

Abstract
Cells establish and sustain structural and functional integrity of the genome to support cellular identity and prevent malignant transformation. In this review, we present a strategic overview of epigenetic regulatory mechanisms including histone modifications and higher order chromatin organization (HCO) that are perturbed in breast cancer onset and progression. Implications for dysfunctions that occur in hormone regulation, cell cycle control and mitotic bookmarking in breast cancer are considered, with an emphasis on epithelial-to-mesenchymal transition and cancer stem cell activities. The architectural organization of regulatory machinery is addressed within the contexts of translating cancer-compromised genomic organization to advances in breast cancer risk assessment, diagnosis, prognosis, and identification of novel therapeutic targets with high specificity and minimal off target effects.
Keywords: Higher order chromatin organization, breast cancer, epithelial to mesenchymal transition, cancer stem cells, hormone regulation, mitotic bookmarking, RUNX
Introduction.
Physiological control of gene expression is dependent on chromatin context and requires timely and dynamic interactions between transcription factors and coregulatory machinery that reside in specialized sub-nuclear microenvironments 1–5. Multiple levels of nuclear organization functionally contribute to biological control and are perturbed in cancer1–47. Morphologically, cancer nuclei are generally larger, more irregularly shaped and have altered sub-nuclear structures 23,31,48. These changes in nuclear structure have long been used by pathologists as a major diagnostic tool to detect tumor cells 23,31. While it is well-known that nuclear morphology is disrupted in cancer cells, emerging evidence supports significant contributions by concomitant changes in higher order chromatin organization (HCO). There is increasing understanding for mechanisms utilized to maintain HCO in normal cells, and the functional consequences of modifications in HCO in cancer onset and progression. Technological advances including high-throughput next generation sequencing49–53 and sophisticated microscopic techniques5,54,55 have revolutionized investigation into genomic organization within the contexts of biological control and pathology.
Cells must maintain genomic structural integrity and functional identity throughout successive generations to prevent malignant transformation56,57. The retention of cell type specific transcription factors and epigenetic histone modifications at target gene loci, designated bookmarking, has been posited to be critical to sustain cellular phenotypes58–60. Bookmarking of chromatin domains has been proposed to play a significant role in re-establishing fidelity for HCO of the genome61. Upon exit from mitosis, the biogenesis of nuclear bodies, that include nucleoli (where ribosomal RNA is transcribed) and histone locus bodies (HLB; where histone mRNA is transcribed), contribute to HCO mediated biological control62,63. These physiologically important examples of regulatory compartmentalization are obligatory for the balance between proliferation and cell lineage specificity. Reprogramming of lineage-committed cells during the initial stages of cancer is associated with loss of critical parameters of normal cellular identity. A cogent hypothesis is that cancer cells hijack an epithelial-to-mesenchymal transition (EMT) in which cells reliquish their epithelial tight junctions and polarity while acquiring mesenchymal characteristics that include migration and invasiveness (Figure 1). Many of the signaling cascades associated with this process are well known64. Signaling pathways that include TGFβ, SNAIL, ZEB, and WNT have been implicated in control that is operative in cancer stem cells (CSCs) 65–67. The cancer stem cell hypothesis postulates that a sub-fraction of tumor cells designated CSCs are competent to proliferate, self-renew, ‘differentiate’, and drive tumor initiation, growth, and recurrence68.
Figure 1. Cancer-compromised higher order chromatin organization.
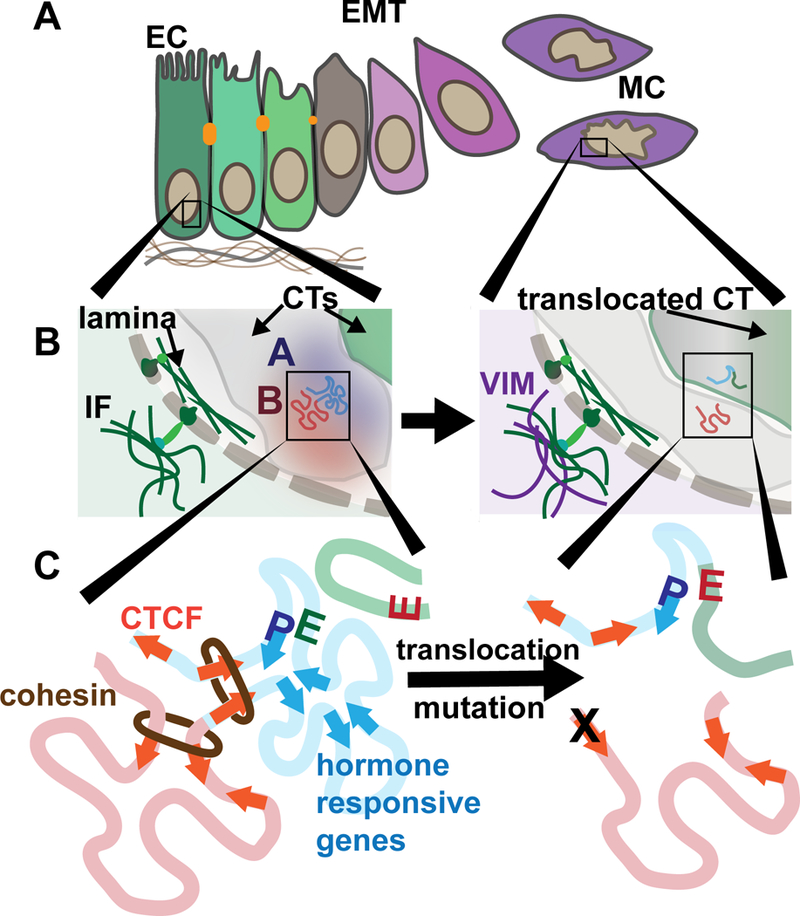
A.) An epithelial-to-mesenchymal transition (EMT) occurs during breast cancer progression during which cells relinquish their epithelial cell (EC) tight junctions and polarity while acquiring mesenchymal cell (MC) characteristics that include migration and invasiveness. B.) An inset of a portion of the nucleus is shown. The nucleo-cytoplasmic link is illustrated wherein forces from within the cytoplasm can be transferred into the nucleus. The intermediate filament (IF) protein, vimentin (VIM), is increased in expression during EMT. Portions of two chromosome territories (CTs) are shown (grey and green). Compartments within one CT are shown. An open, euchromatic, A compartment is blue and closed, heterochromatic, B compartment is red. C) In the loop extrusion model, cohesin extrudes DNA until two convergent CTCF motifs are encountered. Genes that are responsive to a stimulus (e.g. hormones) are enriched to reside within the same TADs. Alterations in the genome that occur within cancer nuclei such as translocations, deletions, and inversions may result in the disruption of proper enhancer (E)- promoter (P) interactions and result in aberrant regulation. Mutation of CTCF binding sites are frequent in cancers and mutation of these sites has been shown to disrupt looping.
In this review, we will present a strategic overview of the principles underlying epigenetics and HCO with consideration for their role(s) in EMT and CSCs during breast cancer initation and progression. The significance of hormone regulation for these pivotal regulatory processes, and the importance of the cell cycle and bookmarking in establishing and maintaining normal breast epithelial cellular identity will be discussed. The implications for these crucial regulatory dimensions of cancer underscore the need for a deeper understanding of mechanisms driving cancer-compromised organization of genome regulatory machinery to inform novel therapeutic strategies.
Higher order chromatin organization is integral to fidelity of genome regulation.
The genome is hierarchically organized at multiple, complex and interdependent levels. At the molecular level, ~146 base pairs of DNA are wrapped around an octameric core of histones (H2A, H2B, H3, H4) termed the nucleosome69. These repeated nucleosomes are configured as a ‘beads-on-a-string’ 10nm chromatin fiber57,70. Repressed chromatin has been posited to form a helical 30nm solenoid-like structure57,70. However, recent studies using small-angle X-ray scattering, cryo-EM, or super-resolution microscopy have not observed these solenoids in vivo71. These studies instead found that the beads-on-a-string structures in nuclei are not uniform, but heterogeneous varying in diameter. The advances in sophisticated microscopic and sequencing techniques have revealed fundamental principles governing higher order levels of chromatin organization that is relevant for cell structure and function. These studies have identified that the chromatin fiber is folded into globular domains designated topologically associating domains (TADs)72–75. TADs then coalesce into two main compartments that are either euchromatic, termed A co-partments, or heterochromatic, termed B compartments76,77. These are in turn comprised of six computationally distinct subcompartments, two that are euchromatic and four that are heterochromatic78. More recently, it has been demonstrated that these subcompartments exist as a spectrum of compartments wherein some TADs associate with both A and B compartments79. As expected, compartments present at the extremes of the spectrum are more indicative of euchromatic or heterochromatic states, respectively. The euchromatic A compartments are noticeably more gene rich, transcriptionally active, marked by active epigenetic signatures, and preferentially accessible to DNaseI than heterochromatic B compartments37,80. At the highest level of organization, chromosomes occupy discrete territories within the nucleus81,82.
The vast majority of the human genome does not encode proteins. Consequently, there has been speculation that these non-coding regions are so-called “junk DNA”83. While there is still no discernable function readily apparent for a portion of what was considered “junk”, increasing evidence has established that many non-coding regions provide regulatory control over gene expression and genome integrity 84. Key to fidelity of regulation, regions of the genome either activate (enhancers) or suppress (silencers) expression of their cognate genes. These elements can be located distal (even up to 1mb) from the genes they regulate. The requirement for long-range interactions of enhancers looping back to interact with their correct promoters is integral and coincident with gene expression. Disruption of physiologically responsive enhancer-promoter interactions has been shown to contribute to cancer onset and progression 85–88. For example, regulatory elements within regions several kb in length that lack protein-coding genes exhibit long-range interactions with both protein and non-protein-coding genes89. These genes include MYC, IGFBP5, KLF4, CCDC26, and DIRC3 and have critical roles in breast cancer90–93.
Enhancers are precluded from interacting with inappropriate promoters by insulator elements bound by chromatin organizer proteins that mediate long-range intra- and interchromosomal interactions94–96. Additionally, these insulators provide barriers against the aberrant spreading of heterochromatin from silencers. In performing these essential functions, insulators organize the genome into the TADs that serve as subnuclear microenvironments94–96. Regions within individual TAD microenvironments are epigenetically marked largely consistently throughout74,75 and contain genes that are expressed at relatively similar levels 97,98. These genes within the same TADs are generally co-regulated and responsive to the same transcriptional stimuli97,99. TADs also function as structural domains to constrain long-range contacts between enhancers and promoters such that they occur almost exclusively within TADs72,100. Given the inextricable link between structure and function within the context of the cell nucleus, it is important to consider the role of HCO in maintaining genomic stability and fidelity101–103, and the resulting disruptions that occur in these TAD microenvironments introduced by translocations, deletions, inversions, and mutations during cancer progression88.
CTCF and/or epigenetic dependent mechanisms contribute to higher order chromatin organization.
CTCF is a major protein involved in insulator function and mediates intra- and interchromosomal looping interactions in vertebrates104. Through interactions with chromatin remodeling proteins, histone modifying enzymes, and transcription factors, CTCF is implicated in a broad spectrum of critical regulatory functions including imprinting105, X chromosome inactivation106, and organizing the major histone locus41. CTCF and its binding sites are mutated in many cancers, including breast cancer, suggesting its functions are perturbed upon malignant transformation107–110.
While the mechanism of how chromatin loops and TADs are established is not fully elucidated, CTCF as well as its interaction with the structural maintenance of chromosome (SMC) cohesin complex are key components of HCO. The best-accepted model to explain TAD formation and maintenance involves a loop-extrusion model111,112. This model proposes that a cohesin ring holds two strands of DNA together and creates loops by actively extruding the DNA. Once cohesin encounters a CTCF motif that is in a convergent orientation, a loop is formed112 (Figure 1). Because CTCF is essential113, investigations have focused on its depletion using the auxin inducible degron (AID) or siRNA methods. Using an RNAi method, it was found that CTCF knockdown slightly decreased intra-TAD contacts while increasing inter-TAD interactions114. Depletion of CTCF using the AID system115 resulted in greater reduction of CTCF and led to a loss in TAD insulation, but does not alter intra-TAD contacts116. In this study, ~20% of TAD boundaries were unaffected by CTCF-independent upon auxin mediated CTCF degradation. In contrast, another study found that while CTCF knockdown reduced genomic occupancy of the cohesin complex, its loss only slightly weakened TAD boundaries and the vast majority of TADs remained unaltered117. Although the segregation of A and B compartments generally occurs at TAD boundaries, knockdown of cohesin and/or CTCF did not affect A and B compartmentalization116,118.
