

Abstract
By a bioinformatics approach, we have identified a novel cysteine knot protein member, VWC2 (von Willebrand factor C domain containing 2) previously known as Brorin. Since Brorin has been proposed to function as a bone morphogenetic protein (BMP) antagonist, we investigated the binding of Brorin/VWC2 to several BMPs; however, none of the BMPs tested were bound to VWC2. Instead, the βA subunit of activin was found as a binding partner among transforming growth factor (TGF)-β superfamily members. Here, we show that Vwc2 gene expression is temporally upregulated early in osteoblast differentiation, VWC2 protein is present in bone matrix, and localized at osteoblasts/osteocytes. Activin A-induced Smad2 phosphorylation was inhibited in the presence of exogenous VWC2 in MC3T3-E1 osteoblast cell line and primary osteoblasts. The effect of VWC2 on ex vivo cranial bone organ cultures treated with activin A was investigated, and bone morphometric parameters decreased by activin A were restored with VWC2. When we further investigated the biological mechanism how VWC2 inhibited the effects of activin A on bone formation, we found that the effects of activin A on osteoblast cell growth, differentiation, and mineralization were reversed by VWC2. Taken together, a novel secretory protein, VWC2 promotes bone formation by inhibiting Activin-Smad2 signaling pathway.
Keywords: Activin A, Bone formation, Calvaria, Osteoblasts, Von Willebrand factor C domain containing 2 (VWC2)
Introduction
The transforming growth factor (TGF)-β superfamily consists of more than 40 members in mammals, and plays pivotal roles in growth, differentiation and death of cells, axis formation, and extracellular matrix protein production [1]. The family is divided into several sub-family members, i.e., TGF-β, bone morphogenetic protein (BMP), and activin. TGF-β is considered as an inhibitor of osteoblast differentiation and mineralization [2, 3], whereas BMP, in general, positively regulates bone formation and healing [4–6]. These signaling pathways appear to counteract the functions of osteoblasts.
Among TGF-β superfamily members, recombinant human BMP-2 and BMP-7 are FDA approved and have been applied to various clinical settings aiming for bone/cartilage repair. However, there are still several challenges in controlling the broad biological functions of BMPs. Thus, it is critical to understand how BMP (and also other TGF -β sub-family) functions are controlled for the development of efficient growth factor-based local treatments. The functions of TGF-β superfamily members are controlled by a variety of modulators at multiple levels, i.e., extracellular, cell surface, and intracellular levels [7]. In the extracellular space, several cysteine knot proteins (CKPs) such as chordin and follistatin [8–10] have been identified as BMP/activin antagonists and CKPs appear to fine-tune TGF-β superfamily signaling(s). We therefore sought to identify such extracellular regulators and investigate their functions that could be useful to develop a promising therapeutic approach leading to an ana-bolic effect in bone.
To identify a new member of CKPs, we first obtained the protein sequence (GenBank #; NP_034023) of mouse CHORDIN, that is known to bind to BMP [11], as a query protein sequence, performed a BLAST search in the mouse genome, and further the PSORT II subcellular localization prediction program as previously reported [12]. This computational screening identified two novel CKP members consisting of the VWC2 family (von Willebrand factor C domain containing 2), i.e., VWC2 and VWC2-like, the structurally closest member of the VWC2. While we investigated the molecular function of VWC2, it was reported that VWC2 functioned as a BMP antagonist. VWC2 modestly decreased BMP-2-or BMP-6-induced alkaline phosphatase activity and inhibited phosphorylation of Smad 1/5/8 only at a later incubation time with BMP-2 and BMP-6 [13]. Several CKP members such as Gremlin, Sclerostin, and Cerberus [14–16] regulate embryonic and organ development by selectively antagonizing the activities of different TGF-β superfamily ligands through the binding, thus similar functions have been speculated for VWC2.
In this study, we first investigated the binding ability of VWC2 to several BMPs, however, we failed to show the binding between VWC2 and BMPs. Instead, we demonstrate that activin is the target of VWC2. Our data indicate that VWC2, a bone matrix-associated secretory protein, enhances cranial bone formation through inhibiting Activin signaling and activin-induced cell functions via direct binding to activin A. These data suggest that the antagonistic effect of VWC2 on BMP signaling does not require the binding to BMPs, but due to a secondary effect through a different TGF-β superfamily signaling pathway(s).
Materials and Methods
Cell Culture
The mouse osteoblastic cell line, MC3T3-E1 cells (sub-clone 4, ATCC CRL-2593), and human embryonic kidney 293 cells were maintained as previously described [17, 18] and used in this study.
Primary Osteoblast Cultures
The C57BL/6 mice (Jackson Laboratory) at postnatal day (P) 7 were euthanized and calvarial bones were dissected. These bones were minced and sequentially digested with collagenase type I (Sigma) in phosphate buffered saline (PBS) at 37 °C for six cycles (5 min for the first cycle, and 10 min for the remaining five cycles). Cells from 3rd, 4th, 5th, and 6th cycles were pooled, collected by centrifugation and cultured as previously reported [19].
Molecular Cloning of Vwc2 and TGF-β Superfamily Members
The cDNA sequences containing the coding region of mouse Vwc2, Tgf-β2, Tgf-β3, Activin-βA, and Activin -βB were isolated by PCR methods. The primers used are shown in Table 1. The PCR products of Vwc2 or each Tgf-β superfamily member was ligated into pcDNA3-HA or pcDNA3.1-V5/His vector, sequenced and the plasmids harboring Vwc2, Tgf-β2, Tgf -β3, Activin-βA, or Activin-βB cDNA followed by HA-or V5/His-tag (pcDNA3-Vwc2-HA and pcDNA3.1-Vwc2-V5/His, pcDNA3.1-Tgf-β2, -Tgf-β3, -Activin-βA, or -Activin-βB-V5/His vectors) were successfully generated. The expression vectors containing Tgf-β1, Bmp- 2, −4, −6, and −7 reported previously [17, 20] were used in this study.
Table 1.
