

Abstract
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease that has significant overlap with frontotemporal dementia (FTD). Mutations in specific genes have been identified that can cause and/or predispose patients to ALS. However, the clinical variability seen in ALS patients suggests that additional genes impact pathology, susceptibility, severity, and/or progression of the disease. To identify molecular pathways involved in ALS, we undertook a meta-analysis of published genetic modifiers both in patients and in model organisms, and undertook bioinformatic pathway analysis. From 72 published studies, we generated a list of 946 genes whose perturbation (1) impacted ALS in patient populations, (2) altered defects in laboratory models, or (3) modified defects caused by ALS gene ortholog loss of function. Herein, these are all called modifier genes. We found 727 modifier genes that encode proteins with human orthologs. Of these, 43 modifier genes were identified as modifiers of more than one ALS gene/model, consistent with the hypothesis that shared genes and pathways may underlie ALS. Further, we used a gene ontology-based bioinformatic analysis to identify pathways and associated genes that may be important in ALS. To our knowledge this is the first comprehensive survey of ALS modifier genes. This work suggests that shared molecular mechanisms may underlie pathology caused by different ALS disease genes. Surprisingly, few ALS modifier genes have been tested in more than one disease model. Understanding genes that modify ALS-associated defects will help to elucidate the molecular pathways that underlie ALS and provide additional targets for therapeutic intervention.
Keywords: ALS, FTD, genetic modifiers, pathway analysis
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a progressive disease that results in selective degeneration and death of upper (cortical) and lower (spinal) motor neurons. First described by Jean-Martin Charcot in 1869 (Rowland, 2001), ALS is characterized by muscle weakness, paralysis, respiratory failure, and death typically within 3–5 years of symptom onset. Within the past two decades, over 20 genes have been identified and/or implicated in ALS (Baker et al., 2006; Brenner et al., 2016; Chaussenot et al., 2014; Chen Y. Z. et al., 2004; Chesi et al., 2013; Chow et al., 2009; Cirulli et al., 2015; Couthouis et al., 2012; Cruts et al., 2006; Daoud et al., 2012; DeJesus-Hernandez et al., 2011; Deng et al., 2011; Deng et al., 1993; Elden et al., 2010; Figlewicz et al., 1994; Freischmidt et al., 2015; Greenway et al., 2006; Gros-Louis et al., 2004; Hutton et al., 1998; Johnson et al., 2014a; Johnson et al., 2010; Johnson et al., 2014b; Kabashi et al., 2008; Kenna et al., 2016; Kim H. J. et al., 2013; Kwiatkowski et al., 2009; Leblond et al., 2014; Leung et al., 2004; Maruyama and Kawakami, 2013; Millecamps et al., 2014; Mitchell et al., 2010; Munch et al., 2005; Munch et al., 2004; Nishimura et al., 2004; Parkinson et al., 2006; Pensato et al., 2015; Rademakers and van Blitterswijk, 2014; Renton et al., 2011; Rosen et al., 1993; Skibinski et al., 2005; Skvortsova et al., 2004; Smith et al., 2014; Sreedharan et al., 2008; Sreedharan and Brown, 2013; Takahashi et al., 2013; Teyssou et al., 2013; Teyssou et al., 2014; Ticozzi et al., 2011; Van Deerlin et al., 2008; Vance et al., 2009; Wu et al., 2012; Yang Y. et al., 2001). Together, mutations in superoxide dismutase 1 (SOD1), TAR DNA binding protein (TARDBP), fused in sarcoma (FUS), and chromosome 9 open reading frame 72 (C9orf72) account for approximately 60–70% of ALS cases with a family history. Other genes, including VAMP-associated protein B (VAPB), valosin-containing protein (VCP), and optineurin (OPTN), account for 30–40% of familial cases. The proteins encoded by these genes are involved in a variety of pathways, including oxidative stress (Barber et al., 2006), protein aggregation (Bruijn et al., 1998), and neuroinflammation (Hooten et al., 2015). However, despite the varied roles of these proteins in healthy cell function, disease alleles of the aforementioned genes can lead to ALS. Even though we have made progress on understanding some aspects of ALS, we do not understand how or why mutations in functionally diverse proteins can cause what appears to be a single disease.
Insights into ALS pathological mechanisms came from the discovery that mutations in a subset of these genes can also cause frontotemporal dementia (FTD), with characteristic degeneration of frontal and temporal lobe neurons (Ratnavalli et al., 2002). ALS and FTD share many pathological hallmarks, including ubiquitinated inclusions, which have been observed in lower motor neurons and cortical neurons of patients with ALS. Furthermore, approximately 50% of ALS patients develop FTD-like symptoms and around 40% of FTD patients develop ALS-like symptoms (Ferrari et al., 2011; Ji et al., 2017; Lomen-Hoerth et al., 2002; Lomen-Hoerth et al., 2003; Strong, 2008). These observations suggest that ALS and FTD are related and may share pathways leading to neurodegeneration (Arai et al., 2006; Leigh et al., 1991; Liscic et al., 2008; Mackenzie and Feldman, 2005). One strategy that can be used to delineate shared pathways, is to find “genetic modifiers” or “modifier genes” of ALS and FTD genes, which can reveal pathological mechanisms.
Broadly defined, “modifier genes” are genes with alleles that ameliorate or exacerbate defects caused by an allele of another gene. Modifier genes, in patients, may influence clinical presentation of disease including disease onset, severity, penetrance, or progression. Classical genetic studies in model organisms have extensively used modifier gene analysis to dissect function and dysfunction, contributing to our understanding of neurodegenerative diseases (Alexander et al., 2014; Dimitriadi and Hart, 2010; Gama Sosa et al., 2012; Plantie et al., 2015; Therrien and Parker, 2014; Verbandt et al., 2016). Large scale forward genetic screens for modifiers are possible in small, genetically tractable organisms, such as S. cerevisiae, C. elegans, and D. melanogaster (Chen X. and Burgoyne, 2012; Sin et al., 2014). These can yield unexpected insights into mechanisms and complement hypothesis-driven studies. Most animal models of ALS compare the consequences of expressing a human protein containing the disease mutation versus the wild type form of the protein. These models have been used to identify genetic modifiers of ALS-associated defects and we surveyed their results. Also, ALS alleles may cause loss of function, which may contribute to disease pathology. Therefore, we surveyed the results of previous studies focused on identifying modifiers of either disease models or ALS-gene ortholog loss of function.
Further, ALS modifier genes have also been identified in human populations and may help explain variation in clinical presentation or disease progression. The site of onset (bulbar or spinal), age of onset, progression rate, and level of cognitive impairment can differ between patients even within the same family (Swinnen and Robberecht, 2014). The variability observed in ALS patients may be, in part, due to a result of different alleles of modifier genes that affect progression, penetrance or onset—even if these modifier alleles do not cause disease per se. Risk genes are also of interest, as they may reveal pathways critical for disease, even if risk genes are neither necessary nor sufficient to cause disease. Genome wide association studies (GWAS) and linkage analysis in humans with ALS have been used to identify genetic modifiers (Giess et al., 2002; Gros-Louis et al., 2004; Lee Y. B. et al., 2013).
Here, we undertook a comprehensive literature search and identified 946 genes that act as modifiers of ALS-associated defects in S. cerevisiae, C. elegans, D. melanogaster, M. musculus, or human patients. As shared mechanisms may underlie ALS, we used a gene ontology bioinformatics approach to identify pathways pertinent to disease. This bioinformatic analysis focused on 727 modifier genes that are orthologous to human genes, some of which have been identified in human studies. The results suggest that shared pathways may underlie ALS, regardless of the disease gene involved.
Experimental Methods
Literature Search
We searched the literature in PubMed from September 7, 2016 - December 31, 2016 and identified studies that reported modifier genes in ALS models or modifiers of ALS ortholog loss of function (Fig. 1). Specifically, we examined papers in PubMed reporting genetic modifiers of SOD1, TDP43, C9orf72, FUS, or VAPB. For SOD1, TDP43, C9orf72, and FUS; two independent co-authors searched the literature. The literature review included, but was not limited to, genome-wide screens and candidate genes reported to modify phenotypes in S. cerevisiae, C. elegans, D. melanogaster, M. musculus, and cell culture models. Additionally, we searched for genetic modifiers of ALS in patients, which was reviewed by Ghasemi and Brown (Ghasemi and Brown, 2017). A database was assembled in Microsoft Excel with the NCBI GeneID, modifier gene name, human ortholog name, ALS model used, type of screen (RNAi knockdown, genome-wide screen) and impact of modifier on phenotype. The total number of modifier genes (946) corresponds to all modifier genes identified in the literature survey; we did not reexamine this list to identify and eliminate orthologous genes independently identified in different model organisms, which would modestly reduce this number. Additionally, using OMIM (Online Mendelian Inheritance of Man) a list of genes known to cause ALS and/or FTD was compiled. At least 2 independent coauthors checked each ALS gene after the database was assembled to ensure the accuracy. All data used in the analysis are included in the manuscript and supplemental files.
Fig. 1.