While CTCF plays a major role in chromatin organization, its absence at many TAD boundaries suggests alternative mechanisms, including epigenetic modifications, for delineation of TAD structures. The fact that TADs are found in species that do not have orthologues of CTCF including Caenorhabditis elegans, Arabidopsis thaliana, Schizosaccharomyces pombe, or Caulobacter crescentus and Escherichia coli provides definitive evidence for CTCF-independent mediation of TAD partitioning, particularly in ancestral genomes119–123. In these species, other insulators, may play an important role in defining TAD boundaries. Alternatively, evidence suggests that the folding of nucleosomes from beads-on-a-string into chromatin domains may be directly related to the differential compaction of chromatin induced by active versus inactive epigenetic states. High levels of acetylation on histone tails results in destabilization of chromatin domains124. This destabilization of chromatin domains could explain the enrichment of epigenetic marks indicative of actively transcribed genes (e.g. housekeeping genes) at TAD boundaries74. In fact, expression data is capable of predicting the three-dimensional folding of the genome125,126. The partitioning of TAD boundaries based upon active expression independently of CTCF binding appears to be more frequent in drosophila melanogaster125,127. TAD boundaries in drosophila are indicative of transitions between open and closed compartmentalization to an even greater extent than in human nuclei125. In fact, the differential packing ability of active and inactive genes was shown to predict TAD boundaries in drosophila based upon polymer simulations127. TADs in drosophila are therefore responsive to transcriptional stimuli (e.g. recovery from heat-shock128 or zygotic genome activation, or transcriptional inhibition129). The fact that the HCO of genomes from lower organisms are more specified by epigenetic states than HCO in the human genome suggests that human cells have more tight control over HCO. Loss of this tight control over epigenetic regulation and HCO are fundamental alterations that occur during breast cancer progression.
Parameters of breast cancer genome topography: Epithelial to mesenchymal transition, cancer stem cells, epigenetics and higher order chromain organization.
Breast cancer is the most common cancer in women, encompassing a diverse array of subtypes with different cellular origins (luminal versus basal) and distinct molecular alterations (e.g., hormonal status including ER, PR, and HER2) that relate to malignancy 130. Gross morphologic alterations in nuclei in breast cancer are indicative of poor prognosis131 and can be used to predict ER status suggesting putative differences in nuclear morphology between these breast cancer subtypes132. Despite considerable advancements deciphering critical genes and pathways driving the various subtypes of breast cancer, the initial molecular events transforming normal cells require more investigation.
During cancer progression, cells lose epithelial-like polarity and acquire mesenchymal-like phenotypes that include increased migration, invasiveness, resistance to chemotherapy, and immune-response in a process termed Epithelial to Mesenchymal Transition (EMT)133. The hallmark of EMT is decreased expression of tight junction proteins including cytokeratin and E-cadherin, and the activation of the mesenchymal genes such as N-cadherin, Vimentin (a cytoskeletal intermediate filament), and Fibronectin64. Due to the importance of EMT in normal development, EMT is precisely regulated by coordinated crosstalk between transcription factors and signaling cascades. For example, E-cadherin expression is downregulated by EMT-inducing transcription factors that are stimulated by Wnt and Notch pathways134. EMT can be activated by extracellular signals, such as cytokines (e.g. TGFβ, BMP, and TNFβ), growth factors (eg. FGF, EGF), and certain extracellular matrix (ECM) proteins135. In turn, the EMT process induces a dynamic reorganization of the cytoskeleton to form membrane protrusions necessary for migration and invasion134. Recent evidence has demonstrated an interaction between cytoskeletal structure, nuclear morphology, and higher order chromatin organization (136–139). For example, the cytoskeletal arrangement of vimentin or actin correlate with nuclear morphology, and depolymerization of vimentin using withaferin A perturbs nuclear morphology140. Proteins that link the cytoskeleton to the nuclear envelope can transfer cytoplasmic forces into the nucleus. Although it is known that actin shuttles into and out of the nucleus, the function of nuclear actin in mediating HCO is unclear. In one study, it was found that cells overexpressing an NLS-containing actin demonstrated decreased expression of adhesive genes, and exhibited altered cytoskeletal and focal adhesion organization and inhibited cell motility relative to cells overexpressing wild type actin141. Moreover, actin or actin related proteins (ARPs) can function in association with chromatin remodelers and/or act as cofactors with other nuclear complexes142,143. Moreover, TGFβ-induced EMT results in genomic instability associated with the suppression of several nuclear envelope proteins that are implicated in the regulation of mitosis144. Together, this evidence suggests a complex interplay between the signaling cascades, cytoskeletal rearrangement, and genome instability induced by EMT and HCO in breast cancer (Figure 1).
Efforts have been made to prevent or revert EMT or CSC properties which can restrain invasion, metastasis, and chemo-resistance. A promising therapeutic strategy is to target the epigenetic properties of cancer cells. For example, 5-azacytidine was shown to block DNA methyltransferase (DNMT) activity leading to hypomethylation and gene de-repression, thereby preventing EMT in vitro145. ZLD1039, an EZH2 inhibitor, also demonstrated a strong anti-cancer effect by inhibiting breast tumor growth and metastasis146. Restoration of factors which function epigenetically is another promising avenue for breast cancer treatment. For example, reduced levels of BRMS1L (breast cancer metastasis suppressor 1 like) is associated with breast cancer metastasis and poor patient survival147. BRMS1L was shown to epigenetically silence the expression of FZD10, a receptor for the Wnt/β-catenin pathway. Therefore, restoring BRMS1L levels can potentially be used to inhibit aberrant Wnt signaling in breast cancer patients.
Although the requirement for EMT to support cancer metastasis has been challenged148,149, it is well acknowledged that EMT is in fact a major driving force in cancer stem cell formation65,150. CSCs are a subpopulation of tumor cells which are capable to form new tumors, self-renew, and “differentiate” into non-stem like cancer cells151. When injected into immunocompromised mice, the CSCs can form tumors with much higher efficiency compared with non-CSC tumor cells152. Multiple lines of evidence have demonstrated that activation of EMT signaling pathways increases the mesenchymal-like CSC population65,150. For example, the E-cadherin promoter is hypermethylated by the EMT-inducing transcription factors Yap, Snail, and Zeb153–155.
RUNX-mediated control of Epithelial-to-Mesenchymal transition and breast cancer stem cells.
Our laboratory has demonstrated that the RUNX1 transciption factor has a key role in supporting the normal breast epithelial phenotype156–158. Depletion of RUNX1, not only initiated EMT 156, but also increased the CSC population in breast cancer cells157, through TGFβ and TGFβ independent mechanisms. This suppression of breast CSCs is regulated through multiple signaling cascades including ZEB1157 and YAP159. The regulation of RUNX1 in suppressing ZEB1 is of particular interest considering that the poised epigenetic state of the ZEB1 promoter has been shown to be crucial for generation of CSCs160. This is even more intriguing considering RUNX1 and ZEB1 are both downstream of TGFβ161. In contrast to RUNX1, RUNX2 (a driver of metastatic bone disease) induces EMT in breast and other cancers162 by upregulating the expression of SNAI2163,164.
RUNX transcription factors directly contribute to chromatin looping by recruiting mediators, chromatin remodelers, and chromatin organizing proteins to regulatory elements of target genes158,165. For example, in hematopoietic stem cells, RUNX1 contributes to the interaction of the CD34 promoter to its distal enhancer 4. Likewise, RUNX2 was shown to bind the promoter of Supt3h and facilitate long-range interactions between the Supt3h and the RUNX2 promoters166. Similar to other transcription factors, RUNX1 has also been shown to be enriched at TAD boundaries and facilitate HCO that is functionally relevant in early stage luminal ER+ BrCa 167. Another level of HCO, that involves all RUNX factors, is their unique protein domain that targets RUNX to subnuclear sites via a nuclear matrix targeting signal (NMTS). This NMTS is essential for assembling multimeric complexes containing KATs, HDACs, and coregulatory factors for signaling pathways critical to cancer progression (e.g. SMADs, WWD, and P53)158. Together these studies suggest RUNX factors are regulators of EMT and can potentially influence HCO in breast cancer.
Hormone signaling and its impact on higher order chromatin organization.
Nuclear hormone receptor (NR) signaling is a major contributor to altered epigenetic and gene expression profiles during breast cancer progression. NRs are ligand-activated transcription factors that drive the development and maintenance of normal cellular phenotypes168, and their dysregulation can result in the loss of key aspects of cellular identity in cancer. Despite the important role these signaling cascades have in modifying the epigenetic landscape in breast cancer cells, the contribution of higher order chromatin organization to these events is less well understood. Open questions remain regarding the contribution of individual NRs to epigenetic signatures and higher order chromatin structures that drive EMT during early and late stage tumor development.
The importance of the biological activity of hormones in breast cancer was indicated by the removal of the ovaries in women, which greatly reduced further metastasis of breast cancer in these patients169. Additionally, it is well appreciated that the active metabolite of estrogen, 17β-estradiol, is required for the development of normal breast tissue and contributes an oncogenic role in breast cancers 170. The invention and application of microarray and next generation sequencing technologies has expanded our understanding and classification of breast cancers 171 and to this end, the intrinsic molecular subtypes of breast cancer are determined by the expression of different genes including hormone receptors 172. Luminal A or B and unclassified/normal-like breast cancers are characterized by the presence of estrogen receptor (ER, Reviewed in 173) and/or progesterone receptor (PR), while triple-negative or basal-like and HER2-enriched subtypes are hormone-receptor negative (Reviewed in 174). Other critical hormone receptors that have been identified in breast cancer are the androgen receptor (AR), glucocorticoid receptor (GR) and thyroid receptor (TR) 175,176.
Hormone signaling is a critical regulator of EMT.
The lack of proper hormone regulation may be one of the key requirements altering cellular identity during breast cancer EMT. The best-studied and arguably most critical hormone in EMT and breast cancer progression is estrogen. Estrogen promotes an epithelial phenotype by suppressing TGFβ, MTA3, and NF-kB. Indicative of EMT, loss of ERα results in altered expression of EGFR, HER2, matrix metalloproteinases and their endogenous inhibitors. Both Snail1 and ZEB1, which are elevated in EMT and in breast CSCs, in turn suppress ERα expression. ERβ has also similarly been shown to suppress EMT. Other hormones also play critical and opposing roles in EMT. For example, growth hormone induces EMT177 whereas prolactin inhibits EMT178. While several EMT inducing genes were increased by PR during mammary alveologenesis179–181, progesterone reversed EMT phenotypes in basal-like breast cancer via a membrane bound PRα mediated pathway 182. Therefore, these studies indicate a complex role for progesterone in normal breast development where it induces EMT versus basal breast cancer where it reverses EMT.
In addition to their roles in EMT and CSCs (discussed above), RUNX factors have been implicated in ER signaling. Loss of function mutations in the DNA binding-Runt homology domain of RUNX1 were detected with a particular frequency in the luminal A ER+ subtype of breast cancer183–186 (Figure 2). Mechanistically, RUNX1 has been shown to recruit and tether ERα to the genome in breast cancer187. A conditional knockout of Runx1 in mice resulted in a significant reduction in ER-positive mature luminal cells. This phenotype can be reversed by Trp53 or Rb1 mutation, suggesting a role for RUNX1 in ER+ luminal breast cancer with background mutations in P53 or RB1188. Loss of RUNX1 in the luminal A subtype of breast cancer was also shown to facilitate estrogen‐induced WNT signaling by suppressing AXIN1189. In contrast, the oncogenic activities of RUNX2 were antagonized by estradiol stimulation190.
Figure 2. Mutations in the DNA binding- Runt homology domain of RUNX1 in breast cancer.