PCR primers used in this study
| Gene | Forward | Reverse |
|---|---|---|
| Vwc2 | 5′-GGGATGCCTAGCTCCTCTGCGATGGCAG-3′ | 5′-CATTTGTCTGCACTCATGTCTCGTGCAC-3′ |
| TGF-β2 | 5′-AACATGCACTACTGTGTGCTGAGCACC-3′ | 5′-GCTGCATTTACAAGACTTGACAATC-3′ |
| TGF-β3 | 5′-CACATGAAGATGCACTTGCAAAGGGCTC-3′ | 5′-GCTGCACTTACACGACTTCACCACCATG-3′ |
| Activin βA | 5′-AGGATGCCCTTGCTTTGGCTGAGAGG-3′ | 5′-GGAGCAGCCACACTCCTCCACAATC-3′ |
| Activin βB | 5′-ACCATGGACGGGCTGCCCGGTCGGGCG-3′ | 5′-GGCGCAGCCACACTCCTCCACGATCATG-3′ |
Reagents and Antibodies
Recombinant human activin A (338-AC) and VWC2 (6147-WF) proteins were obtained from R&D Systems. The anti-bodies used in this study were as follows: anti-V5 (Life Technologies), anti-HA (clone 12CA5, Roche Life Science), anti-HA high affinity (clone 3F10, Roche Life Science), anti-phospho-Smad2 (Ser 465/467, Cell Signaling Technology), anti-Smad2 (Cell Signaling Technology), and anti-Activin A (R&D Systems) antibodies. An affinity-purified polyclonal VWC2 antibody was generated by immunizing rabbits with a synthetic peptide corresponding to the residues 49–64 of mouse VWC2 (i.e., EHASRDSPGRVSELGR) (Bethyl Laboratories, Inc.).
Immunoprecipitation and Western Blot Analysis
To determine the interaction between VWC2 and TGF-β superfamily members, 293 cells were plated and transfected with pcDNA3 -Vwc2-HA together with an empty pcDNA3.1-V5/His A (Invitrogen), pcDNA3.1-Bmp-2-V5/His, pcDNA3.1-Bmp-4-V5/His, pcDNA3.1-Bmp-6-V5/His, pcDNA3.1-Bmp-7-V5/His, pcDNA3.1-Tgf-β1-V5/His, pcDNA3.1-Tgf-β2-V5/His, pcDNA3.1-Tgf-β3-V5/His, pcDNA3.1 -Activin-βA-V5/His, or pcDNA3.1-Activin-βB-V5/His vector using FuGENE6 transfection regent. The binding assay was performed by immunoprecipitation (IP) with the conditioned media collected from the transfected cells and Western blot (WB) analysis as previously described [17].
In Vitro Binding Assay
VWC2 and activin A proteins were incubated in PBS where the total amount of proteins per sample was kept constant by adding bovine serum albumin (BSA). Samples were then immunoprecipitated with anti-VWC2 antibody (1:1000) followed by WB analysis with anti-Activin A antibody.
Real-Time PCR
MC3T3-E1 cells were plated onto 35 mm culture dishes at a density of 2 × 105cells/dish. When cells reached confluence, the growth medium was replaced with the one supplemented with 50 µg/ml of ascorbic acid and 1 mM of β-glycerophosphate (mineralization medium), and maintained for up to 35 days. Total RNA was extracted, cDNA was synthesized and used as PCR templates, and real-time PCR was performed as previously described [17]. The mean fold changes in the expression of Vwc2 (Mm00621766_m1) relative to that of glyceraldehyde-3-phosphate dehydrogenase (Gapdh, ABI assay number: 4308313) were calculated using the values of cDNA derived from the MC3T3-E1 cell culture at Day 0 as a calibrator by means of method as described previously [17]. To investigate the expression levels of osteogenic markers, MC3T3 -E1 cells were plated as described above: the media were changed to the mineralization media; cells were treated with PBS (Control), activin A (250 ng/ml) and VWC2 and activin A, and further cultured up to day 7. Real-time PCR was performed using the following osteogenic markers: Runt-related transcription factor 2 (Runx2)/Core binding factor alpha 1 (Cbfa1) (Runx2, Mm00501578_m1); Osterix (Osx, Mm00504574_ m1); Collagen type 1 alpha 2 chain ( Col1a2, Mm00483888_ m1); Activating transcription factor 4 (Atf4, Mm00515324_ m1); Bone γ-carboxyglutamate protein, related sequence 1 (Osteocalcin/Ocn, Mm01741771_g1).
Extraction of Bone Matrix- and Mineral-Associated Proteins from Bovine Long Bones
Fetal bovine femurs (Aries Scientific Inc.) were used to sequentially extract bone matrix - and mineral-associated proteins. Bone matrix was first fractioned into mineral-nonassociated (G1 fraction) and mineral-associated fractions (E and G2 fractions). The latter (mineral-associated) was further fractioned into EDTA-soluble (E fraction) and EDTA-insoluble fractions (G2 fraction) as previously described [21]. The protein concentration in each fraction was determined by a Detergent Compatible protein assay kit (Bio-Rad). Recombinant mouse VWC2-V5/His protein was generated as previously described [21] and prepared as a positive control. The equal amounts of proteins from each fraction, i.e., bone matrix-associated (G1 representing the matrix molecules not associated with mineral), mineral-associated (E representing soluble matrix molecules associated with mineral), collagen-associated (G2), were prepared and subjected to WB analysis with anti-VWC2 and anti -V5 antibodies. Non-immune rabbit immunoglobulin (NRIg) was used to confirm the specificity of anti-VWC2 antibody.
Immunohistochemistry
The heads of C57BL/6 mouse neonates collected at postnatal day 7 (P7) were removed, fixed with 10% formaldehyde, demineralized with 10% EDTA, sagittally cut in half, dehydrated and embedded in paraffin. Serial sections were obtained and immunohistochemical staining was performed as previously described [ 12] with anti-VWC2 antibody or NRIg (both 1 µg/ml) as a negative control.
Western Blotting
For WB analysis of Smad2, MC3T3-E1 cells or primary osteoblasts were plated onto 6-well culture plates at the density of 2 × 105 cells/well and on the following day, cells were treated with PBS (control), VWC2 (750 ng/ml), activin A (250 ng/ml), or activin A together with various doses (250, 500, or 750 ng/ml) of VWC2 for 15 min. Cells were then rinsed twice with PBS, lysed with RIPA buffer, samples were subjected to WB with anti -phospho-Smad2 antibody, and re-blotted with anti-total Smad2 antibody. Digital images of phosphorylated Smad2 and total Smad2 protein expression were captured using the VersaDoc Imaging System (Bio-Rad). Densitometric analysis was performed using Image-J software. The levels of Smad2 phosphorylation were expressed as the ratio of the intensity of phos-phorylated Smad2 by that of total Smad2. The experiments were performed at least 3 times, the representative blots were shown, and the mean levels of Smad2 phosphorylation (+SD) were presented. Statistical analysis was performed as described below.
