterations in bone marrow lesions in patients with hip osteoarthritis. Arthritis Rheumatol 2016;68:1648-1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henriksen K, Neutzsky-Wulff AV, Bonewald LF, Karsdal MA. Local communication on and within bone controls bone remodeling. Bone 2009;44:1026-1033. [DOI] [PubMed] [Google Scholar]
- 21. Jaiprakash A, Prasadam I, Feng JQ, et al. Phenotypic characterization of osteoarthritic osteocytes from the sclerotic zones: a possible pathological role in subchondral bone sclerosis. Int J Biol Sci 2012;8:406-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moses MA, Wiederschain D, Wu I, et al. Troponin I is present in human cartilage and inhibits angiogenesis. Proc Natl Acad Sci USA 1999;96:2645-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pfander D, Cramer T, Deuerling D, Weseloh G, Swoboda B. Expression of thrombospondin-1 and its receptor CD36 in human osteoarthritic cartilage. Ann Rheum Dis 2000;59:448-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fransès RE, McWilliams DF, Mapp PI, Walsh DA. Osteochondral angiogenesis and increased protease inhibitor expression in OA. Osteoarthritis Cartilage 2010;18:563-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ono T, Takayanagi H. Osteoimmunology in bone fracture healing. Curr Osteoporos Rep 2017;15:367-375. [DOI] [PubMed] [Google Scholar]
- 26. Xiong J, Onal M, Jilka RL, et al. Matrix-embedded cells control osteoclast formation. Nat Med 2011;17:1235-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manilay JO, Zouali M. Tight relationships between B lymphocytes and the skeletal system. Trends Mol Med 2014;20:405-412. [DOI] [PubMed] [Google Scholar]
- 28. Geurts J, Patel A, Hirschmann MT, et al. Elevated marrow inflammatory cells and osteoclasts in subchondral osteosclerosis in human knee osteoarthritis. J Orthop Res 2016;34:262-269. [DOI] [PubMed] [Google Scholar]
- 29. Sun G, Wang Y, Ti Y, et al. Regulatory B cell is critical in bone union process through suppressing proinflammatory cytokines and stimulating Foxp3 in Treg cells. Clin Exp Pharmacol Physiol 2017;44:455-462. [DOI] [PubMed] [Google Scholar]
- 30. Osta B, Benedetti G, Miossec P. Classical and paradoxical effects of TNF-α on bone homeostasis. Front Immunol 2014;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loi F, Córdova LA, Pajarinen J, et al. Inflammation, fracture and bone repair. Bone 2016;86:119-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Apinun J, Sengprasert P, Yuktanandana P, et al. Immune mediators in osteoarthritis: infrapatellar fat pad-infiltrating CD8+ T cells are increased in osteoarthritic patients with higher clinical radiographic grading. Int J Rheumatol 2016;2016:9525724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ponchel F, Burska AN, Hensor EM, et al. Changes in peripheral blood immune cell composition in osteoarthritis. Osteoarthritis Cartilage 2015;23:1870-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Larsson S, Englund M, Struglics A, Lohmander LS. Interleukin-6 and tumor necrosis factor alpha in synovial fluid are associated with progression of radiographic knee osteoarthritis in subjects with previous meniscectomy. Osteoarthritis Cartilage 2015;23:1906-1914. [DOI] [PubMed] [Google Scholar]
- 35. de Lange-Brokaar BJ, Kloppenburg M, Andersen SN, et al. Characterization of synovial mast cells in knee osteoarthritis: association with clinical parameters. Osteoarthritis Cartilage 2016;24:664-671. [DOI] [PubMed] [Google Scholar]
- 36. Kroner J, Kovtun A, Kemmler J, et al. Mast cells are critical regulators of bone fracture-induced inflammation and osteoclast formation and activity. J Bone Miner Res 2017;32:2431-2444. [DOI] [PubMed] [Google Scholar]