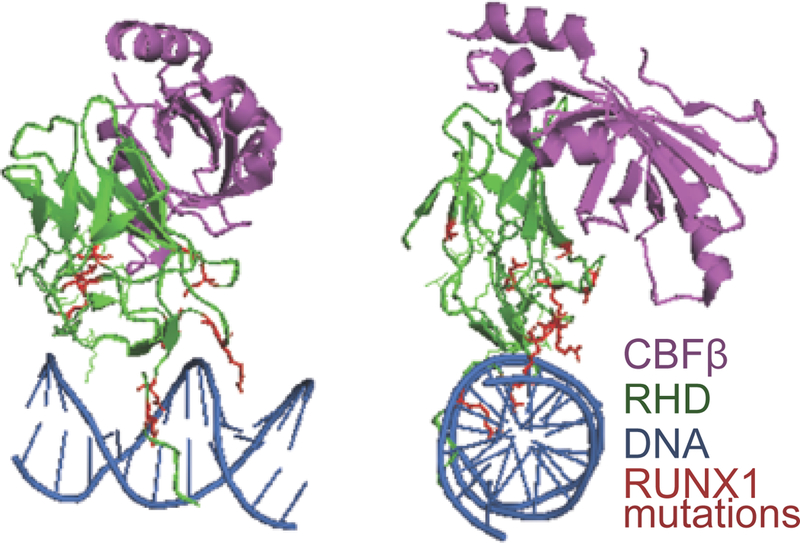
The structure of RUNX1’s Runt homology domain (RHD; rendered based on the Protein Data Bank code 1H9D293 is shown in two orientations rotated 90 degrees relative to each other (front and side). CBF‐β is shown in purple, DNA binding-RHD is in green, and DNA is in blue. Mutations found in the RHD in breast tumor patient samples (red) suggests a loss of RUNX1 function in breast cancer.
Estrogen receptor α coordinates long-range chromatin interactions to drive aberrant transcription in breast cancer.
Estrogen-dependent breast cancer is characterized by abnormally high levels of ERα expression191. ERα acts as a driver of tumorigenesis in about 80% of human breast cancers191. Therefore, endocrine therapies that target ERα are the cornerstone of breast cancer treatment. The tumor-promoting activity of ERα depends on dynamic interaction with dozens of other factors, including pioneer factors and chromatin remodeling complexes, to regulate chromatin structure and gene expression. ERα’s most reliable cofactor, FOXA1, was discovered through the observation that forkhead motifs are heavily enriched within ERα binding sites192. FOXA1 is a pioneer factor, meaning it is able to interact with compacted DNA and unravel it to facilitate the subsequent binding of other transcription factors193. It has been shown to be required for ERα binding in breast cancer cells, and its knockdown slows the growth of the MCF7 cell line194. The transcription factor GATA3 has also been shown to be a key player in estrogen-dependent gene regulation195. Interestingly, GATA3 is required development of normal mammary glands196, suggesting an important role in promoting cellular differentiation, yet silencing of GATA3 inhibits estrogen-dependent breast cancer cell proliferation197. Both FOXA1 and GATA3 are required for establishment of a stable estrogen-responsive transcriptional complex, and they both serve as prognostic indicators for response to antiestrogen therapy192,198.
The organization of the estrogen-dependent breast cancer cell genome is defined by ERα activity. ERα transcriptional activation is mediated through a complex network of ER binding sites located both proximal and distal to transcriptional start sites of target genes199. Many of the distal binding sites have been shown to act as transcriptional enhancers that are involved in long range chromosomal interaction, transcription complex formation, and wide-spanned chromatin rearrangement200,201. Studies of this phenomenon indicate that ERα can regulate a number of its target genes in a relatively confined space, which requires the arrangement of different regulatory regions into a single transcriptional hub. For example, ERα was not only recruited to bind a known target in MCF7 ER-positive breast cancer upon estrogen stimulation, but also resulted in the regulation of enhancer-promoter interactions mediating transcription193. Genes that were contacted by enhancers upon estrogen stimulation contained increased transcriptional activity. With the development of ChIA-PET (a method for determining protein mediated intra-and inter-chromosomal contacts), global ER-mediated chromatin interactions were detected201. This comprehensive chromatin map of ER-alpha revealed that long-range chromatin interactions loop distal promoters together for coordinated transcriptional control. Furthermore, distant estrogen response elements localized in regions frequently amplified in ER positive breast cancers form long-range interactions that support estrogen mediated signaling. These gene clusters potentially predict poor clinical outcomes and drug resistance in breast cancer 202. Estradiol stimulation of MCF7 further demonstrated that hormone-stimulation can function through 3D chromatin organization, its core receptor (ER in this case), epigenetics and gene expression203. Further dissection of this multi-step process (hormone stimulation > receptor activation > recruitment of chromatin remodeling factors > changes in HCO and gene expression) will allow a deeper understanding of the extent to which estrogen-dependent transcriptional dysregulation in breast cancer is influenced by defects in chromatin organization.
In addition to the characterization of estrogen signaling in ER positive breast cancer others have begun to study the effects of other types of hormone-signaling on HCO. Progestin and estradiol influence topologically associated domains (TADs) in the human breast cancer cell-line T47D, that expresses ER and PR97. While the majority of TAD boundaries remain unaltered after 1 hour of progestin stimulation, genes within 20% of TAD regions display differential expression. Regions that were responsive to progestin showed some coincidence with estradiol altered regions, however, elements unique to estradiol stimulation are also detected. Hormone-induced alterations in gene expression and chromatin remodeling, result in simultaneous changes in intra-TAD interactions within TADs that are hormone responsive. Furthermore, stimulation with glucocorticoids, which activate the glucocorticoid receptor also alter long-range chromatin interactions, DNAseI hypersensitivity, and corresponding gene expression programs in murine breast cancer cells204.
Progesterone receptor enhances and blunts estrogen receptor signaling through epigenetic modifications in breast cancer.
The progesterone receptor (PR) is a critical player in progression, therapeutic responsivity and eventual outcome of breast cancers. These receptors when bound to DNA induce assembly of chromatin remodeling complexes and cofactors to induce changes in gene transcription. PR amplifies ER expression in breast cancer cells through direct binding to low-methylated ESR1 promoter. Loss of PR expression results in an increased methylation of the ESR1 promoter and re-expression of PR did not restore ER expression or decrease methylation205. Not surprisingly, methylation of PR-responsive promoters genome-wide impedes PR binding to consensus response elements and subsequent changes in gene expression205. Demethylation of ESR1 in ER negative breast cancer cells can reactivate ERα expression and restore sensitivity206.
In addition to directly increasing ER expression in breast cancer, unliganded PR increases breast cancer cell proliferative response to estrogen and enhances antiestrogens effectiveness through inducing changes in chromatin organization via a scaffolding complex that includes ERα and PELP1 transcriptional co-regulator207. This unliganded PR binds genomic sites and targets a repressive complex containing HP1γ (heterochromatin protein 1 gamma), LSD1 (lysine-specific demethylase 1) among other co-repressors to induce a closed chromatin conformation that precludes gene expression. This includes approximately 20% of hormone-inducible genes in breast cancer cells, keeping these genes silenced prior to hormone treatment. Upon hormone treatment, the liganded PR induces displacement of the repressor complex and allows the recruitment of coactivators needed for chromatin remodeling and increased gene expression207.
Addition of hormone can magnify these nuclear events and also trigger a kinase signaling cascade through activation of cell membrane receptors to amplify these events 208,209. Phosphorylated and under-SUMOylated unliganded PR recruits steroid receptor coactivator 1 (SRC1) to regulate the expression of growth-promoting genes and SUMOylated PR recruits histone deacetylase 3 (HDAC3) to reduce chromatin accessibility and decrease expression of the same genes209. Of note, the liganded PR also can recruit the chromatin remodeling enzyme BRG1 associated with the demethylase repressor complex HPYγ-LSD1 anchored by the histone methyltransferase SUV39H2 to induce heterochromatin. This hormone-dependent transcriptional repression is mediated through BRG1 recruitment to repressed genes involved in cell proliferation and apoptosis. The pioneer factor FOXA1 marks the hormone-repressive promoters enabling BRG1, and not additional associated factors (BAFs), to mediate heterochromatinization210. Knockdown of BRG1 in normal-like mammary epithelial ER-low MCF10A resulted in altered HCO and expression of key extracellular matrix genes that can exert mechanical forces and affect nuclear structure{Barutcu, 2016 #536}. Distinguishing the effects of perturbed BRG1 signaling on HCO in ER positive breast cancers will be of particular interest.
Androgen Receptor signaling in breast cancer is context-dependent.
The androgen receptor (AR) is a well-characterized clinical target in male prostate cancer, however its diagnostic and therapeutic potential in female breast cancer has recently emerged in the literature. AR has clinical implications in both ER-positive and ER-negative breast tumors211. In ER-positive tumors, AR expression was associated with positive clinical outcomes. Higher AR expression was predictive of a more favorable response to ER-targeted therapies, such as tamoxifen and aromatase inhibitors212. There is also preclinical evidence that breast cancers that have become resistant to tamoxifen can be effectively treated with AR-targeted endocrine therapies such as bicalutamide and enzalutamide213.
Triple negative breast cancer (TNBC) has recently been re-organized into several subcategories214. One of these subsets of triple negative breast tumors, termed luminal androgen receptor (LAR) tumors, in which AR has been shown to be a driver of EMT and tumor progression. LAR breast cancer cells are sensitive to androgen therapies, such as bicalutamide and enzalutamide, in vitro and in vivo 215–217. Other molecular subtypes have also exhibited sensitivity to enzalutamide in vivo216. The underlying mechanisms for growth suppression by anti-androgens in these cancers has yet to be fully delineated. However, it has been demonstrated that AR plays a role in promoting growth-factor receptor, PI3K/AKT, and WNT/β-catenin signaling in TNBC cells216,217. While AR is a driver of EMT through these signaling cascades, and HCO may be a critical component of EMT during breast cancer progression (discussed above), the potential for AR to alter HCO during EMT is unexplored.
Thyroid hormone signaling in breast cancer.
The actions of non-steroidal NRs in breast cancers are not well characterized218. Both thyroid hormone receptor alpha (TRα) and thyroid hormone receptor beta (TRβ) are expressed in breast tissue. In BRCA-positive breast cancer TRα and TRβ exhibit opposing roles in prognostic survival; greater expression of TRα strongly correlates with a decrease in overall survival whereas expression of TRβ is associated with improved survival219. Additionally, the isoform of TRα has been observed to be critical as expression of TRα2, a splice variant without a triiodothyronine (T3) binding site, is associated with improved survival220. TRα2 acts antagonistically to TRα1, which exhibits a functional LBD, by binding to TREs and blocking TRα1 from interacting with the chromatin. This blocks thyroid hormone mediated actions arising from TRα1.
Notably, there is compelling evidence that loss of TRβ, a member of the thyroid hormone receptor (TR) family, through genomic modifications and epigenetic silencing is characteristic of breast and other solid tumors221–227. TRβ is silenced or mutated in nearly 60% of invasive breast cancers219,228–231. Of clinical significance, expression of wild-type TRβ is associated with a good prognosis in BRCA-positive breast cancer219 as well as early breast cancer232 and indicates a positive responsivity to chemotherapy230,233. TRβ, both unliganded and liganded, regulates gene expression via interaction with co-regulators and chromatin remodeling complexes234–240. Disruption of TRβ in breast cancer is therefore expected to alter the assembly of co-factors needed for transcriptional programming. In xenograft studies, loss of TRβ in malignant breast cells results in tumor growth and progression whereas restoration of TRβ function reverses these effects and critically blocks estrogen-induced breast cell tumor growth241–245. These observations indicate that not only does TRβ repress tumorigenic signaling, but TRβ may specifically counter ERα tumorigenic signaling in ER+ breast cells. Remarkably, the mechanisms by which TRβ blunts breast tumor growth and protects normal breast epithelial cell function are currently unknown. TRβ, both unliganded and liganded, regulates gene expression via interaction with hormone response elements and recruitment of co-regulators and chromatin remodeling complexes237,239,246–248. The impact that the recruitment of these chromatin remodelers has on HCO requires further investigation. It will be of particular interest to determine whether TRβ signaling counters the estrogen-mediated alterations in HCO discussed above and whether maintainence of the normal mammary epithelial cellular identity requires the long range enhancer-promoter contacts mediated by TRβ.
