

Abstract
β-Mannosidosis is a lysosomal storage disorder characterized by accumulation of disaccharides due to deficiency of the lysosomal enzyme β-mannosidase. The disease is caused by mutations in MANBA and is extremely rare in humans. Although the clinical presentation is heterogeneous, common symptoms include various degrees of developmental delay, behavioral disturbances, hearing loss, and frequent infections. We report a 15-yr-old girl presenting with mild intellectual disability, sensorineural hearing loss, severe behavioral disturbances, dysmorphic traits, and evolving angiokeratomas. Copy-number variation analysis of next-generation sequencing (NGS) data indicated increased coverage in exons 8–11 of MANBA. Low β-mannosidase activity (1 µkatal/kg protein, refv 25–40) established the diagnosis of β-mannosidosis. Whole-genome sequencing (WGS) and cDNA analysis revealed a novel homozygous intragenic inverted duplication in MANBA, where a 13.1-kb region between introns 7 and 11 was duplicated and inserted in an inverted orientation, creating a 67-base nonduplicated gap at the insertion point. Both junctions showed microhomology regions. The inverted duplication resulted in exon skipping of exons 8–9 or 8–10. Our report highlights the importance of copy-number variation analysis of data from NGS and in particular the power of WGS in the identification and characterization of copy-number variants.
Keywords: abnormality of the vasculature of the conjunctiva; aggressive behavior; alacrima; angiokeratoma corporis diffusum; attention-deficit hyperactivity disorder; bilateral sensorineural hearing impairment; broad nasal tip; chronic constipation; hypohidrosis/hyperhidrosis; impulsivity; increased urinary disaccharide excretion; intellectual disability, mild; sleep disturbance; thin upper lip vermilion; upslanted palpebral fissure; wide nasal ridge
INTRODUCTION
β-Mannosidosis (OMIM #248510) is an autosomal recessive lysosomal storage disorder due to deficient activity of the enzyme β-mannosidase (E.C. 3.2.1.25). The disease is well known and relatively common in goats and other kind of cattle, but it is extremely rare in humans. β-Mannosidase is the final exoglycosidase involved in the degradation of N-linked oligosaccharides of glycoproteins, removing β-linked mannose residues (Winchester 2005; Samra and Athar 2008). Affected individuals have pronounced reduction of β-mannosidase activity that can be measured in white blood cells and fibroblasts. Enzyme deficiency in humans leads to lysosomal accumulation of disaccharides (primarily Man(β1 → 4)GlcNac) (Cooper et al. 1988; van Pelt et al. 1990); therefore, the disease belongs to the group of glycoproteinoses. MANBA is located on Chromosome 4q24 and encodes an 879-amino acid lysosomal protein (Alkhayat et al. 1998). So far, only 22 cases of β-mannosidose from 18 families have been reported in humans (Riise Stensland et al. 2008; Labauge et al. 2009; Broomfield et al. 2013). Disease-causing variants include both null mutations (splice, nonsense, small frameshift deletions/insertions) and missense mutations (Riise Stensland et al. 2008; Labauge et al. 2009; Broomfield et al. 2013).
The clinical presentation is heterogeneous, and a wide range of symptoms of different severity has been observed, with no clear genotype–phenotype correlation. Common symptoms include various degrees of developmental delay, behavioral disturbances, hearing loss, and frequent infections (Wenger et al. 1986). Angiokeratomas, facial and skeletal dysmorphic features, hypotonia, peripheral neuropathy, and seizures have also been reported in some of the patients (Cooper et al. 1991; Cherian 2004; Broomfield et al. 2013). Several of these symptoms are typical for glycoproteinoses.
We report a 15-yr-old girl presenting with mild intellectual disability, sensorineural hearing loss, severe behavioral disturbances, dysmorphic traits, and evolving angiokeratomas. A next-generation sequencing (NGS)-panel for lysosomal disorders suggested the diagnosis of β-mannosidosis, which was confirmed enzymatically. Subsequent whole-genome sequencing (WGS) analysis defined at the nucleotide level the presence of a homozygous intragenic inverted duplication in MANBA. This report highlights the importance of copy-number variation analysis of NGS data and in particular the power of WGS in the identification and characterization of copy-number variants.