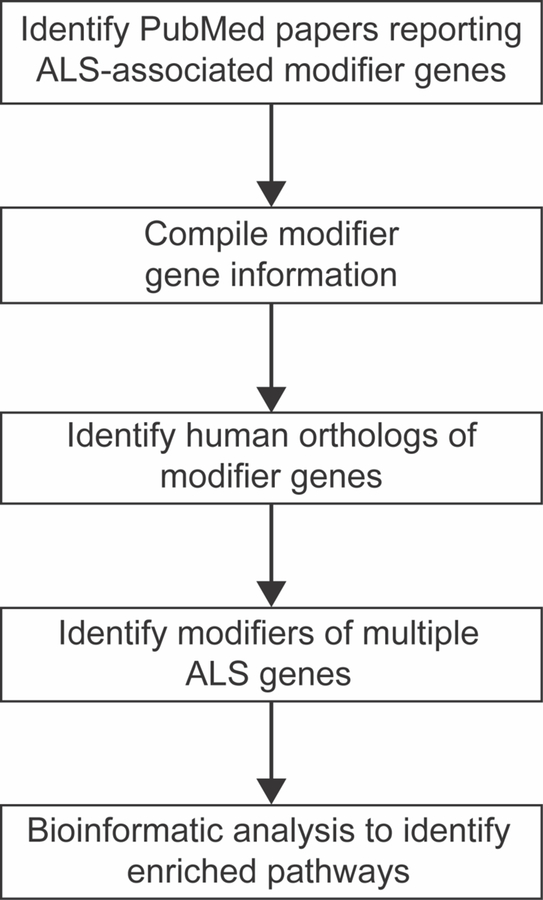
Schematic representation of the workflow used to compile and analyze the list of genetic modifiers of ALS.
Human Ortholog Identification
Human orthologs of modifier genes found in model organisms were identified using best match similarity with BLAST (NCBI) at blast.ncbi.nlm.nih.gov/Blast.cgi based on protein sequences. If there was more than one best match, then up to three were reported in the “other orthologs” column in Supplemental File 1. When genes with identical statistical scores were called as best match, both genes were included in the bioinformatics analysis. For example, Hbr98DE gene is an ortholog of both hnRNPA1 and hnRNPA2B1. Human ortholog identification was verified with DIOPT (http://www.flyrnai.org/diopt) (Hu et al., 2011). If no human ortholog was found, the modifier gene was not included in bioinformatics analysis presented herein.
Gene Ontology Bioinformatic Analysis
Gene ontology (GO) bioinformatic analysis was performed independently for lists of human genes and/or orthologs of modifier genes identified in other species. GO terms that describe Biological Processes are tested to determine if these were over-represented in the curated gene lists, compared to the rest of the transcriptome, using a hypergeometric test implemented in the GOstat package (Falcon and Gentleman, 2007). GO terms with a p-value less than 0.05 after Bonferroni correction were considered overrepresented. In addition, a list of modifier genes associated with more than one ALS genes was assembled and independently subjected to GO analysis. The supplemental files containing lists of modifier genes and other data use in the bioinformatic analysis are available at https://doi.org/10.26300/7asw-k867.
Results
To identify modifier genes associated with ALS, we searched the PubMed literature database at the National Center for Biotechnology Information (NCBI) for modifiers of SOD1, TDP43, FUS, C9orf72, VAPB, and other ALS genes. In total, 72 studies were found reporting modifier genes 1) in ALS models, 2) in human patients, or 3) for loss of function alleles of ALS gene orthologs. The resulting list of genes is available in Supplemental File 1. Here, we provide an abbreviated background for ALS genes that served as the basis for our search, including a brief description of ALS models used in modifier gene studies. For each ALS gene, gene ontology bioinformatic pathway analysis was undertaken and pathways that were enriched in gene ontology analysis are discussed.
SOD1
In 1993, the discovery that point mutations in superoxide dismutase 1 (SOD1) cause ALS revolutionized the field (Deng et al., 1993; Rosen et al., 1993). SOD1 is an evolutionarily conserved, ubiquitously expressed protein that catalyzes breakdown of superoxide radicals into hydrogen peroxide and water. As the second most common gene whose mutation causes familial ALS (fALS), mutations in SOD1 account for approximately 20% of fALS cases and 5% of sporadic ALS (sALS) cases (Kaur et al., 2016). Over 100 mutations have been identified in SOD1, and almost all disease alleles are dominant in patients. From many studies, it seems likely that disease alleles cause a toxic gain of function, but loss of function may contribute to disease pathology (Bruijn et al., 1998; Saccon et al., 2013).
Two non-exclusive hypotheses for SOD1-associated ALS motor neuron degeneration dominate the field: the aggregation hypothesis and the oxidative stress hypothesis. Mutant SOD1 protein aggregates in the cytosol of SOD1 ALS patient cells are thought to confer toxicity or reduce SOD1 enzymatic activity (Stieber et al., 2000; Watanabe M. et al., 2001). Early studies in mice supported a toxic gain of function hypothesis, as SOD1 null mice do not exhibit ALS-like pathology and overexpression of mutant human SOD1 resulted in reduced enzymatic activity (Gurney et al., 1994; Reaume et al., 1996). However, how SOD1 mutations cause ALS is still debated. More recent studies suggest that SOD1 loss of function also contributes to ALS dysfunction and degeneration. One possible mechanism is that mutations in SOD1 cause loss of function by aggregation, causing abnormal buildup of superoxide radicals or hydrogen peroxide, the substrate and byproducts of SOD1 action, respectively (Beckman et al., 1993). SOD1 activity is decreased in patients with ALS, suggesting that SOD1 loss of function may contribute to pathology (Rosen et al., 1993; Watanabe Y. et al., 1997). SOD1-mediated motor neuron death may be caused by a combination of the loss and gain of function consequences of patient alleles of SOD1 (Sahin et al., 2017).
ALS models have been created by overexpressing mutant human SOD1 protein in numerous model organisms and comparing the deleterious consequences of the mutant protein to the consequences of overexpressed wild type human SOD1 protein. Two of the most frequently used patient alleles in SOD1 ALS models are missense mutations that result in a glycine to arginine substitution at position 85 (G85R) or a glycine to alanine substitution at position 93 (G93A). As SOD1 loss of function may also contribute to ALS-associated defects, modifier genes that suppress defects associated with SOD1 loss of function alleles are also of interest. In the literature, we found 33 articles that, in combination, yielded 164 modifier genes in either SOD1 ALS model animals or animals lacking SOD1 ortholog function. These are listed in Supplemental File 1 in the SOD1 tab (Allodi et al., 2016; Bahadorani et al., 2013; Boccitto et al., 2012; Chloupkova et al., 2003; Couillard-Despres et al., 1998; Dadon-Nachum et al., 2015; Dobrowolny et al., 2008; Giess et al., 2002; Hetz et al., 2009; Jablonski et al., 2015; Kieran et al., 2007; Kumimoto et al., 2013; Lambrechts et al., 2003; Lapinskas et al., 1995; Liu et al., 2002; Lobsiger et al., 2005; Lorenzl et al., 2006; Lu et al., 2009; Lunn et al., 2009; Marden et al., 2007; Ohta et al., 2016; Pitzer et al., 2008; Reyes et al., 2010; Riddoch-Contreras et al., 2009; Sharp et al., 2008; Silva et al., 2011; Strain et al., 1998; Teuling et al., 2008; Turner et al., 2014; Wang J. et al., 2009; Yang Y. S. et al., 2009; Zhai et al., 2005).
We hypothesized that shared pathways might link SOD1 modifier genes. To identify these connections, we undertook gene ontology enrichment analysis of the assembled SOD1 modifier gene lists and identified enriched Gene Ontology (GO) pathways. Initially, this analysis was complicated by the diversity of model organisms used for modifier gene identification. To facilitate cross-species comparisons and bioinformatic analysis, the closest human ortholog of each modifier gene was identified based on amino acid similarity using reciprocal BLAST analysis. Proteins that lacked a human ortholog were excluded from bioinformatic analysis. This bioinformatic analysis revealed enrichment of pathways integral for endogenous SOD1 function: “response to reactive oxygen species” (GO:0000302) and “regulation of oxidative stress-induced intrinsic apoptotic signaling” (GO:1902175). The complete SOD1 pathway analysis is presented in Supplemental File 2, SOD1 tab, and top hits are illustrated in Fig. 2A (pathways with odds ratio > 5). The most enriched pathway for SOD1 modifier genes was “neurotransmitter reuptake” (GO:0098810). The SOD1 modifier genes from the literature survey that led to bioinformatic analysis identification of “neurotransmitter reuptake” are shown in Fig. 6.
Fig. 2.