Nuclear receptor crosstalk has implications for breast cancer outcomes and treatment.
In early stage, hormone receptor positive, and dedifferentiated breast cancers, the dynamic gene expression programs are framed by an array of NRs and their cofactors. The studies that have defined NR-regulated transcription and NR-binding events have largely been studied as isolated events using single hormones. This over-simplified approach to understanding nuclear receptor function has become inadequate as it becomes increasingly clear that hormones and NRs do not act alone. Emerging evidence shows that co-expressed NRs exhibit extensive crosstalk with each other in normal tissue and in hormone-driven cancers.
The role of steroid hormone receptor crosstalk in breast cancer has been recently reviewed; specifically interactions between ER and PR, ER and AR, and crosstalk from glucocorticoid receptor249. Briefly, ER and PR have been demonstrated to form protein-protein interactions and PR expression can drive ER-mediated upregulation of over 200 genes in vitro207. Progesterone treatment aids in the recruitment of ER to over 14,000 EREs in T47D cells through a progesterone-dependent protein-protein interaction172. However, progesterone treatment repressed the oncogenic properties of E2 in a xenograft of these cells172,192.
TR/ER interaction at common DNA motifs with opposite transcriptional effects has been described250 and an overlap in estrogen and T3 responsive genes has been noted in breast cancer251. As with PR, TRβ, both unliganded and liganded, regulates gene expression via interaction with co-regulators and chromatin remodeling complexes 234–239. Disruption of TRβ in breast cancer is therefore expected to alter the assembly of co-factors needed for transcriptional programming. Addition of T4 stimulates breast cancer cell proliferation although the effect is likely non-genomic mediated through T4 -αvβ3 integrin and kinase signaling252. In the presence of ER, T3 blunts cell proliferation253. The role of ligand, T3 or T4, has yielded controversial results revealing the complexity of NR cross-talk and interactions that are context dependent.
It is well-established that BRG1 facilitates gene expression control by steroid NRs 210,254,255 and is recruited to ER-responsive promoters 256–257. PR directly interacts with BRG1 in the absence of additional accessory factors to suppress gene expression in breast cancer 210 and thus may inhibit ER activity to diminish resistance to estrogen-based therapy 172. Our recent studies established that TRβ interacts with BRG1258 to synergistically induce changes in chromatin accessibility resulting in decreased expression of an oncogene, RUNX2, in opposition to ERα action. As TRβ and ERα can differentially regulate gene expression mediated through the same DNA binding site and BRG1 cooperatively enhances gene suppression and activation respectively, overlapping genome occupancy by these factors should reveal a subset of coordinately regulated genes central to maintain a normal breast phenotype or tumor suppression program. Our findings point to a convergence of TRβ and ERα signaling whereby TRβ counters ERα genomic occupancy, nuclear organization and transcriptional programs in hormone-dependent cancers. The BRG1 dependent crosstalk between ER and PR as well as TR and ER may be a generalizable mechanism of epigenomic crosstalk between members of the NR superfamily of genes. Given the importance of hormone signaling in regulating the epigenome and gene expression in breast cancer, a deeper understanding of how these signaling cascades impact cellular phenotypes will inform therapeutic strategies. Understanding the role(s) that HCO has in mediating these processes is still in its infancy.
The challenge of cellular division and implications for genomic organization.
Mitosis represents a major reconfiguration of the interphase genome organization every cell cycle. This raises a fundamental question of biological and clinical importance: what mechanisms control reacquisition and preservation of cellular identity during proliferation and growth? As cells prepare for mitosis their chromosomes are packaged into rod-like structures. During prophase TAD structures are lost in a condensin (structural maintenance of chromosomes complex) dependent manner. In early prometaphase a helical arrangement of consecutive 400kb outer loops containing 80kb inner loops emanate from a central spiral-staircase on a condensin scaffold. These loops progressively increase in size to ~12kb during prometaphase, while secondary loops are formed259. During this process, while many protein factors are excluded from the condensing mitotic chromosome, a fraction of transcription factors and chromatin remodelers are retained. This retention of binding during mitosis is termed bookmarking260.
Mitotic Gene Bookmarking in Biological Control.
The first evidence of mitotic bookmarking by a transcription factor was reported in 2003 by our group 261. RUNX2 was shown to remain associated with chromatin throughout mitosis occupying both cell growth-related ribosomal RNA (rRNA) genes that are transcribed by RNA Pol I, as well as cell proliferation and phenotype-related genes regulated by RNA Pol II 262. Consistent with these findings, components of RNA Pol I and II machineries are retained on mitotic chromosomes263–265. Subsequently, our group provided evidence that, during differentiation of multipotent mesenchymal stem cells (MSCs) into myoblasts, osteoblasts or adipocytes, mitotic bookmarking of the ribosomal RNA (rRNA) genes by Myc was replaced by respective lineage-specifying factors MyoD, myogenin, RUNX2, and C/EBPβ. Myc is an activator of rRNA genes during proliferative stage of MSCs. The replacement of Myc by these factors suppresses RNA Pol I-mediated transcriptional control of rRNA genes through an interaction with the upstream binding factor 1 (UBF1; 262,264,266. Concomitantly, these lineage-specifying factors occupy RNA Pol-II regulated genes involved in cell proliferation and fate determination.
Mitotic bookmarking of RNA Pol-II genes by various transcription factors has been demonstrated to be a key component regulating cellular identity in a host of physiological conditions. These include GATA1 in hematopoeisis 267 components of the MHC Class II enhanceosome in B lymphoblastoids 268; FOXA1 in liver development 269, and hepatocyte nuclear factor 1 β (HNF1β) in the early steps of pancreas, kidney, and liver development 270. Clinically relevant mutations found in HNF1β of patients suffering from renal multicystic dysplasia and diabetes; these mutations prevented HNF1β to mitotically bookmark DNA, highlighting clinical relevance of mitotic gene bookmarking 271. Together, these findings identified mitotic gene bookmarking as a wide-spread epigenetic mechanism for coordinate control of cell growth, proliferation and phenotype maintenance.
In pluripotent or totipotent cells or breast cancer cells that have lost aspects of their cellular identity, the presence of both activating and suppressing histone marks at a single genomic locus, designated bivalency, has been posited to be critical for a poised plastic state of chromatin160,272–274. Interestingly, in pluripotent cells bivalent control of a large subset of genes is confined to mitosis, while histone mediated epigenetic suppression is constitutive throughout the cell cycle275. Mitosis restricted presence of activating histone modifications poises phenotypic genes for the potential to subsequently be expressed at lineage commitment. At that time, histone specific repression is relinquished. It has recently been observed that bivalency may be recapitulated when phenotype-specific genes are downregulated in early-stage cancer276. Such oncofetal epigenetic control may reflect loss of cell type specificity and reemergence of progenitor-like properties.
Mitotic bookmarking and nuclear organization.
The retention of factors on mitotic chromatin has been implicated in higher order chromatin organization. For example, it has been posited that the chromatin organizer proteins, CTCF and SMC3, have been shown to be retained on mitotic chromosomes277–279. Analysis of drosophila CTCF (dCTCF) occupancy identified sites that are bound throughout the cell cycle and those that are bound only in interphase or mitosis. dCTCF binding sites that fell within the same class (ie. Throughout the cell cycle versus only in interphase or mitosis) were highly enriched at TAD boundaries280. In contrast, a more recent study demonstrated that CTCF binding is lost in prometaphase. ATAC-seq determined that while CTCF sites became closed during metaphase, transcription start sites were accessible, consistent with the view that transcription factors bookmark. Dekker and colleagues, along with other investigators 37,279,281–284 have found that the histone variants and modifications are maintained during mitosis suggesting a major role for epigenetics in bookmarking. In addition, epigenetic modifying complexes are also maintained on mitotic chromosomes. For example, the polycomb protein PSC is partially retained during mitosis, and its occupancy is enriched at TAD boundaries285. Given the potential role for the segregation of active versus inactive chromatin in delineating TAD structures, bookmarking by epigenetic histone modifications may provide the basis for maintaining cellular identity and HCO.
Mitotic Gene Bookmarking in Cancer.
Given the documented examples of mitotic gene bookmarking thus far, it comes as no surprise that this epigenetic mechanism has significant roles in promoting a cancerous phenotype. For example, in acute myeloid leukemia, bookmarking by RUNX1-ETO (an oncogenic fusion protein between the DNA binding domain of RUNX1 and the entire ETO protein including its NHR domain) has been demonstrated at growth-related rRNA genes, as well as RNA Pol-II genes involved in myeloid cell differentiation. In comparison to normal RUNX1, RUNX1-ETO results in the opposing regulatory effects on mitotically bookmarked genes 286 regulating vital cellular processes such as differentiation, proliferation, apoptosis, and self-renewal to promote leukemogenesis287. Given the roles of RUNX1 in estrogen signaling, and in suppressing EMT and CSC phenotypes in breast cancer, bookmarking by RUNX1 could be a fundamental mechanism maintaining the normal mammary epithelial phenotype.
Reestablishing chromatin domains and nuclear bodies upon exit from mitosis.
The rod-like chromosomes found in mitosis rapidly decondense into chromosome territories (CTs) following completion of cell division and initiation of G1. Within CTs, TADs are decondensed during G1 corresponding with their level of activity79. These TADs are then replicated as units with more active TADs being replicated earlier than those that are less active288. This is consistent with the longstanding evidence that highly transcribed genes tend to replicate earlier in S phase289. This correlation is not absolute and reflect the presence of genes that are minimally expressed within TADs that are predominantly more active and vice versa. TAD structures may therefore be more determinative for replication timing than expression of individual genes.
The differential acetylation of genomic regions of mitotic chromatin may be the primary mechanism by which nuclear bodies are re-established from mitosis into G1 and S phase. Nucleolar organizer regions (NORs) contain the rRNA genes discussed above are present on five different acrocentric chromosomes are bookmarked during mitosis262,264,266. This bookmarking provides a basis for the reassembly of these NOR-bearing chromosomes and biogenesis of nucleoli63,290 (Figure 3). Interestingly, it was discovered that there is a dominant nucleolus that associates with more of these acrocentric chromosomes. Furthermore, particular subsets of these NOR-bearing chromosomes preferentially associated with the same nucleolus291. Epigenetic bookmarking of the histone genes may also be critical for the HCO of the histone locus body wherein the regulation of the histone genes occurs during S phase62,292. The HLB that contains the major histone gene locus is contained within a TAD. In this TAD, three subclusters of histone genes form an active chromatin hub, while two inactive histone genes are excluded. Other regions loop back into this hub suggesting additional potential regulatory roles for HCO in histone gene expression. As expected with the increased proliferative state of breast cancer cells, this region is the most upregulated cluster of genes in breast cancer in vitro and in tumor samples relative to matched controls. In addition, CTCF is present within the HLB and occupies the TAD boundaries around the major histone gene locus, and therefore may play a critical role in the determination of the HCO of this nuclear body41.
Figure 3. Mitotic bookmarking maintains nuclear organization, cellular identity and genome regulation in daughter cells.
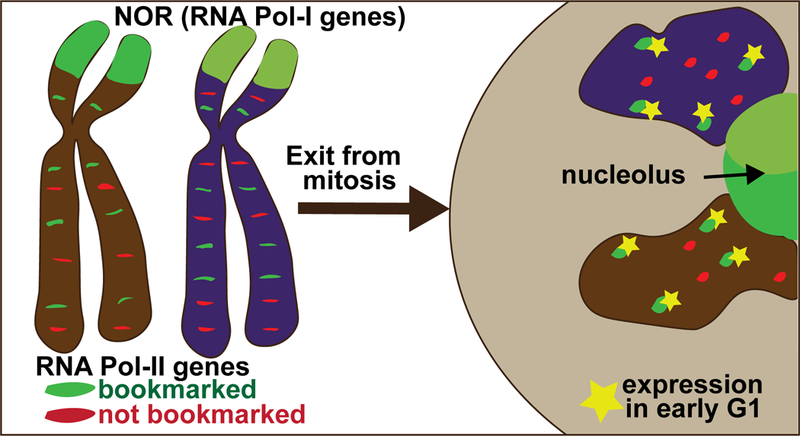
Bookmarking is the retention of transcription factors and epigenetic modifications on mitotic chromosomes. Genes that are bookmarked (green) are active in early G1 compared with genes that are not bookmarked during mitosis (red). Bookmarking of the nucleolar organizer regions (NORs) is key to the biogenesis of nucleoli upon exit from mitosis.