Calvaria Ex Vivo Culture
Calvaria were collected from C57BL/6 neonates at P7, cut into two halves along the sagittal suture, and cultured. On the following day, the media were replaced with mineralization medium, and calvaria were treated with PBS (control), VWC2 (500 ng/ml), activin A (250 ng/ml), or VWC2 and activin A. Five halves of calvaria per group were used (n = 5) and cultured for up to 14 days. The media were refreshed twice a week with the addition of recombinant proteins. At day 14, the cultured calvaria were fixed in 10% paraformaldehyde overnight at 4 °C. Calvaria were demineralized with 10% EDTA for 48 h, embedded into paraffin, sectioned, and sections were subjected to hematoxylin and eosin (H&E) staining. Serial sections were also subjected to Masson Tri-chrome staining to visualize the old bone (red) and new bone (blue) for histomorphometric analysis.
Bone Histomorphometric Analysis
Figure 4A–a illustrates the schematic representative image, which explains how the histomorphometric analysis was performed. The blue area represents new bone; the red area represents old bone; each black dot represents an osteoblast counted on both the endosteal and periosteal surfaces of the calvaria; the double-headed arrows across the section represent calvaria thickness determinations by performing 10 equally spaced linear measures across the field of view using Image-J software straight line tool, and then averaged to obtain the mean thickness. The whole area of parietal bone posterior to the coronal suture was imaged using Olympus DSU microscope within 3 image fields per section and determined as the measurement areas. The total bone area was outlined using the free hand selection in the program. The new bone is differentiated from old bone by its blue color with Trichrome staining. The total and new bone area are expressed as mm 2, and the total and new bone thickness are expressed in mm. A total of 5 half calvarial blocks (n = 5) were used per treatment group, and 5 serial sections were made per block for quantification. The mean value + SD is expressed according to the standardized nomenclature and units [22].
Fig. 4.

Effect of VWC2 on calvarial bone formation. Calvarial bones were dissected from C57BL/6 neonates at P7, cultured, and treated with PBS (control), VWC2 (500 ng/ml), activin A (250 ng/ml), or both VWC2 and activin A. Paraffin-embedded sections were stained with H&E or Masson Trichrome staining. A The representative image of H&E staining (a–d) or Trichrome staining (e–h) per treatment group was shown. Scale bar, 0.2 mm. B Quantitative histomorphometric analysis. a Schematic image of the representative calvarium section by Trichrome staining. The blue area represents new bone area; the red area represents old bone area; each black dot represents an osteoblast (Ob) counted on both the endosteal and periosteal surfaces of the calvarium; the double headed arrows across the section represent thickness determinations by performing 10 equally spaced linear measures across the field of view.b–f Effects of VWC2, activin A, and VWC2/activin A on murine neonatal calvarial bone organ cultures were analyzed by measuring new bone area (b), total bone area (c), new bone thickness (d), total bone thickness (e), and number of osteoblast (Ob)s per bone surface (f) (n = 5 per treatment group). ****P value < 0.0001, ***P value < 0.001, **P value < 0.01, *P value < 0.05
Cell Growth
MC3T3-E1 cells were plated onto 24-well culture plates in triplicate at a density of 4.2 × 104 cells/well (referred to day 0, hereafter) and on the following day, cells were treated with PBS only (control), activin A (250 ng/ml), VWC2 (500 ng/ml), and VWC2 with activin A. Cells were further cultured for up to 14 days. The media were refreshed twice a week and proteins were added every time the media were changed. At days 3, 7, 10, and 14, cells were released by trypsin and cell numbers were counted by hemocytometer. Three independent sets of experiments were performed to confirm the reproducibility and the results were essentially identical. A representative result is shown.
In Vitro Mineralization Assay
MC3T3 -E1 cells were plated onto 35 mm culture dishes at density of 2 × 105 cells/well using α-MEM supplemented with 10% fetal bovine serum (FBS), 100 units/ml peni-cillin, and 100 µg/ml streptomycin until confluence. The medium was then replaced with mineralization medium and cells were treated with PBS (control), VWC2 (500 ng/ml), activin A (250 ng/ml), and VWC2 (500 ng/ml) with activin A (250 ng/ml). Cells were further cultured for 28 days. The media were refreshed twice a week and proteins were added every time the media were changed. After 28 days, cell/matrix layers were stained with 1% Alizarin red S and mineralized nodules were quantified based on the previous report [23]. Briefly, the image of each culture dish was digitally acquired; the background was subtracted; then the image was converted to a binary image by brightness threshold; and the number of mineralized nodules was counted using analyze particle tool in the Image-J software. The experiments were performed at least 6 times, the representative images were shown in Fig. 6A, and the mean numbers of mineralized nodules (+SD) were presented. Statistical analysis was performed as described below.
Fig. 6.

Effect of VWC2 on in vitro mineralization in MC3T3-E1 osteoblasts. a MC3T3-E1 cells were treated with PBS (control), VWC2 (500 ng/ml), activin A (250 ng/ml), or VWC2 and activin A, cultured for 28 days, and stained with Alizarin red S. The experiments were performed at least 6 times and the representative image for each treatment group was shown. b Quantitation of nodule number in each treatment group. Based on three experiments, the mean numbers of mineralized nodules (+SD) were presented with statistical analysis. ***P value < 0.001, *P value < 0.05
Statistical Analysis
One-way ANOVA with Tukey post hoc test or t test (cell growth and osteogenic marker analyses) was used to assess the statistical significance between groups. P value with less than 0.05 was considered significant. The statistical significance level was indicated by asterisks (*) as follows: (*) for P value < 0.05, (**) for P value < 0.01, (***) for P value < 0.001, and (****) for P value < 0.0001. The asterisk above the bar graph indicates the presence of statistical difference between the treatment group with asterisk and untreated control group.
Results
VWC2, a Novel CKP Member that Binds to Activin, but not BMPs
We first tested the binding between VWC2 and several BMP members, since VWC2 was proposed to be a BMP antagonist [13]. However, we found that VWC2 did not bind to BMPs including BMP-2, BMP-4, BMP-6, and BMP-7 (Fig. 1A, upper panel, lanes 2–5). As VWC2 possesses 2 Cysteine-Rich (CR) motifs [13], commonly found in all of CKP members, this strongly indicates that VWC2 has a binding partner in TGF-β superfamily ligands. We next investigated the potential binding of VWC2 to TGF-β superfamily ligand members including TGF-βs and activins. The 293 cells were transfected with TGF-β superfamily ligand expression vector fused to V5/His tag and pcDNA3-Vwc2-HA, and immunoprecipitation (IP)-WB analysis was performed with the conditioned media (Fig. 1a). When IP was performed with anti-V5 antibody followed by WB analysis with anti-HA antibody, thus identifying the VWC2-binding partner(s), distinct immunoreactive bands were observed in the combination of Vwc2-HA and βA subunit isoform of activin-V5 (Fig. 1a, upper panel, lane 9), and to a lesser extent, Vwc2-HA and βB subunit isoform of activin-V5 (Fig. 1a, upper panel, lane 10). The data clearly indicate that Vwc2 specifically binds to activin members. Since it has been shown that activin-βA is the most abundant TGF-β superfamily in bone [24], we further investigated the possibility of direct binding between VWC2 and activin A (homodimer of βA subunit). Recombinant VWC2 and activin A proteins were incubated in vitro, and the binding was assessed. As shown in Fig. 1b, VWC2 directly interacted with activin A (Fig. 1b, lane 3). Our results thus demonstrate that VWC2 is a direct binding protein to mature activin A.