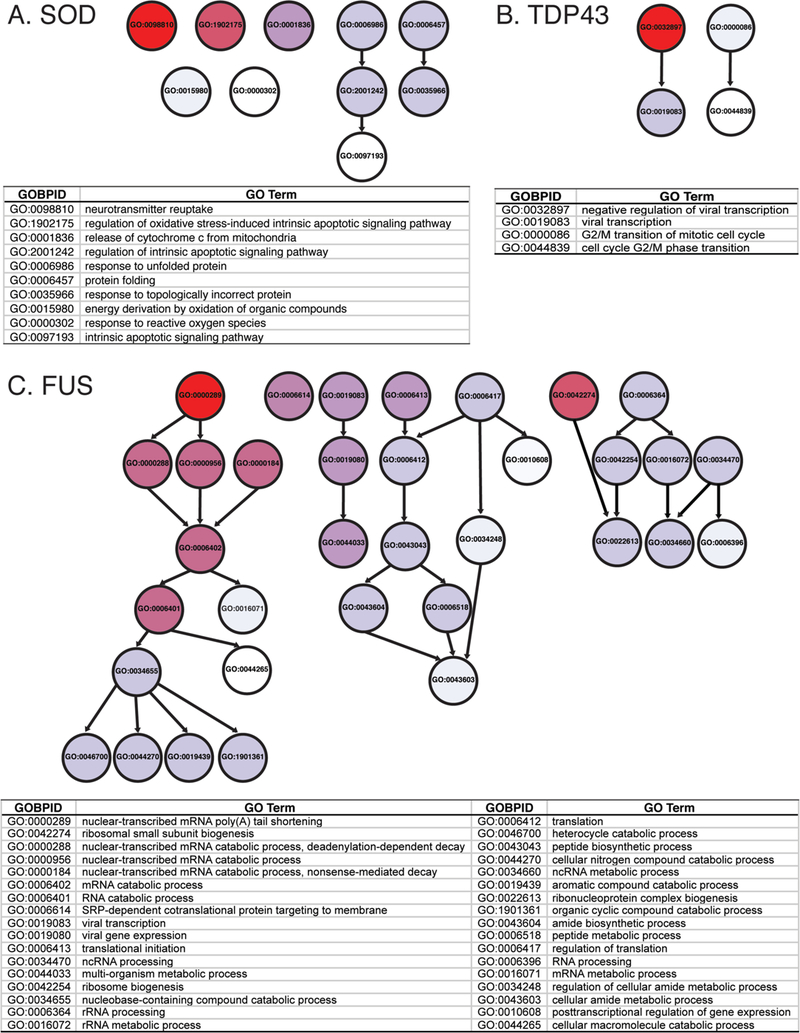
GO terms enriched for SOD1, TDP43 and FUS A) Diagram (top) shows relationship between GO terms enriched in gene ontology analysis of SOD1 genetic modifiers. Arrows indicate related terms that are “nested” inside a broader category. GO terms above odd ratios of 5 or greater are listed (below); the most highly enriched genes are at the top of the list. Darker red hues indicate a higher odds ratio (the magnitude of enrichment). For example, “neurotransmitter reuptake (GO:0098810)” has an odds ratio of 29.86 and is shown in red. This indicates that we observe more genes associated with “neurotransmitter reuptake” in the list of SOD1 genetic modifiers than expected. Though it is enriched in our dataset, “intrinsic apoptotic signaling pathway (GO:0097193)” has an odds ratio of 5.35 and is shown in white. In this case, we still observe more genes than expected with “intrinsic apoptotic signaling pathway” in our dataset, but not to the same extent as “neurotransmitter reuptake” genes. B) GO terms enriched in TDP43 modifiers, presented as in panel A. C) GO terms enriched in FUS modifiers, presented as in panel A.
Fig. 6.
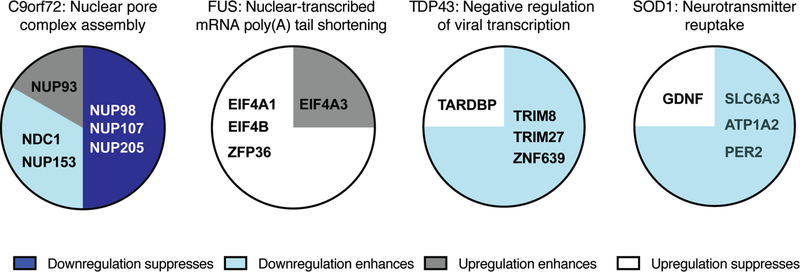
Modifier genes in the most enriched GO term for ALS genes. The most enriched GO term for each ALS genes is listed. Each pie chart contains the names of all human genes (or orthologs) that were associated with the top GO term. Genes are grouped and color coded based on originally reported perturbation of the modifier gene and their impact on ALS-associated defects. For example, knockdown of the NUP98 D. melanogaster ortholog ameliorated ALS-associated defects in a D. melanogaster C9orf72; this gene was classified as “downregulation suppressed”.
TDP43
Ubiquitinated inclusions in affected patient neurons are a frequent pathological hallmark of ALS (Arai et al., 2006; Leigh et al., 1991; Ling et al., 2013; Liscic et al., 2008; Mackenzie and Feldman, 2005; Maekawa et al., 2009; Neumann et al., 2006). In 2006, TAR DNA binding protein 43 (TDP43), was identified as the ubiquitinated protein in intracellular aggregates in both ALS and FTD (Neumann et al., 2006). TDP43, encoded by the TARDBP gene, is a ubiquitously expressed nucleic acid binding protein that play critical roles in RNA splicing and microRNA biogenesis (Buratti and Baralle, 2008). Over 40 missense mutations in TARDBP have been identified in ALS cases (Sreedharan and Brown, 2013). These missense mutations are almost always located in the glycine-rich C-terminal domain of the protein, which has important roles in protein-protein interactions and liquid-liquid phase separation (Wang et al. 2018; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008).
TDP43 is predominantly found in the nucleus, with a minor fraction of the protein cycling through the cytosol. However, cytosolic TDP43 dramatically increases in patients carrying TARDBP fALS alleles, in many sALS patients, and in a large fraction of fALS patients carrying mutations in other causal genes. TDP43 mislocalization may contribute to the degeneration of motor neurons in ALS/FTD. One hypothesis is that mutant TDP43 acts through a gain of toxic function mechanism by aggregating and inhibiting the endogenous function of normal TDP43. In this model, TDP43 loss of function defects would contribute to neurodegeneration. Alternatively, mutations in TDP43 could alter endogenous RNA splicing and microRNA biogenesis via disruption of functional interactions (Conicella et al., 2016) or mutant TDP43 protein may act in an abnormal cellular compartment, resulting in neurodegeneration and indicative of a gain of toxic function mechanism.
We found eleven published studies that, in combination, reported 93 modifier genes of TDP43 ALS/FTD phenotypes (Supplemental File 1, TDP43 Tab), for either mutant TDP43 overexpression, wildtype TDP43 overexpression, or TDP43 ortholog loss of function (Armakola et al., 2012; Chou et al., 2015; Elden et al., 2010; Figley and Gitler, 2013; Jablonski et al., 2015; Kim H. J. et al., 2014; Kim S. H. et al., 2012; Liachko et al., 2013; Sreedharan et al., 2015; Zhan et al., 2013; Zhan et al., 2015). We undertook bioinformatics analysis, as described above, with these TDP43 modifier genes and found only 4 enriched GO pathways with odds ratio above 5 (Fig. 2B). Pathways are listed in Supplemental File 2, TDP43 tab, and include “G/M2 cell cycle regulation” (GO:0000086, GO:0044839) and “regulation of viral transcription” (GO:0019083, GO:0032897), for which TDP43 roles have already been described (Ignatius et al., 1995; Yamashita et al., 2014). Modifier genes that led to bioinformatic analysis identification of “G/M2 cell cycle regulation” are shown in Fig. 6.
FUS
Originally characterized as a liposarcoma oncogene, mutations in the Fused in Sarcoma gene (FUS) were found in a cohort of 197 British ALS patients in 2009. The FUS protein is a ubiquitously expressed RNA-binding protein involved in splicing and stress granule formation (Lagier-Tourenne and Cleveland, 2009). Mutations in FUS cause approximately 4–5% of all familial ALS cases. Patient mutations can be found throughout the FUS protein, but mutation of the C-terminal nuclear localization signal (NLS) is most frequently observed (Ju et al., 2011; Ling et al., 2013). In some cases, FUS mutations result in FTD, and patients with FUS-linked FTD usually show ALS symptoms (Nolan et al., 2016). These FTD patients present with FUS-immunoreactive inclusions; these inclusions are also present in the motor neurons of FUS ALS patients who lack FTD symptoms (Deng et al., 2010; Hewitt et al., 2010; Rademakers et al., 2010).
In most FUS ALS patients examined, mutant FUS is mislocalized from the nucleus and protein aggregates form in the cytoplasm (Dormann et al., 2010; Vance et al., 2009). Furthermore, cytoplasmic FUS incorporates into membraneless organelles - phase separated liquid structures (e.g. stress granules), which may drive mutant FUS aggregation (Bosco et al., 2010; Burke et al., 2015; Patel et al., 2015). Cytoplasmic aggregation of FUS may inhibit the maturation of RNAs integral for the survival of motor neurons, as nuclear FUS is important for mRNA splicing (Colombrita et al., 2015; Sun S. et al., 2015). Alternatively, mutant FUS may act via a gain of function mechanism where patient mutations may subvert DNA repair mechanisms, leading to cumulative increases in DNA damage (Hill et al., 2016).
We found five articles that identified 72 modifiers of FUS (Armakola et al., 2012; Chen Y. et al., 2016; Farg et al., 2013; Ju et al., 2011; Sun Z. et al., 2011) (Supplemental File 1, FUS tab). Many of these suppressor and enhancer genes were identified in genome-wide modifier screens in yeast expressing mutant human FUS at high levels (Sun Z. et al., 2011). Our bioinformatic analysis identified 34 GO terms/pathways that were enriched (Fig. 2C, Supplemental File 2, FUS tab). Many of these are related to cellular pathways associated with normal FUS protein function, including “RNA processing” (GO:0006396) and “translation” (GO:0006412). The FUS modifier genes associated with the most enriched GO term “nuclear-transcribed mRNA poly(A) tail shortening” (GO:0000289) are shown in Fig. 6.
C9orf72
In 2011, expansion of GGGGCC (G4C2) repeats in the non-coding region of chromosome 9 open reading frame 72 (C9orf72) was identified in ALS patients. C9orf72 expansion is one of the most common causes of ALS and FTD and accounts for approximately 40% of fALS cases (Rademakers et al., 2012). The number of G4C2 repeats varies dramatically between patients; Southern blot analysis from one family revealed pathogenic repeats ranging from 700–1,600 (DeJesus-Hernandez et al., 2011; Haeusler et al., 2016). In addition to the typical ALS motor neuron functional defects, C9orf72 ALS patients may have earlier disease onset, cognitive and behavioral impairment, and decreased survival compared to other patients (Rademakers et al., 2012).