Conclusions.
Cells establish and retain structural and functional integrity of the genome to support cellular identity and prevent malignant transformation. Mitotic bookmarking sustains competency for normal biological control, and propetuates gene expression associated with transformed and tumor phenotypes. Regulatory cascades that include RUNX and hormone signaling are altered in EMT and breast CSCs, thereby contributing to breast cancer onset and progression. And downstream, epigenetic mechanisms including histone modifications and higher order chromatin organization are perturbed. In turn, higher order chromatin organization provides a blueprint for control of gene expression within the three dimensional context of nuclear architecture. Elucidation of mechanisms that mediate the genomic organization of regulatory machinery will provide novel insight into control of cancer-compromised gene expression. This understanding can translate to enhanced capabilities for tumor diagnosis, prognosis, and provide options for targeted therapy.
Acknowledgements:
This work was supported by NIH grants NCI P01 CA082834 (G.S.S., J.L.S.), R01 CA139322 (G.S.S.), R37 DE012528 (J.B.L.), NCI 1F32 CA220935 (A.J.F., G.S.S, J.L.S.), U01 CA196383 (J.L.S) and the Charlotte Perelman Fund for Cancer Research (G.S.S.) and an Institutional Research Grant (14-196-01) from the American Cancer Society (project# 033807) to PNG and DJS.
References
- 1.Zaidi SK, Young DW, Choi JY, et al. The dynamic organization of gene-regulatory machinery in nuclear microenvironments. EMBO Rep. 2005;6(2):128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadauke S, Blobel GA. Chromatin loops in gene regulation. Biochim Biophys Acta. 2009;1789(1):17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krivega I, Dean A. Enhancer and promoter interactions-long distance calls. Curr Opin Genet Dev. 2012;22(2):79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levantini E, Lee S, Radomska HS, et al. RUNX1 regulates the CD34 gene in haematopoietic stem cells by mediating interactions with a distal regulatory element. EMBO J. 2011;30(19):4059–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fritz AJ, Barutcu AR, Martin-Buley L, et al. Chromosomes at Work: Organization of Chromosome Territories in the Interphase Nucleus. J Cell Biochem. 2016;117(1):9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stein GS, Berezney R. Nuclear structure and function. J Cell Biochem. 1996;62(2):147–148. [DOI] [PubMed] [Google Scholar]
- 7.Zeng C, van Wijnen AJ, Stein JL, et al. Identification of a nuclear matrix targeting signal in the leukemia and bone-related AML/CBF-alpha transcription factors. Proc Natl Acad Sci U S A. 1997;94(13):6746–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamond AI, Earnshaw WC. Structure and function in the nucleus. Science. 1998;280(5363):547–553. [DOI] [PubMed] [Google Scholar]
- 9.McNeil S, Guo B, Stein JL, et al. Targeting of the YY1 transcription factor to the nucleolus and the nuclear matrix in situ: the C-terminus is a principal determinant for nuclear trafficking. J Cell Biochem. 1998;68(4):500–510. [PubMed] [Google Scholar]
- 10.Verschure PJ, van der Kraan I, Manders EMM, van Driel R. Spatial relationship between transcription sites and chromosome territories. Journal of Cell Biology. 1999;147(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matera AG. Nuclear bodies: multifaceted subdomains of the interchromatin space. Trends Cell Biol. 1999;9(8):302–309. [DOI] [PubMed] [Google Scholar]
- 12.Stein GS, van Wijnen AJ, Stein JL, et al. Intranuclear trafficking of transcription factors: implications for biological control. J Cell Sci. 2000;113 ( Pt 14):2527–2533. [DOI] [PubMed] [Google Scholar]
- 13.Stein GS, Montecino M, van Wijnen AJ, Stein JL, Lian JB. Nuclear structure-gene expression interrelationships: implications for aberrant gene expression in cancer. Cancer Res. 2000;60(8):2067–2076. [PubMed] [Google Scholar]
- 14.Stein GS, van Wijnen AJ, Stein JL, et al. Subnuclear organization and trafficking of regulatory proteins: implications for biological control and cancer. J Cell Biochem Suppl. 2000;Suppl 35:84–92. [PubMed] [Google Scholar]
- 15.Zaidi SK, Javed A, Choi JY, et al. A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J Cell Sci. 2001;114(Pt 17):3093–3102. [DOI] [PubMed] [Google Scholar]
- 16.Barseguian K, Lutterbach B, Hiebert SW, et al. Multiple subnuclear targeting signals of the leukemia-related AML1/ETO and ETO repressor proteins. Proc Natl Acad Sci U S A. 2002;99(24):15434–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington KS, Javed A, Drissi H, et al. Transcription factors RUNX1/AML1 and RUNX2/Cbfa1 dynamically associate with stationary subnuclear domains. J Cell Sci. 2002;115(Pt 21):4167–4176. [DOI] [PubMed] [Google Scholar]
- 18.Spector DL. The dynamics of chromosome organization and gene regulation. Annu Rev Biochem. 2003;72:573–608. [DOI] [PubMed] [Google Scholar]
- 19.Stein GS, Zaidi SK, Braastad CD, et al. Functional architecture of the nucleus: organizing the regulatory machinery for gene expression, replication and repair. Trends Cell Biol. 2003;13(11):584–592. [DOI] [PubMed] [Google Scholar]
- 20.Stein GS, Lian JB, Stein JL, et al. Temporal and spatial parameters of skeletal gene expression: targeting RUNX factors and their coregulatory proteins to subnuclear domains. Connect Tissue Res. 2003;44 Suppl 1:149–153. [PubMed] [Google Scholar]
- 21.Stein GS, Lian JB, Stein JL, et al. Intranuclear organization of RUNX transcriptional regulatory machinery in biological control of skeletogenesis and cancer. Blood Cells Mol Dis. 2003;30(2):170–176. [DOI] [PubMed] [Google Scholar]
- 22.Stein GS, Lian JB, van Wijnen AJ, et al. Nuclear microenvironments support assembly and organization of the transcriptional regulatory machinery for cell proliferation and differentiation. J Cell Biochem. 2004;91(2):287–302. [DOI] [PubMed] [Google Scholar]
- 23.Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4(9):677–687. [DOI] [PubMed] [Google Scholar]
- 24.Stein GS, van Wijnen AJ, Stein JL, et al. An architectural perspective of cell-cycle control at the G1/S phase cell-cycle transition. J Cell Physiol. 2006;209(3):706–710. [DOI] [PubMed] [Google Scholar]
- 25.Zaidi SK, Javed A, Pratap J, et al. Alterations in intranuclear localization of Runx2 affect biological activity. J Cell Physiol. 2006;209(3):935–942. [DOI] [PubMed] [Google Scholar]
- 26.Handwerger KE, Gall JG. Subnuclear organelles: new insights into form and function. Trends Cell Biol. 2006;16(1):19–26. [DOI] [PubMed] [Google Scholar]
- 27.Drobic B, Dunn KL, Espino PS, Davie JR. Abnormalities of chromatin in tumor cells. EXS. 2006(96):25–47. [DOI] [PubMed] [Google Scholar]
- 28.Schneider R, Grosschedl R. Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev. 2007;21(23):3027–3043. [DOI] [PubMed] [Google Scholar]
- 29.Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8(7):574–585. [DOI] [PubMed] [Google Scholar]
- 30.Misteli T Beyond the sequence: cellular organization of genome function. Cell. 2007;128(4):787–800. [DOI] [PubMed] [Google Scholar]
- 31.Zaidi SK, Young DW, Javed A, et al. Nuclear microenvironments in biological control and cancer. Nat Rev Cancer. 2007;7(6):454–463. [DOI] [PubMed] [Google Scholar]
- 32.Matera AG, Izaguire-Sierra M, Praveen K, Rajendra TK. Nuclear bodies: random aggregates of sticky proteins or crucibles of macromolecular assembly? Dev Cell. 2009;17(5):639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dundr M, Misteli T. Biogenesis of nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2(12):a000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lever E, Sheer D. The role of nuclear organization in cancer. J Pathol. 2010;220(2):114–125. [DOI] [PubMed] [Google Scholar]
- 35.Rajapakse I, Groudine M. On emerging nuclear order. J Cell Biol. 2011;192(5):711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy KL, Feinberg AP. Higher order chromatin organization in cancer. Semin Cancer Biol. 2013;23(2):109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gibcus JH, Dekker J. The hierarchy of the 3D genome. Mol Cell. 2013;49(5):773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sleeman JE, Trinkle-Mulcahy L. Nuclear bodies: new insights into assembly/dynamics and disease relevance. Curr Opin Cell Biol. 2014;28:76–83. [DOI] [PubMed] [Google Scholar]
- 39.Hancock R The crowded nucleus. Int Rev Cell Mol Biol. 2014;307:15–26. [DOI] [PubMed] [Google Scholar]
- 40.Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol. 2015;16(4):245–257. [DOI] [PubMed] [Google Scholar]
- 41.Fritz AJ, Ghule PN, Boyd JR, et al. Intranuclear and higher order chromatin organization of the major histone gene cluster in breast cancer. J Cell Physiol. 2018;233(2):1278–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaidi SK, Medina RF, Pockwinse SM, et al. Subnuclear localization and intranuclear trafficking of transcription factors. Methods Mol Biol. 2010;647:77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zaidi SK, Young DW, Choi JY, et al. Intranuclear trafficking: organization and assembly of regulatory machinery for combinatorial biological control. J Biol Chem. 2004;279(42):43363–43366. [DOI] [PubMed] [Google Scholar]
- 44.Sehgal N, Fritz AJ, Vecerova J, et al. Large-scale probabilistic 3D organization of human chromosome territories. Hum Mol Genet. 2016;25(3):419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fritz AJ, Stojkovic B, Ding H, Xu J, Bhattacharya S, Berezney R. Cell type specific alterations in interchromosomal networks across the cell cycle. PLoS Comput Biol. 2014;10(10):e1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sehgal N, Fritz AJ, Morris K, et al. Gene density and chromosome territory shape. Chromosoma. 2014;123(5):499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fritz AJ, Stojkovic B, Ding H, et al. Wide-scale alterations in interchromosomal organization in breast cancer cells: defining a network of interacting chromosomes. Hum Mol Genet. 2014;23(19):5133–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Derenzini M, Trere D, Pession A, Govoni M, Sirri V, Chieco P. Nucleolar size indicates the rapidity of cell proliferation in cancer tissues. J Pathol. 2000;191(2):181–186. [DOI] [PubMed] [Google Scholar]
- 49.Lajoie BR, Dekker J, Kaplan N. The Hitchhiker’s guide to Hi-C analysis: practical guidelines. Methods. 2015;72:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belaghzal H, Dekker J, Gibcus JH. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 2017;123:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58(3):268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schoenfelder S, Javierre BM, Furlan-Magaril M, Wingett SW, Fraser P. Promoter Capture Hi-C: High-resolution, Genome-wide Profiling of Promoter Interactions. J Vis Exp. 2018(136). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barutcu AR, Fritz AJ, Zaidi SK, et al. C-ing the Genome: A Compendium of Chromosome Conformation Capture Methods to Study Higher order Chromatin Organization. J Cell Physiol. 2016;231(1):31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maass PG, Barutcu AR, Weiner CL, Rinn JL. Inter-chromosomal Contact Properties in Live-Cell Imaging and in Hi-C. Mol Cell. 2018;69(6):1039–1045 e1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shachar S, Voss TC, Pegoraro G, Sciascia N, Misteli T. Identification of Gene Positioning Factors Using High-Throughput Imaging Mapping. Cell. 2015;162(4):911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- 57.Hubner MR, Eckersley-Maslin MA, Spector DL. Chromatin organization and transcriptional regulation. Curr Opin Genet Dev. 2013;23(2):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kadauke S, Blobel GA. Mitotic bookmarking by transcription factors. Epigenetics Chromatin. 2013;6(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zaidi SK, Fritz AJ, Tracy KM, et al. Nuclear organization mediates cancer-compromised genetic and epigenetic control. Adv Biol Regul. 2018;69:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zaidi SK, Nickerson JA, Imbalzano AN, Lian JB, Stein JL, Stein GS. Mitotic Gene Bookmarking: An Epigenetic Program to Maintain Normal and Cancer Phenotypes. Mol Cancer Res. 2018;16(11):1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oomen ME, Dekker J. Epigenetic characteristics of the mitotic chromosome in 1D and 3D. Crit Rev Biochem Mol Biol. 2017;52(2):185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duronio RJ, Marzluff WF. Coordinating cell cycle-regulated histone gene expression through assembly and function of the Histone Locus Body. RNA Biol. 2017;14(6):726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindstrom MS, Jurada D, Bursac S, Orsolic I, Bartek J, Volarevic S. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene. 2018;37(18):2351–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015;21(10):1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hong D, Fritz AJ, Zaidi SK, et al. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J Cell Physiol. 2018;233(12):9136–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. [DOI] [PubMed] [Google Scholar]