Fig. 1.
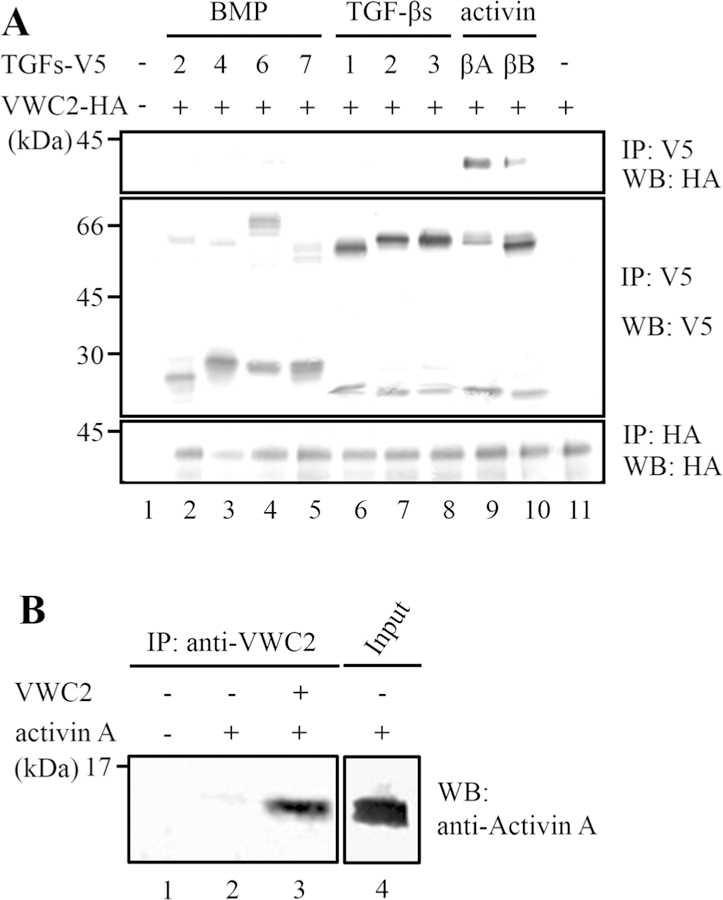
VWC2 is an activin-binding protein. a The expression vectors harboring TGF-β superfamily members including BMP-2, −4, −6, −7, TGF-β1, -β2, -β3, or βA or βB subunit isoform of activin (TGFs-V5) and pcDNA3-Vwc2-HA (Vwc2-HA) were transfected into 293 cells, the conditioned media were collected, and immunoprecipitation (IP)-Western blot (WB) analyses were performed. VWC2 did not bind to any of BMPs (upper panel, lanes 2–5) or TGF -βs (upper panel, lanes 6–8). VWC2 interacted with activin isoforms (upper panel, lanes 9, 10). The large precursor TGF-β superfamily proteins were detected at 45 ~ 70 kDa (middle panel, lanes 2–10), and mature proteins were at < 30 kDa (middle panel, lanes 2–10). b Direct binding of VWC2 to activin A. VWC2 (750 ng indicated by +) and activin A (750 ng indicated by +) proteins were mixed and incubated in vitro, immuno-precipitated with anti-VWC2 antibody, and subjected to WB analysis with anti-Activin A antibody. The expression of activin A used for the in vitro binding experiment as input was analyzed by WB with anti-Activin A antibody (lane 4)
Presence of VWC2 in Bone
Since several CKP members function as BMP antagonists and BMP has a crucial role in bone formation/regeneration and healing, we tested the expression of Vwc2 in osteoblasts. Expression levels of Vwc2 in MC3T3-E1 cells determined by real-time PCR analysis demonstrated that it was highest at day 7 (~ 12 fold of that at day 0, Fig. 2 A) during biomineralization. To investigate the presence of endogenous VWC2 protein in bone matrix, we generated an antibody against VWC2 and applied to protein extracts from fractioned bone matrix (fractions E, G1, and G2). As shown in Fig. 2B, the immunoreactive bands to anti-VWC2 antibody were clearly observed mainly in mineral-associated E fraction and to a lesser extent in collagen-associated G2 fraction, but not in matrix-associated (mineral-nonassociated) G1 fraction of bones (Fig. 2B, upper panel, lanes 1–3), as a ~ 35 kDa protein (expected molecular size for endogenous VWC2 protein). An immunoreactive band for VWC2-V5/His protein (a positive control) was detected at the molecular weight position slightly higher than that of bone VWC2 protein likely due to the presence of V5/His tag (~ 5 kDa) (Fig. 2B, upper panel, lane 4). The immunoreactivity of VWC2 -V5/His fusion protein was verified with anti-V5 antibody (Fig. 2B, lower panel, lane 4). No immunoreactivity was observed when treated with non-immune rabbit Ig (NRIg; Fig. 2B, middle panel). The data confirm the specificity of anti-VWC2 antibody generated, and demonstrate that VWC2 protein is present in bone matrix that is associated with mineral. To identify the localization of VWC2 protein in mouse bone tissues, the same anti-VWC2 antibody was employed to perform immunohistochemistry (Fig. 2C). Immunoreactivity was clearly observed in the developing maxillary bone, especially in the cytoplasm of osteoblasts (Fig. 2C, middle panel, indicated by arrows) and in the bone matrix/osteocytes (Fig. 2C, middle panel, indicated by arrowheads). No immunoreactivity was observed when incubated with NRIg (Fig. 2C, right panel). These data show that VWC2 protein is localized at the bone surface (osteoblasts) and bone matrix-embedded osteocytes.
Fig. 2.