Why G4C2 repeats cause disease is still unclear; studies have suggested the C9orf72 protein has roles in the endolysosomal pathway and vesicle trafficking (Aoki et al., 2017; Corrionero and Horvitz, 2018). Three non-exclusive mechanisms have been proposed: decreased C9orf72 protein expression, toxic expanded G4C2 repeat RNAs, and/or toxic dipeptide repeat (DPR) proteins generated by repeat-associated non-AUG (RAN) translation of G4C2 repeat RNAs (Haeusler et al., 2016).
Considerable evidence suggests that high level expression of either G4C2 repeat-derived RNAs or DPR proteins can be toxic. G4C2 repeat RNA may sequester RNA binding proteins and splicing factors, thus disrupting their normal functions and causing neurodegeneration (Lee Y. B. et al., 2013; Mori et al., 2016; Xu et al., 2013). This model is supported by the observation that overexpression of Pur-α, an RNA-binding protein that physically interacts with repeat RNAs, suppresses G4C2-mediated neurodegeneration in mouse neuronal cells and D. melanogaster (Xu et al., 2013). However, DPR proteins are also toxic. These are produced through RAN translation of G4C2 repeat RNA, which occurs in the absence of an AUG initiation codon and from both sense and antisense G4C2 repeat strands (Zu et al., 2013). Different DPR proteins have varying levels of toxicity: the arginine-rich DPRs, poly(GR) and poly(PR) are most toxic (Jovicic et al., 2015; Kwon et al., 2014; Wen et al., 2014), poly(GA) is moderately toxic, and poly(GP) and poly(PA) are the least toxic (Freibaum et al., 2015; Wen et al., 2014).
We found eight articles that identified modifier genes for G4C2 RNA and/or DPR toxicity (Supplemental file 1, C9orf72 tab) (Boeynaems et al., 2016; Freibaum et al., 2015; Jovicic et al., 2015; Kramer et al., 2016; Lee K. H. et al., 2016; Mori et al., 2016; Xu et al., 2013; Zhang et al., 2015). Multiple unbiased genetic screens were undertaken in D. melanogaster and S. cerevisiae for modifiers of poly(PR) toxicity (Boeynaems et al., 2016; Jovicic et al., 2015). Screens in a D. melanogaster eye poly(PR) model yielded modifiers encoding proteins that directly interact with poly(GR) and poly (PR) peptides (Lee K. H. et al., 2016). In a candidate-based screen using D. melanogaster expressing (G4C2)30 repeats, RanGAP was identified as a suppressor of neurodegeneration (Zhang et al., 2015). No modifiers of C9orf72 loss of function have been reported.
From these eight studies, we assembled a list of 285 genetic modifiers with human orthologs of G4C2 toxicity (Supplemental File 1, C9orf72 tab). Gene ontology bioinformatic analysis revealed 98 enriched GO pathways (Supplemental File 2, C9orf72 tab). These include “nuclear pore assembly”, “protein import”, and “protein export” (Fig. 3, Fig. 4). Additionally, “gene silencing by miRNA” (GO:0035195) and metabolism-associated pathways were enriched in this dataset. Genes associated with the most enriched pathway in our bioinformatics analysis, “nuclear pore complex assembly” (GO:0051292), are presented in Fig. 6.
Fig. 3.
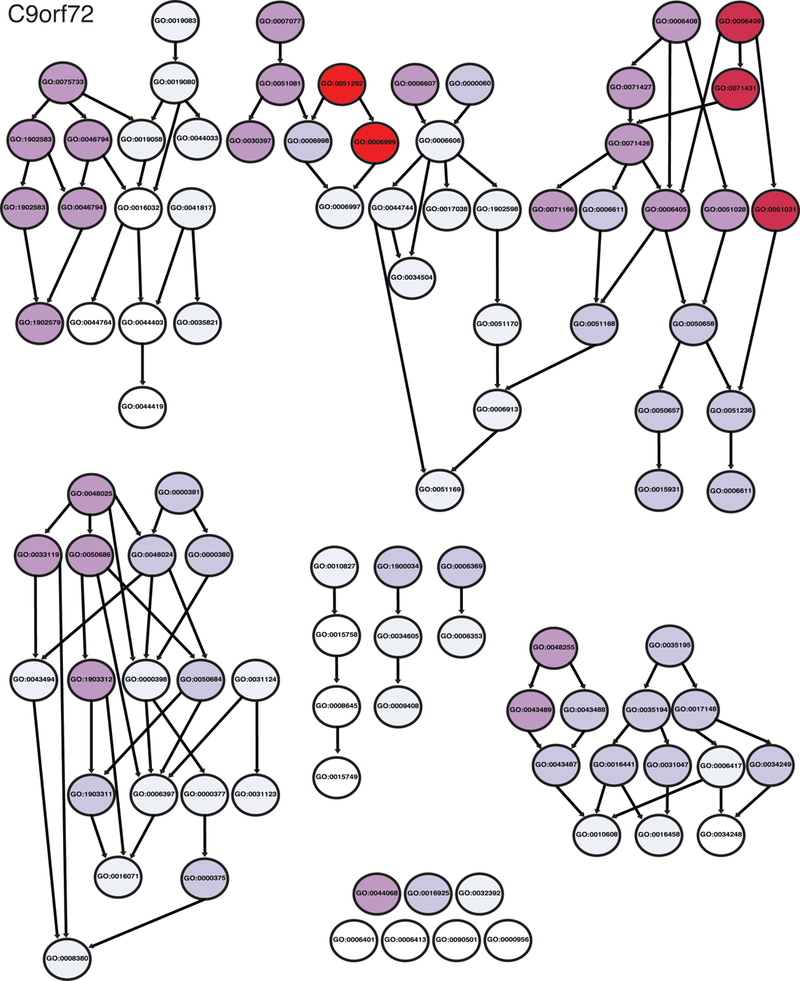
Diagram of GO terms enriched for C9orf72. Illustration of the relationship between GO terms enriched in gene ontology analysis of C9orf72 genetic modifiers. Arrows indicate related terms that are “nested” inside a broader category. Darker red hues are GO terms that were more enriched in the modifier list.
Fig. 4.

List of GO terms enriched for C9orf72. GO terms with odd ratios of 5 or more are listed; the most highly enriched genes are at the top of the list.
VAPB and other ALS genes
Most studies that report ALS modifier genes focus on SOD1, TDP43, C9orf72, or FUS. Mutations in other genes also lead to ALS and insights into disease pathogenesis may arise from analysis of these other disease genes. In 2004, a P56S mutation in the Vesicle-Associated Membrane Protein-Associated Protein B/C (VAPB) gene was identified in seven different Brazilian families with afflicted individuals showing ALS and/or late-onset spinal muscular atrophy (Nishimura et al., 2004). VAPB protein interacts with SNARE proteins and regulates vesicular transport. Although the severity, presentation, and progression of disease varies between families, the VAPB P56S mutation was dominant (Nishimura et al., 2004). The P56S mutation lies in the VAPB protein Major Sperm Protein (MSP) domain, which likely mediates protein dimerization and other protein-protein interactions.
The functional consequences of VAPB P56S that lead to ALS are poorly understood, but both gain of function (Kuijpers et al., 2013; Ratnaparkhi et al., 2008) and loss of function mechanisms (Kabashi et al., 2013) have been proposed. In normal cells, VAPB mediates membrane interactions between mitochondria and the endoplasmic reticulum, which are critical for mitochondrial calcium regulation and ATP production (Stoica et al., 2014; Stoica et al., 2016). Mutations in either TDP43 or FUS can disrupt VAPB function, ultimately leading to disrupted mitochondrial calcium uptake and decreasing ATP production (Stoica et al., 2014; Stoica et al., 2016). We found two articles describing genetic modifiers of VAPB (Deivasigamani et al., 2014; Sanhueza et al., 2015), as well as one article describing genetic modifiers of OPTN (Akizuki et al., 2013) and one article describing VCP modifier genes (Ritson et al., 2010) (Supplemental File 1, Other tab). We searched for modifier genes of other fALS-linked genes, but did not uncover additional modifier studies in the published literature. VAPB modifiers were identified in two different D. melanogaster screens. Deivasigamani et al. upregulated or downregulated D. melanogaster VAPB (dVAP) ortholog levels, which results in altered bristles (Deivasigamani et al., 2014). Sanhueza et al., found that high level expression of dVAP[P58S] in the D. melanogaster eye leads to reduced eye size and used this observation to identify 85 modifier genes (Sanhueza et al., 2015). Only one pathway, “single-organism cellular localization” (GO:1902580), was significantly enriched in our gene ontology bioinformatics.
Modifier genes associated with more than one ALS gene
If patient alleles in the genes listed above lead to a single disease, which we call ALS, then one would expect commonalities in disease mechanism and pathological processes. Accordingly, one might expect common pathways to arise from modifier gene analyses. This hypothesis is supported by previous work demonstrating that some modifier genes impact ALS-associated defects in more than one ALS model. In total, 946 modifier genes were identified from the literature with 727 corresponding human orthologs (Supplemental File 1). To look for commonalities between ALS modifier genes, we compiled a list of modifier genes with impact on more than one ALS causal gene (Table 1). For example, if a gene modified defects in both an SOD1 ALS model and a TDP43 ALS model, it was included in Table 1 (and in Supplemental File 1, multiple ALS genes). In addition, Table 1 includes modifier genes identified in ALS patient GWAS or genetic studies that have been validated in ALS models, as these are likely relevant to disease. In total, 43 modifier genes have functional impact on more than one ALS gene and are listed in Table 1.