- 69.Marino-Ramirez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Rev Proteomics. 2005;2(5):719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maeshima K, Imai R, Tamura S, Nozaki T. Chromatin as dynamic 10-nm fibers. Chromosoma. 2014;123(3):225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, O’Shea CC. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017;357(6349). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dixon JR, Gorkin DU, Ren B. Chromatin Domains: The Unit of Chromosome Organization. Mol Cell. 2016;62(5):668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dekker J, Heard E. Structural and functional diversity of Topologically Associating Domains. FEBS Lett. 2015;589(20 Pt A):2877–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sexton T, Yaffe E, Kenigsberg E, et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148(3):458–472. [DOI] [PubMed] [Google Scholar]
- 76.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518(7539):331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagano T, Lubling Y, Varnai C, et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017;547(7661):61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fortin JP, Hansen KD. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 2015;16:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cremer T, Cremer M. Chromosome territories. Cold Spring Harb Perspect Biol. 2010;2(3):a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fritz AJ, Sehgal N, Pliss A, Xu J, Berezney R. Chromosome territories and the global regulation of the genome Genes, Chromosomes and Cancer 2018;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Doolittle WF. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci U S A. 2013;110(14):5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Polychronopoulos D, King JWD, Nash AJ, Tan G, Lenhard B. Conserved non-coding elements: developmental gene regulation meets genome organization. Nucleic Acids Res. 2017;45(22):12611–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andrey G, Mundlos S. The three-dimensional genome: regulating gene expression during pluripotency and development. Development. 2017;144(20):3646–3658. [DOI] [PubMed] [Google Scholar]
- 86.Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era. Cell. 2014;157(1):13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mora A, Sandve GK, Gabrielsen OS, Eskeland R. In the loop: promoter-enhancer interactions and bioinformatics. Brief Bioinform. 2016;17(6):980–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lupianez DG, Spielmann M, Mundlos S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 2016;32(4):225–237. [DOI] [PubMed] [Google Scholar]
- 89.Dryden NH, Broome LR, Dudbridge F, et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014;24(11):1854–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dubik D, Dembinski TC, Shiu RP. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987;47(24 Pt 1):6517–6521. [PubMed] [Google Scholar]
- 91.Ghoussaini M, Edwards SL, Michailidou K, et al. Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat Commun. 2014;4:4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu F, Li J, Chen H, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30(18):2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guan Y, Kuo WL, Stilwell JL, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007;13(19):5745–5755. [DOI] [PubMed] [Google Scholar]
- 94.Yang J, Corces VG. Chromatin insulators: a role in nuclear organization and gene expression. Adv Cancer Res. 2011;110:43–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krivega M, Dean A. Insulators organize chromatin: emerging rules of the game. Mol Cell. 2011;44(1):1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Phillips-Cremins JE, Corces VG. Chromatin insulators: linking genome organization to cellular function. Mol Cell. 2013;50(4):461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Le Dily F, Bau D, Pohl A, et al. Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev. 2014;28(19):2151–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nora EP, Lajoie BR, Schulz EG, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485(7398):381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dowen JM, Fan ZP, Hnisz D, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159(2):374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schoenfelder S, Furlan-Magaril M, Mifsud B, et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015;25(4):582–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Canela A, Maman Y, Jung S, et al. Genome Organization Drives Chromosome Fragility. Cell. 2017;170(3):507–521 e518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lang F, Li X, Zheng W, et al. CTCF prevents genomic instability by promoting homologous recombination-directed DNA double-strand break repair. Proc Natl Acad Sci U S A. 2017;114(41):10912–10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Engreitz JM, Agarwala V, Mirny LA. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PLoS One. 2012;7(9):e44196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Franco MM, Prickett AR, Oakey RJ. The role of CCCTC-binding factor (CTCF) in genomic imprinting, development, and reproduction. Biol Reprod. 2014;91(5):125. [DOI] [PubMed] [Google Scholar]
- 106.Galupa R, Heard E. X-chromosome inactivation: new insights into cis and trans regulation. Curr Opin Genet Dev. 2015;31:57–66. [DOI] [PubMed] [Google Scholar]
- 107.Oh S, Oh C, Yoo KH. Functional roles of CTCF in breast cancer. BMB Rep. 2017;50(9):445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Katainen R, Dave K, Pitkanen E, et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet. 2015;47(7):818–821. [DOI] [PubMed] [Google Scholar]
- 109.Guo YA, Chang MM, Huang W, et al. Mutation hotspots at CTCF binding sites coupled to chromosomal instability in gastrointestinal cancers. Nat Commun. 2018;9(1):1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dai J, Zhu M, Wang C, et al. Systematical analyses of variants in CTCF-binding sites identified a novel lung cancer susceptibility locus among Chinese population. Sci Rep. 2015;5:7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sanborn AL, Rao SS, Huang SC, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015;112(47):E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016;15(9):2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moore JM, Rabaia NA, Smith LE, et al. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS One. 2012;7(4):e34915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zuin J, Dixon JR, van der Reijden MI, et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111(3):996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6(12):917–922. [DOI] [PubMed] [Google Scholar]
- 116.Nora EP, Goloborodko A, Valton AL, et al. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell. 2017;169(5):930–944 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kubo N, Ishii H, Gorkin D, et al. Preservation of Chromatin Organization after Acute Loss of CTCF in Mouse Embryonic Stem Cells. bioRxiv. 2017. [Google Scholar]
- 118.Schwarzer W, Abdennur N, Goloborodko A, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017;551(7678):51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Crane E, Bian Q, McCord RP, et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523(7559):240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dong P, Tu X, Chu PY, et al. 3D Chromatin Architecture of Large Plant Genomes Determined by Local A/B Compartments. Mol Plant. 2017;10(12):1497–1509. [DOI] [PubMed] [Google Scholar]
- 121.Mizuguchi T, Fudenberg G, Mehta S, et al. Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature. 2014;516(7531):432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Le TB, Laub MT. Transcription rate and transcript length drive formation of chromosomal interaction domain boundaries. EMBO J. 2016;35(14):1582–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lioy VS, Cournac A, Marbouty M, et al. Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell. 2018;172(4):771–783 e718. [DOI] [PubMed] [Google Scholar]
- 124.Pepenella S, Murphy KJ, Hayes JJ. Intra- and inter-nucleosome interactions of the core histone tail domains in higher order chromatin structure. Chromosoma. 2014;123(1–2):3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rowley MJ, Nichols MH, Lyu X, et al. Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol Cell. 2017;67(5):837–852 e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rennie S, Dalby M, van Duin L, Andersson R. Transcriptional decomposition reveals active chromatin architectures and cell specific regulatory interactions. Nat Commun. 2018;9(1):487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ulianov SV, Khrameeva EE, Gavrilov AA, et al. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res. 2016;26(1):70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li L, Lyu X, Hou C, et al. Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol Cell. 2015;58(2):216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hug CB, Grimaldi AG, Kruse K, Vaquerizas JM. Chromatin Architecture Emerges during Zygotic Genome Activation Independent of Transcription. Cell. 2017;169(2):216–228 e219. [DOI] [PubMed] [Google Scholar]
- 130.Dai X, Li T, Bai Z, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res. 2015;5(10):2929–2943. [PMC free article] [PubMed] [Google Scholar]
- 131.Wolberg WH, Street WN, Mangasarian OL. Importance of nuclear morphology in breast cancer prognosis. Clin Cancer Res. 1999;5(11):3542–3548. [PubMed] [Google Scholar]
- 132.Rawat RR, Ruderman D, Macklin P, Rimm DL, Agus DB. Correlating nuclear morphometric patterns with estrogen receptor status in breast cancer pathologic specimens. NPJ Breast Cancer. 2018;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Skrypek N, Goossens S, De Smedt E, Vandamme N, Berx G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017;33(12):943–959. [DOI] [PubMed] [Google Scholar]
- 134.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. [DOI] [PubMed] [Google Scholar]
- 136.Ramdas NM, Shivashankar GV. Cytoskeletal control of nuclear morphology and chromatin organization. J Mol Biol. 2015;427(3):695–706. [DOI] [PubMed] [Google Scholar]
- 137.Spichal M, Fabre E. The Emerging Role of the Cytoskeleton in Chromosome Dynamics. Front Genet. 2017;8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rothballer A, Kutay U. The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma. 2013;122(5):415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gerlitz G, Bustin M. The role of chromatin structure in cell migration. Trends Cell Biol. 2011;21(1):6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Keeling MC, Flores LR, Dodhy AH, Murray ER, Gavara N. Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci Rep. 2017;7(1):5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sharili AS, Kenny FN, Vartiainen MK, Connelly JT. Nuclear actin modulates cell motility via transcriptional regulation of adhesive and cytoskeletal genes. Sci Rep. 2016;6:33893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dion V, Shimada K, Gasser SM. Actin-related proteins in the nucleus: life beyond chromatin remodelers. Curr Opin Cell Biol. 2010;22(3):383–391. [DOI] [PubMed] [Google Scholar]
- 143.Klages-Mundt NL, Kumar A, Zhang Y, Kapoor P, Shen X. The Nature of Actin-Family Proteins in Chromatin-Modifying Complexes. Front Genet. 2018;9:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Comaills V, Kabeche L, Morris R, et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep. 2016;17(10):2632–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee E, Wang J, Yumoto K, et al. DNMT1 Regulates Epithelial-Mesenchymal Transition and Cancer Stem Cells, Which Promotes Prostate Cancer Metastasis. Neoplasia. 2016;18(9):553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Song X, Gao T, Wang N, et al. Selective inhibition of EZH2 by ZLD1039 blocks H3K27 methylation and leads to potent anti-tumor activity in breast cancer. Sci Rep. 2016;6:20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gong C, Qu S, Lv XB, et al. BRMS1L suppresses breast cancer metastasis by inducing epigenetic silence of FZD10. Nat Commun. 2014;5:5406. [DOI] [PubMed] [Google Scholar]
- 148.Fischer KR, Durrans A, Lee S, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zheng X, Carstens JL, Kim J, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22(5–6):396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23(10):1124–1134. [DOI] [PubMed] [Google Scholar]
- 152.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lim SO, Gu JM, Kim MS, et al. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135(6):2128–2140, 2140 e2121–2128. [DOI] [PubMed] [Google Scholar]