Expression of Vwc2 in osteoblasts and bone. A The expression of Vwc2 in MC3T3-E1 osteoblastic cells during biomineralization was determined by quantitative real-time PCR analysis. Note that the expression was temporally upregulated at day 7 (early differentiation stage) and decreased thereafter. B Presence of VWC2 protein in bone. Bone fractions (G1, E, and G2) and purified VWC2-V5/His protein (VWC2-V5) were subjected to Western blot (WB) analysis using anti-VWC2, anti-V5 antibodies, and non-immune rabbit immuno-globulin (NRIg). The immunoreactive band to anti-VWC2 antibody was observed in E & G2 fractions in bone (upper panel, lanes 2,3). G1 fraction contains the matrix molecules not associated with mineral, E fraction has soluble matrix molecules associated with mineral, and G2 has collagen-associated. C Immunolocalization of VWC2 in mouse developing maxillary bone. Serial sections were stained using anti-VWC2 antibody (a, b) or NRIg (negative control, c). Panel (b) shows higher magnification of the open boxed area in (a). Note that VWC2 is present in osteoblasts (indicated by arrows) and osteocytes (arrowheads). Scale bar, 0.05 mm
Effect of VWC2 on Activin A-Induced Smad2 Phosphorylation in MC3T3-E1 and Primary Osteoblast Cells
Activin transduces signals by binding with high affinity to Activin type II receptor (ACVR2) followed by the recruitment of type I receptor, ACVR1b (also known as ALK4). Phosphorylated/activated ACVR1b then phosphorylates Smad2/3 and transduces the specific signals [25–27]. Since VWC2 binds to activin A (Fig. 1), and MC3T3-E1 osteoblastic cells express Vwc2 (Fig. 2A) and activin-βA (data not shown), we next investigated the effect of VWC2 on activin A-induced Smad2 phosphorylation in osteoblasts (Fig. 3). The data indicated that activin A-induced Smad2 phosphorylation (Fig. 3a, lane 3 and Fig. 3b, lane 3) was inhibited by VWC2 protein in a dose dependent manner in both MC3T3-E1 (Fig. 3a, lanes 4–6) and primary osteoblasts (Fig. 3b, lanes 4–6). The densitometric analysis showed that significantly higher level of phosphorylated Smad2 was observed by activin A treatment in MC3T3-E1 (Fig. 3a, lane 3) and primary osteoblasts (Fig. 3b, lane 3). This increased Smad2 phosphorylation was reduced by VWC2 treatment in a dose-dependent manner and the decrease of Smad2 phosphorylation level by a higher dose of VWC2 (750 ng/ml) was statistically different in MC3T3-E1 (Fig. 3a, lane 6) and primary osteoblasts (Fig. 3b, lane 6).
Fig. 3.

Effect of VWC2 on activin A-induced Smad2 phosphorylation. MC3T3-E1 cells (a) or mouse primary osteoblasts (b) were treated with VWC2 (200, 500, or 750 ng/ml indicated by +, ++, or +++, respectively) and activin A (250 ng/ml indicated by +) for 15 min and cell lysates were subjected to Western blot (WB) analysis with anti-phospho-Smad2 (pSmad2; upper panels in A, B) or anti-total Smad2 antibody (Smad2; lower panels in a, b). Experiments were performed at least three times and the representative results are shown. The intensities of the expression levels of phosphorylated Smad2 and total Smad2 were measured based on three experiments, the ratio of Smad2 phosphorylation was calculated, expressed as the mean + SD (left; MC3T3-E1 cells, right; mouse primary osteoblasts) and statistically analyzed. **P value < 0.01, *P value < 0.05
Taken together, our data clearly demonstrate that VWC2 is a direct binding partner of activin A and functions as an activin antagonist in osteoblasts.
Effect of VWC2 on Calvarial Bone Formation in Ex Vivo
As activin has been shown to inhibit mineralization in vitro [28], we next investigated the effect of VWC2 on activin-mediated inhibition in bone formation using calvarial bone ex vivo organ cultures. Calvaria were treated with PBS as a control, VWC2 (500 ng/ml), activin A (250 ng/ml), and VWC2 and activin A, and further cultured for 14 days. Samples were prepared for histological analysis with H&E (Fig. 4A, a–d). When calvaria were treated with activin A, the calvarial bones appeared to be thinner than the control (Fig. 4A–c vs. 4A–a), and VWC2 treatment rescued activin A-induced reduction of calvarial bone formation (Fig. 4A–d). Interestingly, VWC2 treatment alone enhanced the thickness of calvarial bone (Fig. 4A–b).
Masson Trichrome staining was performed using the serial sections and the representative image of each treatment group (i.e., control, VWC2, activin A, VWC2/activin A) with Trichrome staining was shown (Fig. 4A, e–h) to visualize the old bone (red) and new bone (blue). Figure 4B–a shows the schematic presentation of the representative image by Trichrome staining. To quantitatively measure the bone morphometric parameters, bone histomorphometric analysis was performed (Fig. 4B, b–f). Although activin A completely suppressed newly formed bone area as compared to untreated controls (Fig. 4B–b, column 3 vs. 1), VWC2 treatment significantly increased newly formed bone area inhibited by activin A (Fig. 4B–b, column 4 vs. 3). Activin A also decreased total bone area significantly by 35% as compared to untreated controls (Fig. 4B–c, column 3 vs. 1). VWC2 treatment together with activin A increased total bone area significantly by 235% as compared to activin A (Fig. 4B–c, column 4 vs. 3). As activin A abolished newly formed bone (Fig. 4B–b, column 3), newly formed bone thickness was absent. However, the suppression of newly formed bone thickness was significantly increased and dramatically rescued by VWC2 treatment (Fig. 4B–d, column 4 vs. 3). Activin A decreased total bone thickness significantly by 30% as compared to untreated controls (Fig. 4B–e, column 3 vs. 1), and VWC2 with activin A increased the total bone thickness significantly by 194% as compared to activin A (Fig. 4B–e, column 4 vs. 3). Activin A alone increased the number of osteoblasts per bone surface significantly by 118% as compared to untreated controls (Fig. 4B–f, column 3 vs. 1). VWC2 treatment inhibited activin A-induced cell growth by 26% (Fig. 4B–f, column 4 vs. 3) to untreated control levels (Fig. 4B–f, column 4 vs. 1). In addition, all bone morphometric parameters tested (total and new bone areas, total and new bone thickness, and osteoblast number) were significantly increased with VWC2 treatment alone as compared to untreated controls (Fig. 4 B, b–f, column 2 vs. 1). Our data clearly show that VWC2 antagonizes activin A-induced suppression of new bone synthesis and increase of osteoblast cell growth in calvarial bone organ culture likely through inhibiting activin A bioactivity.
Effect of VWC2 on Activin A-Mediated Cell Functions
To further gain the biological insights into how VWC2 antagonizes activin A at a cellular level, the effects of VWC2 on activin A-induced osteoblastic cell growth and subsequent differentiation were analyzed. Since activin has the mitogenic effect on MC3T3-E1 cells [29], we first investigated the effect of VWC2 on activin A-induced MC3T3-E1 osteoblast cell growth. MC3T3-E1 cells were treated with VWC2, activin A, or VWC2 and activin A, and further cultured for 14 days. Cell numbers were counted at intervals and compared between the treatment groups. The results demonstrated that activin A treatment accelerated growth of osteoblasts at days 7, 10, and 14 as compared to untreated controls (Fig. 5a), which is consistent with our data that activin A increased osteoblast number in calvarial organ culture (Fig. 4B–f, lane 3). VWC2 inhibited activin A-stimulated cell growth significantly at days 7, 10, and 14 (Fig. 5a).