Table 1.
Genetic modifiers that may modify multiple ALS genes. This list includes the human orthologs that were identified as modifiers of more than one ALS-causal genes (e.g. SRRT orthologs were reported as modifiers in both SOD1 and TDP43 models). Additionally, human genes reported as modifiers through GWAS or linkage analysis studies are also included in this list.
Two genes, KPNB1 and TARDBP, were identified as genetic modifiers in more than two ALS models. KPNB1 was reported as a modifier of C9orf72, VAPB, and TDP43 ALS models. In an RNAi screen conducted in D. melanogaster C9orf72 model of ALS, decreased KPNB1 function enhanced PR25-mediated eye degeneration (Boeynaems et al., 2016). In HeLa cells, KNPB1 knockdown enhanced the cytosolic localization of TDP43 (Kim S. H. et al., 2012). Additionally, overexpression of the D. melanogaster ortholog of KPNB1, Fs(2)Ket, resulted in suppression of the rough eye phenotype in a VAPB model of ALS. TARDBP was reported as a modifier of C9orf72, VAPB, and VCP. RNAi knockdown of TARDBP in a C9orf72 model suppressed a viability defect and the rough-eye phenotype in a D. melanogaster model (Lee K. H. et al., 2016). TARDBP has also been reported as a suppressor of VCP-related degeneration (Ritson et al., 2010) and acts as a suppressor in an overexpression model of VAPB (Deivasigamani et al., 2014). While TARDBP was intentionally selected for assessment in these studies, the nuclear pore complex protein, KPNB1, was independently identified in less biased screens.
To identify common pathways associated with modifier genes, we undertook bioinformatic analysis with the genes listed in Table 1. Forty-two GO terms were enriched in this analysis. Pathways enriched in in this modifier gene list included GO terms associated with protein transport or metabolic processes. The most enriched GO term was “protein import into nucleus, translocation” (GO:0000060).
Discussion
Modifier gene studies have the potential to dramatically increase our understanding of ALS pathogenesis and to provide insight into variation in patient symptoms, penetrance of disease, and disease progression. Modifier gene studies can provide insights into pathways associated with neuronal dysfunction and neurodegeneration in ALS. Furthermore, common genetic modifiers may link ALS caused by mutations in different genes, suggesting a common mechanism of motor neuron degeneration. Additionally, modifier genes can be used to identify pathways and targets for therapeutic intervention. Many genetic modifiers of ALS have been discovered through hypothesis-driven experiments, forward genetic screens, or genetic studies in human populations. But, to our knowledge, a comprehensive listing and analysis of modifier genes pertinent to ALS has not been undertaken previously.
It is likely that both loss and gain of function mechanisms contribute to ALS pathology. Therefore, we included modifiers of both loss- and gain of function in our survey, as well as overexpression of wildtype or mutant protein. In total, we compiled a list of 727 modifier genes with human orthologs. Many modifier genes were found in S. cerevisiae, C. elegans, D. melanogaster, M. musculus, and cell culture models of ALS. Additionally, genetic modifiers were identified in ALS patients through linkage analysis and genome wide association studies. In total, we identified 72 articles in the published literature that reported 727 modifier genes with human orthologs for SOD1, TDP43, FUS, C9orf72, VAPB, VCP, or OPTN. We searched for modifiers of other ALS-linked genes, but did not find any in the published literature. Interestingly, the 727 genes identified as modifiers of ALS corresponds to roughly 5% of human genes, consistent with the complexity of this disease. We appreciate the enormous effort these original studies represent and we hope to highlight the importance of these studies and leverage their results to identify common pathways pertinent to ALS. We note that additional modifier genes have been reported in subsequent studies, including those by Kramer et al. 2018 (Kramer et al., 2018).
Many of the pathways that were enriched in our bioinformatics analysis are associated with the endogenous functions of genes implicated in ALS and are established as dysregulated in ALS patients, including “response to reactive oxygen species” (GO:0000302) in SOD1 and “RNA processing “(GO:0006396) in FUS. Interestingly, metabolic processes were identified as enriched pathways in all of our modifier gene lists. This commonality highlights the importance of previous studies demonstrating that metabolism is affected in ALS patients (Mattiazzi et al., 2002).
FUS and TARDBP encode RNA-associated proteins and it has been suggested that they act in the same pathways in ALS pathogenesis (Honda et al., 2013). The analysis of FUS and TARDBP modifier genes could be interpreted to support this hypothesis. When GO pathways are examined, 2 of the 12 enriched GO terms found from the list of TDP43 modifier were also included in the list of 60 FUS-modifier enriched GO terms: “cellular macromolecule metabolic process” and “viral transcription”. There were over 2000 GO pathway terms available in our bioinformatic analysis; the small overlap we observed between FUS and TDP43 is significant, but may reflect the importance of RNA-binding proteins in these processes.
The comprehensive literature search reveals that relatively few ALS modifier genes have been tested in other models of ALS or have been identified in more than one independent modifier screen. Overall, only 43 modifier genes are reported to modify more than one ALS gene. Of these 43 genes, KNBP1 and TARDBP were reported to be modifiers in more than two ALS models. An inherent bias against the publication of “failure to suppress cross-species” may partially account for this, as well as a bias against reporting negative results, or a lack of motivation to re-test modifier genes identified in other species/models. To fully understand why mutations in specific genes cause ALS and to identify therapeutic targets, we suggest that modifier genes should be tested in multiple ALS models. This should expose commonalities and differences between ALS caused by mutations in different genes and inform the selection of therapeutic targets.
ALS modifier gene studies have already increased our understanding of pathways that may be dysregulated in this devastating disease. We provide the first comprehensive review of published ALS modifier genes and undertook bioinformatic analysis. These data suggest that common pathways may underlie ALS caused by mutations in different genes. We expect that as additional modifier genes are identified and tested in additional models of ALS, more commonalities between the different ALS genes will be found and additional therapeutic targets will be developed for the treatment of this disease.
Supplementary Material
Supplemental File 1. List of genes reported to modify ALS-associated defects. The page labeled “Modifiers of multiple” contains the genes that were found to modify two or more ALS genes. Pages are labeled with the name of the ALS gene that the genetic modifier was observed to modify. Each tab contains the following information: the originally reported gene, the human ortholog, and the reference.
Supplemental File 2. Gene ontology analysis results listing enriched GO terms for each of the analyses reported in the main text and the Figures. Each page lists the gene ontology term, the odds ratio, p-value, expected counts (the number of times a gene should be in this category given the number of genes on our list), the count (the number of times a gene is associated with the specific GO term here), and size (the number of genes associated with the called GO term).
Supplemental File 3. Genes associated with enriched GO terms. The modifier genes associated with each enriched GO term are reported from the original analysis output.
Fig. 5.
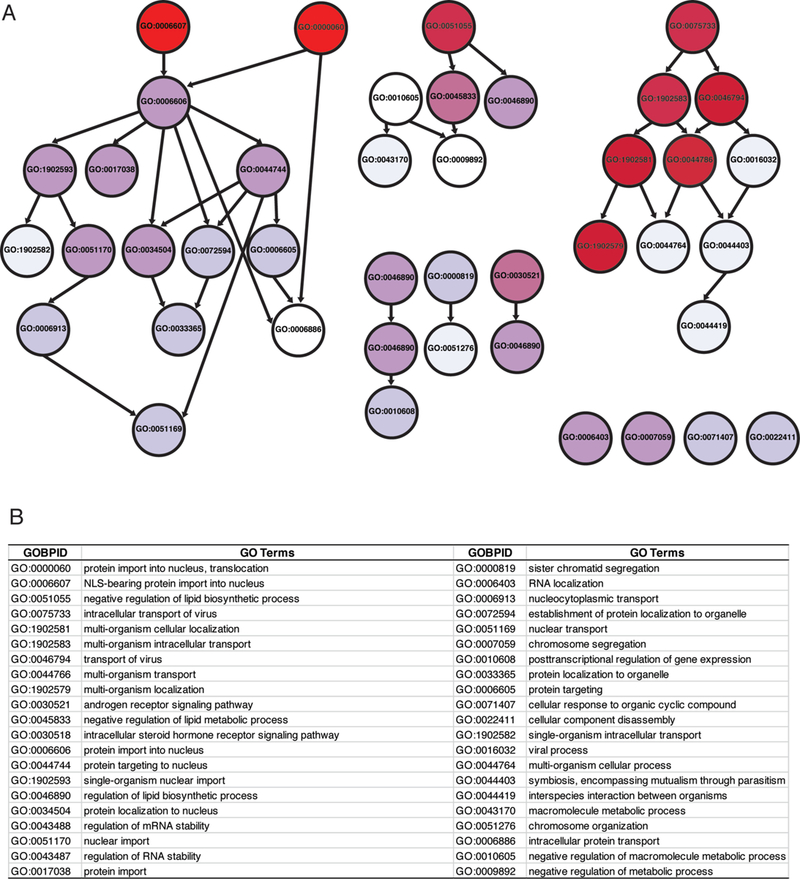
GO terms enriched for genes reported to be modifiers of multiple ALS genes. Diagram (top) shows relationship between GO terms enriched in gene ontology analysis. Arrows indicate related terms that are “nested” inside a broader category. Darker red hues are GO terms that were more enriched in the modifier list. GO terms above odd ratios of 5 or greater are listed (below); the most highly enriched genes are at the top of the list.
Acknowledgements
We thank Robert H. Brown Jr. and the ALS@Brown community for helpful discussions.