- 154.Espada J, Peinado H, Lopez-Serra L, et al. Regulation of SNAIL1 and E-cadherin function by DNMT1 in a DNA methylation-independent context. Nucleic Acids Res. 2011;39(21):9194–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fukagawa A, Ishii H, Miyazawa K, Saitoh M. deltaEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer Med. 2015;4(1):125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hong D, Messier TL, Tye CE, et al. Runx1 stabilizes the mammary epithelial cell phenotype and prevents epithelial to mesenchymal transition. Oncotarget. 2017;8(11):17610–17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hong D, Fritz AJ, Finstad KH, et al. Suppression of Breast Cancer Stem Cells and Tumor Growth by the RUNX1 Transcription Factor. Mol Cancer Res. 2018;16(12):1952–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hong D, Fritz AJ, Gordon JA, et al. RUNX1-dependent mechanisms in biological control and dysregulation in cancer. J Cell Physiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kulkarni M, Tan TZ, Syed Sulaiman NB, et al. RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget. 2018;9(18):14175–14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chaffer CL, Marjanovic ND, Lee T, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154(1):61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Joseph JV, Conroy S, Tomar T, et al. TGF-beta is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014;5:e1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cohen-Solal KA, Boregowda RK, Lasfar A. RUNX2 and the PI3K/AKT axis reciprocal activation as a driving force for tumor progression. Mol Cancer. 2015;14:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chimge NO, Baniwal SK, Little GH, et al. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: the role of SNAI2. Breast Cancer Res. 2011;13(6):R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Niu DF, Kondo T, Nakazawa T, et al. Transcription factor Runx2 is a regulator of epithelial-mesenchymal transition and invasion in thyroid carcinomas. Lab Invest. 2012;92(8):1181–1190. [DOI] [PubMed] [Google Scholar]
- 165.Chuang LS, Ito K, Ito Y. RUNX family: Regulation and diversification of roles through interacting proteins. Int J Cancer. 2013;132(6):1260–1271. [DOI] [PubMed] [Google Scholar]
- 166.Barutcu AR, Tai PW, Wu H, et al. The bone-specific Runx2-P1 promoter displays conserved three-dimensional chromatin structure with the syntenic Supt3h promoter. Nucleic Acids Res. 2014;42(16):10360–10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Barutcu AR, Hong D, Lajoie BR, et al. RUNX1 contributes to higher order chromatin organization and gene regulation in breast cancer cells. Biochim Biophys Acta. 2016;1859(11):1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sever R, Glass CK. Signaling by nuclear receptors. Cold Spring Harb Perspect Biol. 2013;5(3):a016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Beatson GT. On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment, with Illustrative Cases. Trans Med Chir Soc Edinb. 1896;15:153–179. [PMC free article] [PubMed] [Google Scholar]
- 170.Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res Treat. 1999;53(3):217–227. [DOI] [PubMed] [Google Scholar]
- 171.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mohammed H, Russell IA, Stark R, et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature. 2015;523(7560):313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87(3):905–931. [DOI] [PubMed] [Google Scholar]
- 174.Barnard ME, Boeke CE, Tamimi RM. Established breast cancer risk factors and risk of intrinsic tumor subtypes. Biochim Biophys Acta. 2015;1856(1):73–85. [DOI] [PubMed] [Google Scholar]
- 175.Cochrane DR, Bernales S, Jacobsen BM, et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014;16(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sorrentino G, Ruggeri N, Zannini A, et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat Commun. 2017;8:14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Brittain AL, Basu R, Qian Y, Kopchick JJ. Growth Hormone and the Epithelial-to-Mesenchymal Transition. J Clin Endocrinol Metab. 2017;102(10):3662–3673. [DOI] [PubMed] [Google Scholar]
- 178.Nouhi Z, Chughtai N, Hartley S, Cocolakis E, Lebrun JJ, Ali S. Defining the role of prolactin as an invasion suppressor hormone in breast cancer cells. Cancer Res. 2006;66(3):1824–1832. [DOI] [PubMed] [Google Scholar]
- 179.Brisken C, Heineman A, Chavarria T, et al. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000;14(6):650–654. [PMC free article] [PubMed] [Google Scholar]
- 180.Fernandez-Valdivia R, Mukherjee A, Creighton CJ, et al. Transcriptional response of the murine mammary gland to acute progesterone exposure. Endocrinology. 2008;149(12):6236–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fernandez-Valdivia R, Mukherjee A, Ying Y, et al. The RANKL signaling axis is sufficient to elicit ductal side-branching and alveologenesis in the mammary gland of the virgin mouse. Dev Biol. 2009;328(1):127–139. [DOI] [PubMed] [Google Scholar]
- 182.Zuo L, Li W, You S. Progesterone reverses the mesenchymal phenotypes of basal phenotype breast cancer cells via a membrane progesterone receptor mediated pathway. Breast Cancer Res. 2010;12(3):R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kas SM, de Ruiter JR, Schipper K, et al. Insertional mutagenesis identifies drivers of a novel oncogenic pathway in invasive lobular breast carcinoma. Nat Genet. 2017;49(8):1219–1230. [DOI] [PubMed] [Google Scholar]
- 184.Pereira B, Chin SF, Rueda OM, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ciriello G, Gatza ML, Beck AH, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163(2):506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rooney N, Riggio AI, Mendoza-Villanueva D, Shore P, Cameron ER, Blyth K. Runx Genes in Breast Cancer and the Mammary Lineage. Adv Exp Med Biol. 2017;962:353–368. [DOI] [PubMed] [Google Scholar]
- 187.Stender JD, Kim K, Charn TH, et al. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010;30(16):3943–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.van Bragt MP, Hu X, Xie Y, Li Z. RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. Elife. 2014;3:e03881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chimge NO, Little GH, Baniwal SK, et al. RUNX1 prevents oestrogen-mediated AXIN1 suppression and beta-catenin activation in ER-positive breast cancer. Nat Commun. 2016;7:10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chimge NO, Baniwal SK, Luo J, et al. Opposing effects of Runx2 and estradiol on breast cancer cell proliferation: in vitro identification of reciprocally regulated gene signature related to clinical letrozole responsiveness. Clin Cancer Res. 2012;18(3):901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Muscat GE, Eriksson NA, Byth K, et al. Research resource: nuclear receptors as transcriptome: discriminant and prognostic value in breast cancer. Mol Endocrinol. 2013;27(2):350–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Carroll JS. Mechanisms of oestrogen receptor (ER) gene regulation in breast cancer. Eur J Endocrinol. 2016;175(1):R41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Carroll JS, Liu XS, Brodsky AS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122(1):33–43. [DOI] [PubMed] [Google Scholar]
- 194.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43(1):27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zwart W, Theodorou V, Kok M, Canisius S, Linn S, Carroll JS. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. The EMBO journal. 2011;30(23):4764–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Asselin-Labat ML, Sutherland KD, Barker H, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 2007;9(2):201–209. [DOI] [PubMed] [Google Scholar]
- 197.Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, Brown M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007;67(13):6477–6483. [DOI] [PubMed] [Google Scholar]
- 198.Kong SL, Li G, Loh SL, Sung WK, Liu ET. Cellular reprogramming by the conjoint action of ERalpha, FOXA1, and GATA3 to a ligand-inducible growth state. Molecular systems biology. 2011;7:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liu MH, Cheung E. Estrogen receptor-mediated long-range chromatin interactions and transcription in breast cancer. Mol Cell Endocrinol. 2014;382(1):624–632. [DOI] [PubMed] [Google Scholar]
- 200.Pan YF, Wansa KD, Liu MH, et al. Regulation of estrogen receptor-mediated long range transcription via evolutionarily conserved distal response elements. The Journal of biological chemistry. 2008;283(47):32977–32988. [DOI] [PubMed] [Google Scholar]
- 201.Fullwood MJ, Liu MH, Pan YF, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462(7269):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hsu PY, Hsu HK, Lan X, et al. Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer. Cancer Cell. 2013;24(2):197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mourad R, Hsu PY, Juan L, et al. Estrogen induces global reorganization of chromatin structure in human breast cancer cells. PLoS One. 2014;9(12):e113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Stavreva DA, Coulon A, Baek S, et al. Dynamics of chromatin accessibility and long-range interactions in response to glucocorticoid pulsing. Genome Res. 2015;25(6):845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Verde G, De Llobet LI, Wright RHG, et al. Unliganded Progesterone Receptor Governs Estrogen Receptor Gene Expression by Regulating DNA Methylation in Breast Cancer Cells. Cancers (Basel). 2018;10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ferguson AT, Lapidus RG, Baylin SB, Davidson NE. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 1995;55(11):2279–2283. [PubMed] [Google Scholar]
- 207.Daniel AR, Gaviglio AL, Knutson TP, et al. Progesterone receptor-B enhances estrogen responsiveness of breast cancer cells via scaffolding PELP1- and estrogen receptor-containing transcription complexes. Oncogene. 2015;34(4):506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hilton HN, Clarke CL, Graham JD. Estrogen and progesterone signalling in the normal breast and its implications for cancer development. Mol Cell Endocrinol. 2018;466:2–14. [DOI] [PubMed] [Google Scholar]
- 209.Daniel AR, Lange CA. Protein kinases mediate ligand-independent derepression of sumoylated progesterone receptors in breast cancer cells. Proc Natl Acad Sci U S A. 2009;106(34):14287–14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nacht AS, Pohl A, Zaurin R, et al. Hormone-induced repression of genes requires BRG1-mediated H1.2 deposition at target promoters. EMBO J. 2016;35(16):1822–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Iacopetta D, Rechoum Y, Fuqua SA. The Role of Androgen Receptor in Breast Cancer. Drug discovery today Disease mechanisms. 2012;9(1–2):e19–e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hu R, Dawood S, Holmes MD, et al. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin Cancer Res. 2011;17(7):1867–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.D’Amato NC, Gordon MA, Babbs B, et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol Cancer Res. 2016;14(11):1054–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Judes G, Dagdemir A, Karsli-Ceppioglu S, et al. Molecular and Epigenetic Biomarkers in Luminal Androgen Receptor: A Triple Negative Breast Cancer Subtype. OMICS. 2016;20(10):610–613. [DOI] [PubMed] [Google Scholar]
- 216.Barton VN, D’Amato NC, Gordon MA, et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015;14(3):769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ni M, Chen Y, Lim E, et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer cell. 2011;20(1):119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Doan TB, Graham JD, Clarke CL. Emerging functional roles of nuclear receptors in breast cancer. J Mol Endocrinol. 2017;58(3):R169–r190. [DOI] [PubMed] [Google Scholar]
- 219.Heublein S, Mayr D, Meindl A, et al. Thyroid Hormone Receptors Predict Prognosis in BRCA1 Associated Breast Cancer in Opposing Ways. PLoS One. 2015;10(6):e0127072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Jerzak KJ, Cockburn J, Pond GR, et al. Thyroid hormone receptor alpha in breast cancer: prognostic and therapeutic implications. Breast cancer research and treatment. 2015;149(1):293–301. [DOI] [PubMed] [Google Scholar]
- 221.Kim WG, Cheng SY. Thyroid hormone receptors and cancer. Biochim Biophys Acta. 2013;1830(7):3928–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Aranda A, Martinez-Iglesias O, Ruiz-Llorente L, Garcia-Carpizo V, Zambrano A. Thyroid receptor: roles in cancer. Trends Endocrinol Metab. 2009;20(7):318–324. [DOI] [PubMed] [Google Scholar]
- 223.Jezequel P, Campone M, Gouraud W, et al. bc-GenExMiner: an easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res Treat. 2012;131(3):765–775. [DOI] [PubMed] [Google Scholar]
- 224.Jezequel P, Frenel JS, Campion L, et al. bc-GenExMiner 3.0: new mining module computes breast cancer gene expression correlation analyses. Database (Oxford). 2013;2013:bas060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Silva J, Dominguez G, Gonzalez-Sancho J, et al. Expression of thyroid hormone receptor/erbA gene is altered in human breast cancer. In. Vol 21 Oncogene 2002:4307–4316. [DOI] [PubMed] [Google Scholar]