Fig. 5.

Effect of VWC2 on activin A-induced cell functions. a Effect of VWC2 on activin A-induced osteoblastic cell growth. MC3T3-E1 cells were plated in triplicate, and on the following day, cells were treated with PBS (control), activin A (250 ng/ml), VWC2 (500 ng/ml), or both VWC2 and activin A, and further cultured up to 14 days. Cell numbers were counted at each time point indicated and expressed as the mean ± SD. ***P value < 0.001, **P value < 0.01, *P value < 0.05. b Effect of VWC2 on osteoblast differentiation treated with activin A. MC3T3-E1 cells were treated with PBS (control), activin A (250 ng/ml), and VWC2 together with activin A, and further cultured for 7 days. Total RNA was extracted and the expression of osteoblastic markers was analyzed by real-time PCR. The normalized values are shown as mean + S.D. based on triplicate assays and statistically analyzed. **P value < 0.01, *P value < 0.05. Runx2; runtrelated transcription factor 2, Osx; Osterix, Col1a2; Collagen type 1 alpha 2 chain, Atf4; Activating transcription factor 4, Ocn; Osteocalcin
Since our data demonstrated that accelerated osteoblast cell growth by activin A was inhibited by VWC2 in MC3T3-E1 cell cultures (Fig. 5a) and in calvarial organ cultures (Fig. 4A–f), and activin A is known to inhibit the fetal rat calvarial osteoblast differentiation at the early phase of culture [30], we next investigated the subsequent osteoblast differentiation by VWC2 and activin A. The levels of osteogenic markers at the early stage of MC3T3-E1 cells (day 7) were investigated by real-time PCR analysis (Fig. 5b). The results demonstrated that activin A treatment upregulated Runx2 and Col1a2, but not Atf4 expression, and down-regulated Osx expression. The down-regulation of Osx by activin A was markedly rescued by VWC2 treatment, and the upregu-lation of Col1a2 by activin A was significantly decreased by VWC2. However, the upregulated Runx2 expression by activin A was not changed by VWC2.
Since our data showed that VWC2 partially rescued the inhibition of osteoblast differentiation induced by activin A (Fig. 5b) and previous studies have demonstrated that activin A inhibited osteoblast mineralization [28], we investigated the effect of VWC2 on osteoblast mineralization in cell cultures (Fig. 6). Our results showed that cells treated with VWC2 for 28 days had more mineralized nodules (Fig. 6a, indicated by VWC2) than those of control group (Fig. 6a, control). Activin A inhibited in vitro mineralization (Fig. 6a, activin A), and when VWC2 was combined with activin A, more mineralized nodules were observed than activin A alone (Fig. 6a, VWC2/activin A). Quantification of mineralized nodules (Fig. 6b) revealed that cells treated with VWC2 had significantly higher numbers of nodules than control. Activin A-treated cells had significantly less mineralized nodules than control. Treatment with VWC2 combined with activin A produced significantly higher numbers of nodules than activin A treatment.
Our data demonstrated that VWC2 antagonizes activin A-mediated suppression of bone formation likely through inhibiting activin A-mediated osteoblast cell growth, differentiation, and subsequent mineralization.
Discussion
Activin A (homodimer of βA subunit) is one of the most abundant TGF-β superfamily members in bovine bone [24] and the βA subunit is the most abundant isoform expressed in human bone [28]. Activin A also has a high binding affinity to MC3T3-E1 osteoblastic cells (Kd = 0.26 nM) [29]. Previous studies have clearly demonstrated that activin A inhibits osteoblast differentiation and mineralization [28, 30]. In addition, blockade of Activin signaling using extracellular domain of ACVR2A fused to Fc domain of IgG1 (ACVR2A-Fc) has been shown to increase bone formation, bone mass, and bone strength in normal mice as well as ovariectomized mice [31]. Our results are in agreement with these previous findings, i.e., VWC2, which inhibits activin A-induced Smad2 signaling (Fig. 3), antagonizes the suppression of bone formation by activin A (Fig. 4). As ACVR2A is known to interact with several TGF-β superfamily ligands including BMP-6/7/9/10, Growth Differentiation Factor (GDF)-5 as well as βA and βB subunit of activin [32–36], suppression of more than one growth factor activity by ACVR2A-Fc may lead to unwanted outcomes in vivo. Since our results demonstrate that VWC2 selectively binds to activin members, the control of activin A signaling and function by VWC2 appears to be more specific, thus potentially a promising therapeutic approach for increases in bone formation.
Our data demonstrated that the levels of new bone synthesis/thickness by VWC2 with activin A were increased more significantly than those in untreated controls (Fig. 4 B–b and B–d, column 4 vs. 1), suggesting that VWC2 fully rescued activin A-induced decrease of new bone synthesis/thickness, and that VWC2 alone likely had anabolic effect on bone in addition to the suppression of activin function. The latter was well supported by our other bone histomorphometric parameter data where VWC2 alone increased new/total bone areas and thickness (Fig. 4B–b–e, column 2) as well as in vitro mineralization assay (Fig. 6).
It is not fully clear at this point as to why VWC2 and activin A treatment highly suppressed the osteoblast growth in vitro at days 7–14 lesser than the level of untreated controls (Fig. 5a). Since it is generally accepted that when cells including osteoblasts differentiate, the rate of proliferation decreases [37], the additional suppression of osteoblast cell growth by VWC2 with activin A observed in our study may associate with enhanced osteoblast differentiation. To support this notion, our data also showed that the expression of Osterix, a master regulator crucial for bone formation [38], was significantly upregulated by VWC2 and activin A treatment (Fig. 5b), which may in part explain the additional effect of VWC2 on osteoblastogenesis. Nonetheless, we believe that such upregulations of master genes result in higher bone formation found in calvarial ex vivo culture (Fig. 4).
We showed that Vwc2 is expressed in MC3T3-E1 cells (Fig. 2A), VWC2 protein is localized in osteoblasts/osteocytes in murine maxillary bone (Fig. 2C) and endogenous VWC2 protein is present in the mineral-associated fraction of bovine bone (Fig. 2B). It is thus reasonable to speculate that VWC2 may be secreted from osteoblasts and/or osteocytes, embedded together with mineral, and then released when osteoclastic activity is high, resulting in attenuation of activin function. In support of this notion, previous studies have shown that activin is a multifunctional growth factor on both osteoblastic and osteoclastic activities. It has been demonstrated that activin enhances osteoclast -like cell formation in murine bone marrow cultures [39], and the effect of activin is synergized with receptor activator of NF-κB ligand (RANKL) on osteoclast-like cell induction/differentiation [40] possibly through stimulation of RANK expression [41]. These findings may well explain our results that there was a decrease in total bone area and thickness when treated with activin A (Fig. 4A–c, e, lane 3) as compared to control.