Funding
Research reported in this publication was supported in part by the ALS Finding a Cure Foundation (to J.R.F., D.L., R.A.R., K.A.W., A.C.H), ALS Association Grant ID 15-IIP-203 (to A.C.H.), National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) Award Number R01GM118530 (to N.L.F) and a starter grant 17-IIP-342 from the ALS Association (to N.L.F.). A.H. and V.H.R. were supported in part by the Robert J. and Nancy D. Carney Institute for Brain Science Graduate Award. K.S.Y, K.H.H., K.R., and V.H.R. was supported in part by a NIMH training grant to the Neuroscience Graduate Program at Brown University (T32MH020068). K.S.Y was also supported in part by a National Institute of Neurological Disorders and Stroke (NINDS) training grant to the Neuroscience Graduate Program at Brown University (T32NS62443). Z. W. was partially supported by R01GM122083, P20GM109035, and P20GM109035. V. H. R. was partially supported by a grant from the NINDS (F31NS110301).
Abbreviations
- ALS
Amyotrophic Lateral Sclerosis
- C9orf72
Chromosome 9 open reading frame 72
- fALS
Familial ALS
- FTD
Frontotemporal Dementia
- FUS
Fused in sarcoma
- OPTN
Optineurin
- sALS
Sporadic ALS
- SOD1
Superoxide dismutase 1
- TDP43
TAR DNA binding protein 43
- VCP
Valosin-containing protein
- VAPB
Vesicle-Associated Membrane Protein-Associated Protein B/C
Footnotes
Availability of data and material
All data used in the analysis are included in the manuscript and supplemental files.
Competing interests
The authors report no competing interests.
References
- ((NCBI) NCBI, Basic Local Alignment Search Tool In: (US), N.L.o.M. (Ed.). National Center for Biotechnology Information, Bethesda (MD). [Google Scholar]
- Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, Takahashi R, 2013. Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J Neurochem 126, 699–704. [DOI] [PubMed] [Google Scholar]
- Alexander AG, Marfil V, Li C, 2014. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front Genet 5, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allodi I, Comley L, Nichterwitz S, Nizzardo M, Simone C, Benitez JA, Cao M, Corti S, et al. , 2016. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci Rep 6, 25960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AGL, Varela MA, Sathyaprakash C, et al. , 2017. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, et al. , 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351, 602–611. [DOI] [PubMed] [Google Scholar]
- Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, Fang X, Shorter J, et al. , 2012. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 44, 1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadorani S, Mukai ST, Rabie J, Beckman JS, Phillips JP, Hilliker AJ, 2013. Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol Aging 34, 2322–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, et al. , 2006. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. [DOI] [PubMed] [Google Scholar]
- Barber SC, Mead RJ, Shaw PJ, 2006. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta 1762, 1051–1067. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Carson M, Smith CD, Koppenol WH, 1993. ALS, SOD and peroxynitrite. Nature 364, 584. [DOI] [PubMed] [Google Scholar]
- Boccitto M, Lamitina T, Kalb RG, 2012. Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS One 7, e33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovicic A, De Baets G, Scheveneels W, et al. , 2016. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6, 20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ Jr., Sapp P, McKenna-Yasek D, et al. , 2010. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19, 4160–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner D, Muller K, Wieland T, Weydt P, Bohm S, Lule D, Hubers A, Neuwirth C, et al. , 2016. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, e28. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, et al. , 1998. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE, 2008. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci 13, 867–878. [DOI] [PubMed] [Google Scholar]
- Burke KA, Janke AM, Rhine CL, Fawzi NL, 2015. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol Cell 60, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaussenot A, Le Ber I, Ait-El-Mkadem S, Camuzat A, de Septenville A, Bannwarth S, Genin EC, Serre V, et al. , 2014. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol Aging 35, 2884 e2881–2884. [DOI] [PubMed] [Google Scholar]
- Chen X, Burgoyne RD, 2012. Identification of common genetic modifiers of neurodegenerative diseases from an integrative analysis of diverse genetic screens in model organisms. BMC Genomics 13, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Deng J, Wang P, Yang M, Chen X, Zhu L, Liu J, Lu B, et al. , 2016. PINK1 and Parkin are genetic modifiers for FUS-induced neurodegeneration. Hum Mol Genet [DOI] [PMC free article] [PubMed]
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, et al. , 2004. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74, 1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi A, Staahl BT, Jovicic A, Couthouis J, Fasolino M, Raphael AR, Yamazaki T, Elias L, et al. , 2013. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci 16, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chloupkova M, LeBard LS, Koeller DM, 2003. MDL1 is a high copy suppressor of ATM1: Evidence for a role in resistance to oxidative stress. Journal of Molecular Biology 331, 155–165. [DOI] [PubMed] [Google Scholar]
- Chou CC, Alexeeva OM, Yamada S, Pribadi A, Zhang Y, Mo B, Williams KR, Zarnescu DC, et al. , 2015. PABPN1 suppresses TDP-43 toxicity in ALS disease models. Hum Mol Genet 24, 5154–5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, et al. , 2009. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 84, 85–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, et al. , 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Onesto E, Buratti E, de la Grange P, Gumina V, Baralle FE, Silani V, Ratti A, 2015. From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models. Biochim Biophys Acta 1849, 1398–1410. [DOI] [PubMed] [Google Scholar]
- Conicella AE, Zerze GH, Mittal J, Fawzi NL, 2016. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 24, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Horvitz HR, 2018. A C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis. Curr Biol [DOI] [PubMed]
- Couillard-Despres S, Zhu QZ, Wong PC, Price DL, Cleveland DW, Julien JP, 1998. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proceedings of the National Academy of Sciences of the United States of America 95, 9626–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, et al. , 2012. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21, 2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, et al. , 2006. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. [DOI] [PubMed] [Google Scholar]
- Dadon-Nachum M, Ben-Yaacov K, Ben-Zur T, Barhum Y, Yaffe D, Perlson E, Offen D, 2015. Transplanted modified muscle progenitor cells expressing a mixture of neurotrophic factors delay disease onset and enhance survival in the SOD1 mouse model of ALS. J Mol Neurosci 55, 788–797. [DOI] [PubMed] [Google Scholar]
- Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A, Tranchant C, Dion P, et al. , 2012. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging 33, 839 e835–839. [DOI] [PubMed] [Google Scholar]
- Deivasigamani S, Verma HK, Ueda R, Ratnaparkhi A, Ratnaparkhi GS, 2014. A genetic screen identifies Tor as an interactor of VAPB in a Drosophila model of amyotrophic lateral sclerosis. Biol Open 3, 1127–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, et al. , 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, et al. , 2011. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, et al. , 1993. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261, 1047–1051. [DOI] [PubMed] [Google Scholar]
- Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, Mishra M, Ajroud-Driss S, et al. , 2010. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriadi M, Hart AC, 2010. Neurodegenerative disorders: insights from the nematode Caenorhabditis elegans. Neurobiol Dis 40, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolny G, Aucello M, Molinaro M, Musaro A, 2008. Local expression of mIgf-1 modulates ubiquitin, caspase and CDK5 expression in skeletal muscle of an ALS mouse model. Neurol Res 30, 131–136. [DOI] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, et al. , 2010. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, et al. , 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R, 2007. Using GOstats to test gene lists for GO term association. Bioinformatics 23, 257–258. [DOI] [PubMed] [Google Scholar]
- Farg MA, Soo KY, Warraich ST, Sundaramoorthy V, Blair IP, Atkin JD, 2013. Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum Mol Genet 22, 717–728. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Kapogiannis D, Huey ED, Momeni P, 2011. FTD and ALS: a tale of two diseases. Curr Alzheimer Res 8, 273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DA, Krizus A, Martinoli MG, Meininger V, Dib M, Rouleau GA, Julien JP, 1994. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet 3, 1757–1761. [DOI] [PubMed] [Google Scholar]
- Figley MD, Gitler AD, 2013. Yeast genetic screen reveals novel therapeutic strategy for ALS. Rare Dis 1, e24420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, et al. , 2015. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, et al. , 2015. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 18, 631–636. [DOI] [PubMed] [Google Scholar]
- Gama Sosa MA, De Gasperi R, Elder GA, 2012. Modeling human neurodegenerative diseases in transgenic systems. Hum Genet 131, 535–563. [DOI] [PubMed] [Google Scholar]
- Ghasemi M, Brown RH Jr., 2017. Genetics of Amyotrophic Lateral Sclerosis Cold Spring Harb Perspect Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giess R, Holtmann B, Braga M, Grimm T, Muller-Myhsok B, Toyka KV, Sendtner M, 2002. Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: Potential impact of CNTF as a candidate modifier gene. American Journal of Human Genetics 70, 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, et al. , 2006. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38, 411–413. [DOI] [PubMed] [Google Scholar]
- Gros-Louis F, Andersen PM, Dupre N, Urushitani M, Dion P, Souchon F, D’Amour M, Camu W, et al. , 2009. Chromogranin B P413L variant as risk factor and modifier of disease onset for amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America 106, 21777–21782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros-Louis F, Lariviere R, Gowing G, Laurent S, Camu W, Bouchard JP, Meininger V, Rouleau GA, et al. , 2004. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J Biol Chem 279, 45951–45956. [DOI] [PubMed] [Google Scholar]
- Gurney M, Pu H, Chiu A, Dal Canto M, Polchow C, Alexander D, Caliendo J, Hentati A, et al. , 1994. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. [DOI] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Rothstein JD, 2016. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci 17, 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, et al. , 2009. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 23, 2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt C, Kirby J, Highley JR, Hartley JA, Hibberd R, Hollinger HC, Williams TL, Ince PG, et al. , 2010. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 67, 455–461. [DOI] [PubMed] [Google Scholar]