- 226.Wojcicka A, Piekielko–Witkowska A, Kedzierska H, et al. Epigenetic Regulation of Thyroid Hormone Receptor Beta in Renal Cancer. PLoS One. 2014;9(5):e97624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Park JW, Zhao L, Willingham M, Cheng S-y. Oncogenic mutations of thyroid hormone receptor β. Oncotarget. 2015;6(10):8115–8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ling Y, Li Q, Yang H, et al. Loss of heterozygosity in thyroid hormone receptor beta in invasive breast cancer. Tumori. 2015;101(5):572–577. [DOI] [PubMed] [Google Scholar]
- 229.Ling Y, Xu X, Hao J, et al. Aberrant methylation of the THRB gene in tissue and plasma of breast cancer patients. Cancer Genet Cytogenet. 2010;196(2):140–145. [DOI] [PubMed] [Google Scholar]
- 230.Gu G, Gelsomino L, Covington KR, et al. Targeting thyroid hormone receptor beta in triple-negative breast cancer. Breast Cancer Res Treat. 2015;150(3):535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Li Z, Meng Z, Chandrasekaran R, et al. Biallelic inactivation of the thyroid hormone receptor beta 1 gene in early stage breast cancer. In. Vol 62 Cancer Res 2002:1939–1943. [PubMed] [Google Scholar]
- 232.Jerzak KJ, Cockburn JG, Dhesy-Thind SK, et al. Thyroid hormone receptor beta-1 expression in early breast cancer: a validation study. Breast Cancer Res Treat. 2018;171(3):709–717. [DOI] [PubMed] [Google Scholar]
- 233.Gu G, Dustin D, Fuqua SAW. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr Opin Pharmacol. 2016;31:97–103. [DOI] [PubMed] [Google Scholar]
- 234.Shi YB. Unliganded thyroid hormone receptor regulates metamorphic timing via the recruitment of histone deacetylase complexes. Curr Top Dev Biol. 2013;105:275–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Shi YB, Matsuura K, Fujimoto K, Wen L, Fu L. Thyroid hormone receptor actions on transcription in amphibia: The roles of histone modification and chromatin disruption. Cell Biosci. 2012;2(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Heimeier RA, Hsia VS, Shi YB. Participation of Brahma-related gene 1 (BRG1)-associated factor 57 and BRG1-containing chromatin remodeling complexes in thyroid hormone-dependent gene activation during vertebrate development. Mol Endocrinol. 2008;22(5):1065–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ramadoss P, Abraham BJ, Tsai L, et al. Novel mechanism of positive versus negative regulation by thyroid hormone receptor beta1 (TRbeta1) identified by genome-wide profiling of binding sites in mouse liver. J Biol Chem. 2014;289(3):1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ayers S, Switnicki MP, Angajala A, Lammel J, Arumanayagam AS, Webb P. Genome-wide binding patterns of thyroid hormone receptor beta. PLoS One. 2014;9(2):e81186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Grontved L, Waterfall JJ, Kim DW, et al. Transcriptional activation by the thyroid hormone receptor through ligand-dependent receptor recruitment and chromatin remodelling. Nat Commun. 2015;6:7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gillis NE, Taber TH, Bolf EL, et al. Thyroid Hormone Receptor beta Suppression of RUNX2 Is Mediated by Brahma-Related Gene 1-Dependent Chromatin Remodeling. Endocrinology. 2018;159(6):2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Martinez-Iglesias O, Garcia-Silva S, Tenbaum S, et al. Thyroid hormone receptor β1 acts as a potent suppressor of tumor invasiveness and metastasis. In. Vol 69 Cancer Res 2009:501–509. [DOI] [PubMed] [Google Scholar]
- 242.Park J, Zhao L, Cheng S. Inhibition of estrogen-dependent tumorigenesis by the thyroid hormone receptor B in xenograft models. In. Vol 3 Am J Cancer Res 2013:302–311. [PMC free article] [PubMed] [Google Scholar]
- 243.Kim WG, Zhao L, Kim DW, Willingham MC, Cheng SY. Inhibition of tumorigenesis by the thyroid hormone receptor beta in xenograft models. Thyroid. 2014;24(2):260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Kim WG, Zhu X, Kim DW, Zhang L, Kebebew E, Cheng SY. Reactivation of the silenced thyroid hormone receptor beta gene expression delays thyroid tumor progression. Endocrinology. 2013;154(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Peng X, Zhang Y, Sun Y, et al. Overexpressing modified human TRβ1 suppresses the proliferation of breast cancer MDA-MB-468 cells. Oncol Lett. 2018;16(1):785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Cheng S-Y, Leonard JL, Davis PJ. Molecular Aspects of Thyroid Hormone Actions. Endocr Rev. 2010;31(2):139–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Brent GA. Mechanisms of thyroid hormone action. The Journal of Clinical Investigation. 2012;122(9):3035–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Vella KR, Hollenberg AN. The actions of thyroid hormone signaling in the nucleus. Mol Cell Endocrinol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Truong TH, Lange CA. Deciphering Steroid Receptor Crosstalk in Hormone-Driven Cancers. Endocrinology. 2018;159(12):3897–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Glass CK, Holloway JM, Devary OV, Rosenfeld MG. The thyroid hormone receptor binds with opposite transcriptional effects to a common sequence motif in thyroid hormone and estrogen response elements. Cell. 1988;54(3):313–323. [DOI] [PubMed] [Google Scholar]
- 251.Figueiredo NB, Cestari SH, Conde SJ, et al. Estrogen-responsive genes overlap with triiodothyronine-responsive genes in a breast carcinoma cell line. ScientificWorldJournal. 2014;2014:969404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Davis PJ, Goglia F, Leonard JL. Nongenomic actions of thyroid hormone. Nat Rev Endocrinol. 2016;12(2):111–121. [DOI] [PubMed] [Google Scholar]
- 253.Cestari SH, Figueiredo NB, Conde SJ, et al. Influence of estradiol and triiodothyronine on breast cancer cell lines proliferation and expression of estrogen and thyroid hormone receptors. Arq Bras Endocrinol Metabol. 2009;53(7):859–864. [DOI] [PubMed] [Google Scholar]
- 254.Trotter KW, Archer TK. The BRG1 transcriptional coregulator. Nucl Recept Signal. 2008;6:e004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Trotter KW, Fan HY, Ivey ML, Kingston RE, Archer TK. The HSA domain of BRG1 mediates critical interactions required for glucocorticoid receptor-dependent transcriptional activation in vivo. Mol Cell Biol. 2008;28(4):1413–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.DiRenzo J, Shang Y, Phelan M, et al. BRG-1 Is Recruited to Estrogen-Responsive Promoters and Cooperates with Factors Involved in Histone Acetylation. Mol Cell Biol. 2000;20(20):7541–7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.García-Pedrero JM, Kiskinis E, Parker MG, Belandia B. The SWI/SNF Chromatin Remodeling Subunit BAF57 Is a Critical Regulator of Estrogen Receptor Function in Breast Cancer Cells. J Biol Chem. 2006;281(32):22656–22664. [DOI] [PubMed] [Google Scholar]
- 258.Gillis NE, Taber TH, Bolf EL, et al. Thyroid Hormone Receptor β Suppression of RUNX2 is Mediated by Brahma Related Gene 1 Dependent Chromatin Remodeling. Endocrinology. 2018:en.2018–00128-en.02018–00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gibcus JH, Samejima K, Goloborodko A, et al. A pathway for mitotic chromosome formation. Science. 2018;359(6376). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zaidi SK, Grandy RA, Lopez-Camacho C, et al. Bookmarking target genes in mitosis: a shared epigenetic trait of phenotypic transcription factors and oncogenes? Cancer Res. 2014;74(2):420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zaidi SK, Young DW, Pockwinse SM, et al. Mitotic partitioning and selective reorganization of tissue-specific transcription factors in progeny cells. Proc Natl Acad Sci U S A. 2003;100(25):14852–14857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Young DW, Hassan MQ, Yang XQ, et al. Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2. Proc Natl Acad Sci U S A. 2007;104(9):3189–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Roussel P, Andre C, Comai L, HernandezVerdun D. The rDNA transcription machinery is assembled during mitosis in active NORs and absent in inactive NORs. Journal of Cell Biology. 1996;133(2):235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Young DW, Hassan MQ, Pratap J, et al. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 2007;445(7126):442–446. [DOI] [PubMed] [Google Scholar]
- 265.Teves SS, An L, Hansen AS, Xie L, Darzacq X, Tjian R. A dynamic mode of mitotic bookmarking by transcription factors. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ali SA, Zaidi SK, Dacwag CS, et al. Phenotypic transcription factors epigenetically mediate cell growth control. Proc Natl Acad Sci U S A. 2008;105(18):6632–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kadauke S, Udugama MI, Pawlicki JM, et al. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012;150(4):725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Arampatzi P, Gialitakis M, Makatounakis T, Papamatheakis J. Gene-specific factors determine mitotic expression and bookmarking via alternate regulatory elements. Nucleic Acids Res. 2013;41(4):2202–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013;27(3):251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lerner J, Bagattin A, Verdeguer F, et al. Human mutations affect the epigenetic/bookmarking function of HNF1B. Nucleic Acids Res. 2016;44(17):8097–8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Zaidi SK, Young DW, Montecino MA, et al. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat Rev Genet. 2010;11(8):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kinkley S, Helmuth J, Polansky JK, et al. reChIP-seq reveals widespread bivalency of H3K4me3 and H3K27me3 in CD4(+) memory T cells. Nat Commun. 2016;7:12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. [DOI] [PubMed] [Google Scholar]
- 274.Bapat SA, Jin V, Berry N, et al. Multivalent epigenetic marks confer microenvironment-responsive epigenetic plasticity to ovarian cancer cells. Epigenetics. 2010;5(8):716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Grandy RA, Whitfield TW, Wu H, et al. Genome-Wide Studies Reveal that H3K4me3 Modification in Bivalent Genes Is Dynamically Regulated during the Pluripotent Cell Cycle and Stabilized upon Differentiation. Mol Cell Biol. 2016;36(4):615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Messier TL, Boyd JR, Gordon JA, Stein JL, Lian JB, Stein GS. Oncofetal Epigenetic Bivalency in Breast Cancer Cells: H3K4 and H3K27 Tri-Methylation as a Biomarker for Phenotypic Plasticity. J Cell Physiol. 2016;231(11):2474–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Burke LJ, Zhang R, Bartkuhn M, et al. CTCF binding and higher order chromatin structure of the H19 locus are maintained in mitotic chromatin. EMBO J. 2005;24(18):3291–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008;22(22):3089–3114. [DOI] [PubMed] [Google Scholar]
- 279.Liu Y, Pelham-Webb B, Di Giammartino DC, et al. Widespread Mitotic Bookmarking by Histone Marks and Transcription Factors in Pluripotent Stem Cells. Cell Rep. 2017;19(7):1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Shen W, Wang D, Ye B, Shi M, Zhang Y, Zhao Z. A possible role of Drosophila CTCF in mitotic bookmarking and maintaining chromatin domains during the cell cycle. Biol Res. 2015;48:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295(5558):1306–1311. [DOI] [PubMed] [Google Scholar]
- 282.Naumova N, Imakaev M, Fudenberg G, et al. Organization of the mitotic chromosome. Science. 2013;342(6161):948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kelly TK, Miranda TB, Liang G, et al. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol Cell. 2010;39(6):901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008;10(1):102–109. [DOI] [PubMed] [Google Scholar]
- 285.Follmer NE, Wani AH, Francis NJ. A polycomb group protein is retained at specific sites on chromatin in mitosis. PLoS Genet. 2012;8(12):e1003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bakshi R, Zaidi SK, Pande S, et al. The leukemogenic t(8;21) fusion protein AML1-ETO controls rRNA genes and associates with nucleolar-organizing regions at mitotic chromosomes. J Cell Sci. 2008;121(Pt 23):3981–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23(24):4255–4262. [DOI] [PubMed] [Google Scholar]
- 288.Pope BD, Ryba T, Dileep V, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515(7527):402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Rhind N, Gilbert DM. DNA replication timing. Cold Spring Harb Perspect Biol. 2013;5(8):a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.McCann KL, Baserga SJ. Driving nucleolar assembly. Genes Dev. 2014;28(3):211–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Pliss A, Fritz AJ, Stojkovic B, et al. Non-Random Patterns in the Distribution of NOR-Bearing Chromosome Territories in Human Fibroblasts: A Network Model of Interactions. J Cell Physiol. 2015;230(2):427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ghule PN, Seward DJ, Fritz AJ, et al. Higher order genomic organization and regulatory compartmentalization for cell cycle control at the G1/S-phase transition. J Cell Physiol. 2018;233(10):6406–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Bravo J, Li Z, Speck NA, Warren AJ. The leukemia-associated AML1 (Runx1)--CBF beta complex functions as a DNA-induced molecular clamp. Nat Struct Biol. 2001;8(4):371–378. [DOI] [PubMed] [Google Scholar]