We have previously reported and partially characterized another CKP member, VWC2-like, which is the structurally closest member among CKP family to VWC2 (60 and 55% identity in human and mouse, respectively). VWC2-like binds to the activin βA subunit (data not shown), suggesting that VWC2 and VWC2-like may possess a common structural motif to interact with activin A. In addition to the structural similarity, VWC2-like protein treatment promotes MC3T3-E1 osteoblastic cell mineralization in vitro possibly through the upregulation of Osx expression [18]. As a marked upregulation of Osx gene expression was observed by VWC2 treatment with activin A (Fig. 5b), our current and previous findings indicate the presence of a specific signaling pathway which controls Osx gene expression and that VWC2-like and VWC2 might have a common molecular target to enhance Osx expression.
At this point, it is still unclear regarding the role of VWC2 in skeletal development in vivo, as there have not been any reports of Vwc2 (or Brorin)-deficient mouse model. Recently it has been reported that inhibition of zebrafish brorin mRNA by morpholino oligonucleotides showed developmental defects in brain [42]. Although this is the first report to show the role of VWC2/Brorin in normal tissue development using in vivo model, there was no information about any skeletal changes in the model. Further investigation into the role of Vwc2 in skeletal development using an animal model, particularly in relation to Activin signaling, is now of considerable interest.
In summary, to the best of our knowledge, our study was the first to show that VWC2 is an activin antagonist through its direct binding to activin A. Attenuation of bone formation by activin A is rescued by VWC2 treatment, at least in part by osteoblast cell growth and differentiation levels, thereby providing a mechanism for VWC2-induced anabolic effects on bone.
Acknowledgements
We thank Dr. Philip Trackman (Boston University) for his valuable comments on the manuscript. This study was supported by grants from the NIH (NIDCR; DE019527 and NIAMS; AR057451 to Y.Mo., NIDCR; DE020843 to Y.Mi.). The nucleotide sequences for mouse Vwc2 gene have been deposited in the GenBank database under GenBank Accession number; DQ421811.
Footnotes
Compliance with Ethical Standards
Conflict of interest Ahmad Almehmadi, Yoshio Ohyama, Masaru Kaku, Ahmed Alamoudi, Dina Husein, Michitsuna Katafuchi, Yuji Mishina, Yoshiyuki Mochida declared that no competing financial interest.
Human and Animal Rights and Informed Consent The use of animals and all animal procedures in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of Boston University Medical campus (approved protocol number: AN-15053), and all efforts were made to minimize suffering animals. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
References
- 1.Shi Y, Massague J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113(6):685–700 [DOI] [PubMed] [Google Scholar]
- 2.Spinella-Jaegle S, Roman-Roman S, Faucheu C, Dunn FW, Kawai S, Gallea S, Stiot V, Blanchet AM, Courtois B, Baron R, Rawadi G (2001) Opposite effects of bone morphogenetic protein-2 and transforming growth factor-beta1 on osteoblast differentiation. Bone 29(4):323–330 [DOI] [PubMed] [Google Scholar]
- 3.Harris SE, Bonewald LF, Harris MA, Sabatini M, Dallas S, Feng JQ, Ghosh-Choudhury N, Wozney J, Mundy GR (1994) Effects of transforming growth factor beta on bone nodule formation and expression of bone morphogenetic protein 2, osteocalcin, osteopontin, alkaline phosphatase, and type I collagen mRNA in long-term cultures of fetal rat calvarial osteoblasts. J Bone Miner Res 9(6):855–863. 10.1002/jbmr.5650090611 [DOI] [PubMed] [Google Scholar]
- 4.Chen D, Zhao M, Mundy GR (2004) Bone morphogenetic proteins. Growth Factors 22(4):233–241 [DOI] [PubMed] [Google Scholar]
- 5.Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V (2006) BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet 38(12):1424–1429 [DOI] [PubMed] [Google Scholar]
- 6.Kamiya N, Mishina Y (2011) New insights on the roles of BMP signaling in bone-A review of recent mouse genetic studies. Bio-factors 37(2):75–82. 10.1002/biof.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19(23):2783–2810 [DOI] [PubMed] [Google Scholar]
- 8.Avsian-Kretchmer O, Hsueh AJ (2004) Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol Endocrinol 18(1):1–12 [DOI] [PubMed] [Google Scholar]
- 9.Harrison CA, Gray PC, Vale WW, Robertson DM (2005) Antagonists of activin signaling: mechanisms and potential biological applications. Trends Endocrinol Metab 16(2):73–78 [DOI] [PubMed] [Google Scholar]
- 10.Harrison CA, Wiater E, Gray PC, Greenwald J, Choe S, Vale W (2004) Modulation of activin and BMP signaling. Mol Cell Endocrinol 225(1–2):19–24 [DOI] [PubMed] [Google Scholar]
- 11.Larrain J, Bachiller D, Lu B, Agius E, Piccolo S, De Robertis EM (2000) BMP-binding modules in chordin: a model for signalling regulation in the extracellular space. Development 127(4):821–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mochida Y, Parisuthiman D, Kaku M, Hanai J, Sukhatme VP, Yamauchi M (2006) Nephrocan, a novel member of the small leucine -rich repeat protein family, is an inhibitor of transforming growth factor-beta signaling. J Biol Chem 281(47):36044–36051 [DOI] [PubMed] [Google Scholar]
- 13.Koike N, Kassai Y, Kouta Y, Miwa H, Konishi M, Itoh N (2007) Brorin, a novel secreted bone morphogenetic protein antagonist, promotes neurogenesis in mouse neural precursor cells. J Biol Chem 282(21):15843–15850 [DOI] [PubMed] [Google Scholar]
- 14.Shi W, Zhao J, Anderson KD, Warburton D (2001) Gremlin negatively modulates BMP-4 induction of embryonic mouse lung branching morphogenesis. Am J Physiol Lung Cell Mol Physiol 280(5):L1030–L1039 [DOI] [PubMed] [Google Scholar]
- 15.Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N (2003) Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem 278(26):24113–24117. 10.1074/jbc.M301716200 [DOI] [PubMed] [Google Scholar]