- Hill SJ, Mordes DA, Cameron LA, Neuberg DS, Landini S, Eggan K, Livingston DM, 2016. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc Natl Acad Sci U S A 113, E7701–E7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda D, Ishigaki S, Iguchi Y, Fujioka Y, Udagawa T, Masuda A, Ohno K, Katsuno M, et al. , 2013. The ALS/FTLD-related RNA-binding proteins TDP-43 and FUS have common downstream RNA targets in cortical neurons. FEBS Open Bio 4, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooten KG, Beers DR, Zhao W, Appel SH, 2015. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 12, 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE, 2011. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, et al. , 1998. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. [DOI] [PubMed] [Google Scholar]
- Ignatius SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB, 1995. Cloning and Characterization of a Novel Cellular Protein, Tdp-43, That Binds to Human-Immunodeficiency-Virus Type-1 Tar DNA-Sequence Motifs. Journal of Virology 69, 3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski AM, Lamitina T, Liachko NF, Sabatella M, Lu J, Zhang L, Ostrow LW, Gupta P, et al. , 2015. Loss of RAD-23 Protects Against Models of Motor Neuron Disease by Enhancing Mutant Protein Clearance. J Neurosci 35, 14286–14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji AL, Zhang X, Chen WW, Huang WJ, 2017. Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J Med Genet 54, 145–154. [DOI] [PubMed] [Google Scholar]
- Johnson JO, Glynn SM, Gibbs JR, Nalls MA, Sabatelli M, Restagno G, Drory VE, Chio A, et al. , 2014a. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 137, e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, et al. , 2010. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, et al. , 2014b. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17, 664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, et al. , 2015. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18, 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, Bosco DA, Hayward LJ, et al. , 2011. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol 9, e1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, El Oussini H, Bercier V, Gros-Louis F, Valdmanis PN, McDearmid J, Mejier IA, Dion PA, et al. , 2013. Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet 22, 2350–2360. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, et al. , 2008. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40, 572–574. [DOI] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, et al. , 2014. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 81, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur SJ, McKeown SR, Rashid S, 2016. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 577, 109–118. [DOI] [PubMed] [Google Scholar]
- Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, van Rheenen W, van Eijk KR, et al. , 2016. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet 48, 1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Woods I, Villunger A, Strasser A, Prehn JH, 2007. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci U S A 104, 20606–20611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, et al. , 2013. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, et al. , 2014. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 46, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Zhan L, Hanson KA, Tibbetts RS, 2012. High-content RNAi screening identifies the Type 1 inositol triphosphate receptor as a modifier of TDP-43 localization and neurotoxicity. Hum Mol Genet 21, 4845–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer NJ, Carlomagno Y, Zhang YJ, Almeida S, Cook CN, Gendron TF, Prudencio M, Van Blitterswijk M, et al. , 2016. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 353, 708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer NJ, Haney MS, Morgens DW, Jovicic A, Couthouis J, Li A, Ousey J, Ma R, et al. , 2018. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet 50, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers M, van Dis V, Haasdijk ED, Harterink M, Vocking K, Post JA, Scheper W, Hoogenraad CC, et al. , 2013. Amyotrophic lateral sclerosis (ALS)-associated VAPB-P56S inclusions represent an ER quality control compartment. Acta Neuropathol Commun 1, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumimoto EL, Fore TR, Zhang B, 2013. Transcriptome Profiling Following Neuronal and Glial Expression of ALS-Linked SOD1 in Drosophila. G3 (Bethesda) 3, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, et al. , 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, et al. , 2014. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW, 2009. Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, et al. , 2003. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet 34, 383–394. [DOI] [PubMed] [Google Scholar]
- Lapinskas PJ, Cunningham KW, Liu XF, Fink GR, Culotta VC, 1995. Mutations in PMR1 suppress oxidative damage in yeast cells lacking superoxide dismutase. Mol Cell Biol 15, 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Kaneb HM, Dion PA, Rouleau GA, 2014. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol 262 Pt B, 91–101. [DOI] [PubMed] [Google Scholar]
- Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, et al. , 2016. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 167, 774–788 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Li YR, Ingre C, Weber M, Grehl T, Gredal O, de Carvalho M, Meyer T, et al. , 2011. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet 20, 1697–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, et al. , 2013. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5, 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE, Gallo JM, Weller RO, et al. , 1991. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 114 ( Pt 2), 775–788. [DOI] [PubMed] [Google Scholar]
- Leung CL, He CZ, Kaufmann P, Chin SS, Naini A, Liem RK, Mitsumoto H, Hays AP, 2004. A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol 14, 290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko NF, McMillan PJ, Guthrie CR, Bird TD, Leverenz JB, Kraemer BC, 2013. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann Neurol 74, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW, 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ, 2008. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol 15, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RG, Li BL, Flanagan SW, Oberley LW, Gozal D, Qiu MS, 2002. Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival. Journal of Neurochemistry 80, 488–500. [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Garcia ML, Ward CM, Cleveland DW, 2005. Altered axonal architecture by removal of the heavily phosphorylated neurofilament tail domains strongly slows superoxide dismutase 1 mutant-mediated ALS. Proc Natl Acad Sci U S A 102, 10351–10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen-Hoerth C, Anderson T, Miller B, 2002. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. [DOI] [PubMed] [Google Scholar]
- Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B, 2003. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 60, 1094–1097. [DOI] [PubMed] [Google Scholar]
- Lorenzl S, Narr S, Angele B, Krell HW, Gregorio J, Kiaei M, Pfister HW, Beal MF, 2006. The matrix metalloproteinases inhibitor Ro 28–2653 [correction of Ro 26–2853] extends survival in transgenic ALS mice. Exp Neurol 200, 166–171. [DOI] [PubMed] [Google Scholar]
- Lu L, Wang S, Zheng L, Li X, Suswam EA, Zhang X, Wheeler CG, Nabors LB, et al. , 2009. Amyotrophic lateral sclerosis-linked mutant SOD1 sequesters Hu antigen R (HuR) and TIA-1-related protein (TIAR): implications for impaired post-transcriptional regulation of vascular endothelial growth factor. J Biol Chem 284, 33989–33998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn JS, Sakowski SA, Kim B, Rosenberg AA, Feldman EL, 2009. Vascular endothelial growth factor prevents G93A-SOD1-induced motor neuron degeneration. Dev Neurobiol 69, 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Feldman HH, 2005. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol 64, 730–739. [DOI] [PubMed] [Google Scholar]
- Maekawa S, Leigh PN, King A, Jones E, Steele JC, Bodi I, Shaw CE, Hortobagyi T, et al. , 2009. TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology 29, 672–683. [DOI] [PubMed] [Google Scholar]
- Marden JJ, Harraz MM, Williams AJ, Nelson K, Luo M, Paulson H, Engelhardt JF, 2007. Redox modifier genes in amyotrophic lateral sclerosis in mice. J Clin Invest 117, 2913–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Kawakami H, 2013. Optineurin and amyotrophic lateral sclerosis. Geriatr Gerontol Int 13, 528–532. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G, 2002. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem 277, 29626–29633. [DOI] [PubMed] [Google Scholar]
- Millecamps S, De Septenville A, Teyssou E, Daniau M, Camuzat A, Albert M, LeGuern E, Galimberti D, et al. , 2014. Genetic analysis of matrin 3 gene in French amyotrophic lateral sclerosis patients and frontotemporal lobar degeneration with amyotrophic lateral sclerosis patients. Neurobiol Aging 35, 2882 e2813–2885. [DOI] [PubMed] [Google Scholar]
- Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, Habgood J, Panoutsou S, et al. , 2010. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci U S A 107, 7556–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Nihei Y, Arzberger T, Zhou Q, Mackenzie IR, Hermann A, Hanisch F, German Consortium for Frontotemporal Lobar D, et al. , 2016. Reduced hnRNPA3 increases C9orf72 repeat RNA levels and dipeptide-repeat protein deposition. EMBO Rep 17, 1314–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, Sedlmeier R, Meyer T, et al. , 2005. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol 58, 777–780. [DOI] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, et al. , 2004. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, et al. , 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, et al. , 2004. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan M, Talbot K, Ansorge O, 2016. Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Soucy G, Phaneuf D, Audet J-N, Gros-Louis F, Rouleau GA, Blasco H, Corcia P, et al. , 2016. Sex-dependent effects of chromogranin B P413L allelic variant as disease modifier in amyotrophic lateral sclerosis. Human Molecular Genetics, ddw304. [DOI] [PMC free article] [PubMed]
- Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, Morrison KE, Pall HS, et al. , 2006. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077. [DOI] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, et al. , 2015. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162, 1066–1077. [DOI] [PubMed] [Google Scholar]
- Pensato V, Tiloca C, Corrado L, Bertolin C, Sardone V, Del Bo R, Calini D, Mandrioli J, et al. , 2015. TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: identification of novel mutations. J Neurol 262, 1376–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzer C, Kruger C, Plaas C, Kirsch F, Dittgen T, Muller R, Laage R, Kastner S, et al. , 2008. Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain 131, 3335–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantie E, Migocka-Patrzalek M, Daczewska M, Jagla K, 2015. Model organisms in the fight against muscular dystrophy: lessons from Drosophila and Zebrafish. Molecules 20, 6237–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Neumann M, Mackenzie IR, 2012. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 8, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Stewart H, Dejesus-Hernandez M, Krieger C, Graff-Radford N, Fabros M, Briemberg H, Cashman N, et al. , 2010. Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, van Blitterswijk M, 2014. Excess of rare damaging TUBA4A variants suggests cytoskeletal defects in ALS. Neuron 84, 241–243. [DOI] [PubMed] [Google Scholar]
- Ratnaparkhi A, Lawless GM, Schweizer FE, Golshani P, Jackson GR, 2008. A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS One 3, e2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnavalli E, Brayne C, Dawson K, Hodges JR, 2002. The prevalence of frontotemporal dementia. Neurology 58, 1615–1621. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, et al. , 1996. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13, 43–47. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, et al. , 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes NA, Fisher JK, Austgen K, VandenBerg S, Huang EJ, Oakes SA, 2010. Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis. J Clin Invest 120, 3673–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddoch-Contreras J, Yang SY, Dick JR, Goldspink G, Orrell RW, Greensmith L, 2009. Mechano-growth factor, an IGF-I splice variant, rescues motoneurons and improves muscle function in SOD1(G93A) mice. Exp Neurol 215, 281–289. [DOI] [PubMed] [Google Scholar]
- Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, Tang W, Winton MJ, et al. , 2010. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci 30, 7729–7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, et al. , 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. [DOI] [PubMed] [Google Scholar]
- Rowland LP, 2001. How amyotrophic lateral sclerosis got its name - The clinical-pathologic genius of Jean-Martin Charcot. Archives of Neurology 58, 512–515. [DOI] [PubMed] [Google Scholar]
- Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P, 2013. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin A, Held A, Bredvik K, Major P, Achilli TM, Kerson AG, Wharton K, Stilwell G, et al. , 2017. Human SOD1 ALS Mutations in a Drosophila Knock-In Model Cause Severe Phenotypes and Reveal Dosage-Sensitive Gain- and Loss-of-Function Components. Genetics 205, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Chai A, Smith C, McCray BA, Simpson TI, Taylor JP, Pennetta G, 2015. Network analyses reveal novel aspects of ALS pathogenesis. PLoS Genet 11, e1005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PS, Akbar MT, Bouri S, Senda A, Joshi K, Chen HJ, Latchman DS, Wells DJ, et al. , 2008. Protective effects of heat shock protein 27 in a model of ALS occur in the early stages of disease progression. Neurobiol Dis 30, 42–55. [DOI] [PubMed] [Google Scholar]
- Silva MC, Fox S, Beam M, Thakkar H, Amaral MD, Morimoto RI, 2011. A genetic screening strategy identifies novel regulators of the proteostasis network. PLoS Genet 7, e1002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin O, Michels H, Nollen EA, 2014. Genetic screens in Caenorhabditis elegans models for neurodegenerative diseases. Biochim Biophys Acta 1842, 1951–1959. [DOI] [PubMed] [Google Scholar]
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, et al. , 2005. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37, 806–808. [DOI] [PubMed] [Google Scholar]
- Skvortsova V, Shadrina M, Slominsky P, Levitsky G, Kondratieva E, Zherebtsova A, Levitskaya N, Alekhin A, et al. , 2004. Analysis of heavy neurofilament subunit gene polymorphism in Russian patients with sporadic motor neuron disease (MND). Eur J Hum Genet 12, 241–244. [DOI] [PubMed] [Google Scholar]
- Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, et al. , 2014. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, et al. , 2008. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Brown RH Jr., 2013. Amyotrophic lateral sclerosis: Problems and prospects. Ann Neurol 74, 309–316. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Neukomm LJ, Brown RH Jr., Freeman MR, 2015. Age-Dependent TDP-43-Mediated Motor Neuron Degeneration Requires GSK3, hat-trick, and xmas-2. Curr Biol 25, 2130–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieber A, Gonatas JO, Gonatas NK, 2000. Aggregates of mutant protein appear progressively in dendrites, in periaxonal processes of oligodendrocytes, and in neuronal and astrocytic perikarya of mice expressing the SOD1(G93A) mutation of familial amyotrophic lateral sclerosis. J Neurol Sci 177, 114–123. [DOI] [PubMed] [Google Scholar]
- Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, Vizcay-Barrena G, Lin WL, et al. , 2014. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 5, 3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R, Paillusson S, Gomez-Suaga P, Mitchell JC, Lau DH, Gray EH, Sancho RM, Vizcay-Barrena G, et al. , 2016. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep 17, 1326–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strain J, Lorenz CR, Bode J, Garland S, Smolen GA, Tall DT, Vickery LE, Culotta VC, 1998. Suppressors of superoxide dismutase (SOD1) deficiency in Saccharomyces cerevisiae - Identification of proteins predicted to mediate iron-sulfur cluster assembly. Journal of Biological Chemistry 273, 31138–31144. [DOI] [PubMed] [Google Scholar]
- Strong MJ, 2008. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 9, 323–338. [DOI] [PubMed] [Google Scholar]
- Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, et al. , 2015. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 6, 6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD, 2011. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol 9, e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, Robberecht W, 2014. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10, 661–670. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, Belzil VV, Dion PA, et al. , 2013. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet 93, 900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuling E, van Dis V, Wulf PS, Haasdijk ED, Akhmanova A, Hoogenraad CC, Jaarsma D, 2008. A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum Mol Genet 17, 2849–2862. [DOI] [PubMed] [Google Scholar]
- Teyssou E, Takeda T, Lebon V, Boillee S, Doukoure B, Bataillon G, Sazdovitch V, Cazeneuve C, et al. , 2013. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 125, 511–522. [DOI] [PubMed] [Google Scholar]
- Teyssou E, Vandenberghe N, Moigneu C, Boillee S, Couratier P, Meininger V, Pradat PF, Salachas F, et al. , 2014. Genetic analysis of SS18L1 in French amyotrophic lateral sclerosis. Neurobiol Aging 35, 1213 e1219–1213 e1212. [DOI] [PubMed] [Google Scholar]
- Therrien M, Parker JA, 2014. Worming forward: amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front Genet 5, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticozzi N, Vance C, Leclerc AL, Keagle P, Glass JD, McKenna-Yasek D, Sapp PC, Silani V, et al. , 2011. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet B Neuropsychiatr Genet 156B, 285–290. [DOI] [PubMed] [Google Scholar]
- Turner BJ, Alfazema N, Sheean RK, Sleigh JN, Davies KE, Horne MK, Talbot K, 2014. Overexpression of survival motor neuron improves neuromuscular function and motor neuron survival in mutant SOD1 mice. Neurobiol Aging 35, 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, et al. , 2008. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, et al. , 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbandt S, Cammue BP, Thevissen K, 2016. Yeast as a model for the identification of novel survival-promoting compounds applicable to treat degenerative diseases. Mech Ageing Dev [DOI] [PubMed]
- Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL, 2009. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet 5, e1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Brent JR, Tomlinson A, Shneider NA, McCabe BD, 2011. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Invest 121, 4118–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Conicella AE, Schmidt HB, Martin EW, Rhoads SN, Reeb AN, Nourse A, Ramirez Montero D, et al. 2018. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J 37:e97452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD, 2001. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8, 933–941. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Kuno N, Kono Y, Nanba E, Ohama E, Nakashima K, Takahashi K, 1997. Absence of the mutant SOD1 in familial amyotrophic lateral sclerosis (FALS) with two base pair deletion in the SOD1 gene. Acta Neurol Scand 95, 167–172. [DOI] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, et al. , 2014. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, et al. , 2012. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, et al. , 2013. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 110, 7778–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Nonaka T, Hirai S, Miwa A, Okado H, Arai T, Hosokawa M, Akiyama H, et al. , 2014. Distinct pathways leading to TDP-43-induced cellular dysfunctions. Hum Mol Genet 23, 4345–4356. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, et al. , 2001. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nature Genetics 29, 160–165. [DOI] [PubMed] [Google Scholar]
- Yang YS, Harel NY, Strittmatter SM, 2009. Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. J Neurosci 29, 13850–13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, et al. , 2008. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63, 538–542. [DOI] [PubMed] [Google Scholar]
- Zhai J, Lin H, Canete-Soler R, Schlaepfer WW, 2005. HoxB2 binds mutant SOD1 and is altered in transgenic model of ALS. Hum Mol Genet 14, 2629–2640. [DOI] [PubMed] [Google Scholar]
- Zhan L, Hanson KA, Kim SH, Tare A, Tibbetts RS, 2013. Identification of genetic modifiers of TDP-43 neurotoxicity in Drosophila. PLoS One 8, e57214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L, Xie Q, Tibbetts RS, 2015. Opposing roles of p38 and JNK in a Drosophila model of TDP-43 proteinopathy reveal oxidative stress and innate immunity as pathogenic components of neurodegeneration. Hum Mol Genet 24, 757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, et al. , 2015. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, et al. , 2013. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110, E4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File 1. List of genes reported to modify ALS-associated defects. The page labeled “Modifiers of multiple” contains the genes that were found to modify two or more ALS genes. Pages are labeled with the name of the ALS gene that the genetic modifier was observed to modify. Each tab contains the following information: the originally reported gene, the human ortholog, and the reference.
Supplemental File 2. Gene ontology analysis results listing enriched GO terms for each of the analyses reported in the main text and the Figures. Each page lists the gene ontology term, the odds ratio, p-value, expected counts (the number of times a gene should be in this category given the number of genes on our list), the count (the number of times a gene is associated with the specific GO term here), and size (the number of genes associated with the called GO term).
Supplemental File 3. Genes associated with enriched GO terms. The modifier genes associated with each enriched GO term are reported from the original analysis output.