- 16.Piccolo S, Agius E, Leyns L, Bhattacharyya S, Grunz H, Bouw-meester T, De Robertis EM (1999) The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature 397(6721):707–710. 10.1038/17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mochida Y, Parisuthiman D, Yamauchi M (2006) Biglycan is a positive modulator of BMP-2 induced osteoblast differentiation. Adv Exp Med Biol 585:101–113 [DOI] [PubMed] [Google Scholar]
- 18.Ohyama Y, Katafuchi M, Almehmadi A, Venkitapathi S, Jaha H, Ehrenman J, Morcos J, Aljamaan R, Mochida Y (2012) Modulation of matrix mineralization by Vwc2-like protein and its novel splicing isoforms. Biochem Biophys Res Commun 418(1):12–16. 10.1016/j.bbrc.2011.12.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronen-berg HM, Feng JQ, Mishina Y (2008) BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 135(22):3801–3811. 10.1242/dev.025825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atsawasuwan P, Mochida Y, Katafuchi M, Kaku M, Fong KS, Csiszar K, Yamauchi M (2008) Lysyl oxidase binds transforming growth factor-beta and regulates its signaling via amine oxidase activity. J Biol Chem 10.1074/jbc.M803142200 [DOI] [PMC free article] [PubMed]
- 21.Mochida Y, Kaku M, Yoshida K, Katafuchi M, Atsawasuwan P, Yamauchi M (2011) Podocan -like protein: a novel small leucine-rich repeat matrix protein in bone. Biochem Biophys Res Commun 410(2):333–338. 10.1016/j.bbrc.2011.05.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28(1):2–17. 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vayrynen JP, Vornanen JO, Sajanti S, Bohm JP, Tuomisto A, Makinen MJ (2012) An improved image analysis method for cell counting lends credibility to the prognostic significance of T cells in colorectal cancer. Virchows Arch 460(5):455–465. 10.1007/s00428-012-1232-0 [DOI] [PubMed] [Google Scholar]
- 24.Ogawa Y, Schmidt DK, Nathan RM, Armstrong RM, Miller KL, Sawamura SJ, Ziman JM, Erickson KL, de Leon ER, Rosen DM et al. (1992) Bovine bone activin enhances bone morphogenetic protein-induced ectopic bone formation. J Biol Chem 267(20):14233–14237 [PubMed] [Google Scholar]
- 25.Carcamo J, Weis FM, Ventura F, Wieser R, Wrana JL, Attisano L, Massague J (1994) Type I receptors specify growth -inhibitory and transcriptional responses to transforming growth factor beta and activin. Mol Cell Biol 14(6):3810–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Attisano L, Wrana JL, Montalvo E, Massague J (1996) Activation of signalling by the activin receptor complex. Mol Cell Biol 16(3):1066–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebrun JJ, Vale WW (1997) Activin and inhibin have antagonistic effects on ligand-dependent heteromerization of the type I and type II activin receptors and human erythroid differentiation. Mol Cell Biol 17(3):1682–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eijken M, Swagemakers S, Koedam M, Steenbergen C, Derkx P, Uitterlinden AG, van der Spek PJ, Visser JA, de Jong FH, Pols HA, van Leeuwen JP (2007) The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. Faseb J 21(11):2949–2960 [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto M, Shoda A, Inoue S, Yamada R, Kondo T, Sakurai T, Ueno N, Muramatsu M (1992) Functional regulation of osteoblastic cells by the interaction of activin-A with follistatin. J Biol Chem 267(7):4999–5004 [PubMed] [Google Scholar]
- 30.Ikenoue T, Jingushi S, Urabe K, Okazaki K, Iwamoto Y (1999) Inhibitory effects of activin-A on osteoblast differentiation during cultures of fetal rat calvarial cells. J Cell Biochem 75(2):206–214 [DOI] [PubMed] [Google Scholar]
- 31.Pearsall RS, Canalis E, Cornwall-Brady M, Underwood KW, Haigis B, Ucran J, Kumar R, Pobre E, Grinberg A, Werner ED, Glatt V, Stadmeyer L, Smith D, Seehra J, Bouxsein ML (2008) A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc Natl Acad Sci USA 105(19):7082–7087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebisawa T, Tada K, Kitajima I, Tojo K, Sampath TK, Kawabata M, Miyazono K, Imamura T (1999) Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J Cell Sci 112(Pt 20):3519–3527 [DOI] [PubMed] [Google Scholar]
- 33.Macias -Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL (1998) Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J Biol Chem 273(40):25628–25636 [DOI] [PubMed] [Google Scholar]
- 34.Townson SA, Martinez-Hackert E, Greppi C, Lowden P, Sako D, Liu J, Ucran JA, Liharska K, Underwood KW, Seehra J, Kumar R, Grinberg AV (2012) Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J Biol Chem 287(33):27313–27325. 10.1074/jbc.M112.377960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishitoh H, Ichijo H, Kimura M, Matsumoto T, Makishima F, Yamaguchi A, Yamashita H, Enomoto S, Miyazono K (1996) Identification of type I and type II serine/threonine kinase receptors for growth/differentiation factor-5. J Biol Chem 271(35):21345–21352 [DOI] [PubMed] [Google Scholar]
- 36.Chapman SC, Woodruff TK (2001) Modulation of activin signal transduction by inhibin B and inhibin-binding protein (INhBP). Mol Endocrinol 15(4):668–679. 10.1210/mend.15.4.0616 [DOI] [PubMed] [Google Scholar]
- 37.Lian JB, Stein GS (1992) Concepts of osteoblast growth and differentiation: basis for modulation of bone cell development and tissue formation. Crit Rev Oral Biol Med 3(3):269–305 [DOI] [PubMed] [Google Scholar]
- 38.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108(1):17–29 [DOI] [PubMed] [Google Scholar]
- 39.Sakai R, Eto Y, Ohtsuka M, Hirafuji M, Shinoda H (1993) Activin enhances osteoclast-like cell formation in vitro. Biochem Biophys Res Commun 195(1):39–46 [DOI] [PubMed] [Google Scholar]
- 40.Fuller K, Bayley KE, Chambers TJ (2000) Activin A is an essential cofactor for osteoclast induction. Biochem Biophys Res Commun 268(1):2–7 [DOI] [PubMed] [Google Scholar]
- 41.Sugatani T, Alvarez UM, Hruska KA (2003) Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J Cell Biochem 90(1):59–67. 10.1002/jcb.10613 [DOI] [PubMed] [Google Scholar]
- 42.Miyake A, Mekata Y, Fujibayashi H, Nakanishi K, Konishi M, Itoh N (2017) Brorin is required for neurogenesis, gliogenesis, and commissural axon guidance in the zebrafish forebrain. PLoS ONE 12(4):e0176036 10.1371/journal.pone.0176036 [DOI] [PMC free article] [PubMed] [Google Scholar]