RESULTS
Clinical Presentation and Family History
The patient is now a 15-yr-old girl born to consanguineous parents (second cousins once removed) of Norwegian ancestry. She has two older brothers. Pregnancy was uneventful until delivery by acute caesarean section in week 38 due to fetal distress; her Apgar scores were 9–10. Her birth weight was 2980 g, length 47 cm, and occipitofrontal head circumference 34.5 cm. Bottle feeding was initiated because of poor sucking. She showed globally delayed development from the newborn period, including muscular hypotonia, sitting at the age of 1 yr, crawling at the age of 17 mo, and walking at the age of 18 mo. At the age of 1 yr, a diagnosis of unilateral cerebral palsy (CP) was given because of hypertonia, hyperreflexia, and reduced function of her right arm. However, functions improved over the next years and the diagnosis was withdrawn. She still preferably uses her left arm. Cerebral MRI at the age of 3 showed delayed myelination but no structural abnormalities and no focal pathology that could explain unilateral CP. Because of her improvement in motor function, a repeat MRI was not performed.
Strabismus and hypermetropia was noted from the age of 1. At the age of 2 yr, sensorineural hearing loss was recognized and corrected with hearing aids. She suffered from recurrent infections in her childhood. Her parents commented early on that her facial features differed from parents and siblings. Dysmorphic features include upslanting palpebral fissures, blepharophimosis, broad nasal ridge and nasal tip, thin lips, and posteriorly rotated ears (Fig. 1B). A diagnosis of mild intellectual disability was given after cognitive testing in early school years. She displayed hyperactive behavior since early childhood and now has a formal diagnosis of attention-deficit hyperactivity disorder (ADHD). Her sleep pattern has always been disturbed. Severe behavioral disturbances in the form of self-biting, impulsiveness, improper language, aggression, and rubbing her glasses until they break have emerged over the years.
Figure 1.

Clinical manifestations. (A) Flushing of the scalp/face. (B) Facial features with upslanting, narrow palpebral fissures, periorbital fullness, broad nasal root and tip, and thin lips. (C) Telangiectatic and tortuous conjunctival vessels. (D) Angiokeratomas on the tips of the fingers (evolving). (E) Close-up on angiokeratomas (thigh). (F) Multiple angiokeratomas on the dorsal side of both thighs.
At 12 yr of age, autonomic features like no tear production, high pain threshold, constipation, and urinary incontinence as well as flushing of the skin on the scalp were identified (Fig. 1A). A new clinical examination at age 13 revealed numerous pinpoint red/dark spots—some macular, some papular—that did not blanch on pressure, on her thighs, lower torso, fingertips, and vermillion border of the lips (Fig. 1D–F). Varicose veins were found on both thighs, and eye examination showed dilated and tortuous conjunctival vessels (Fig. 1C) but normal appearance of the retina including the vasculature.
Biochemical and Molecular Studies
Initial genetic investigations undertaken at 1 yr of age showed normal G-band analysis, 46, XX, as well as normal results regarding Southern blot for fragile X syndrome (FMR1) and multiplex ligation-dependent probe amplification (MLPA) analysis for DiGeorge syndrome (P023B MRC Holland), for microdeletions/duplications causing mental retardation (MLPA P064 and P096, MRC Holland), and for subtelomere areas (MLPA P036B and P070, MRC Holland). Screening for inborn errors of metabolism in urine showed normal results for amino acids and organic acids. At 5 yr of age, chromosomal microarray (Genome-Wide Human SNP array 6.0, Affymetrix) detected six large regions of homozygosity, including one in 4q24, where MANBA is located (data not shown).
At the age of 13 yr, genetic investigations with an NGS panel showed neither clinically significant SNVs nor small indels in any of the genes associated with lysosomal disorders. However, copy-number variation analysis showed increased coverage (average fold change +1.73) in exons 8–11 of MANBA (data not shown). This finding prompted the analysis of β-mannosidase in isolated lymphocytes which showed very low activity (1 µkatal/kg protein, reference value 25–40).
cDNA analysis of the patient revealed two abnormal MANBA transcripts lacking exons 8–9 and 8–10, respectively. No wild-type transcript was found, suggesting that the genetic variant was in homozygous form (Fig. 2). The patient's mother showed wild-type transcript in addition to the two truncated variants, as expected for a heterozygous carrier (Fig. 2).
Figure 2.
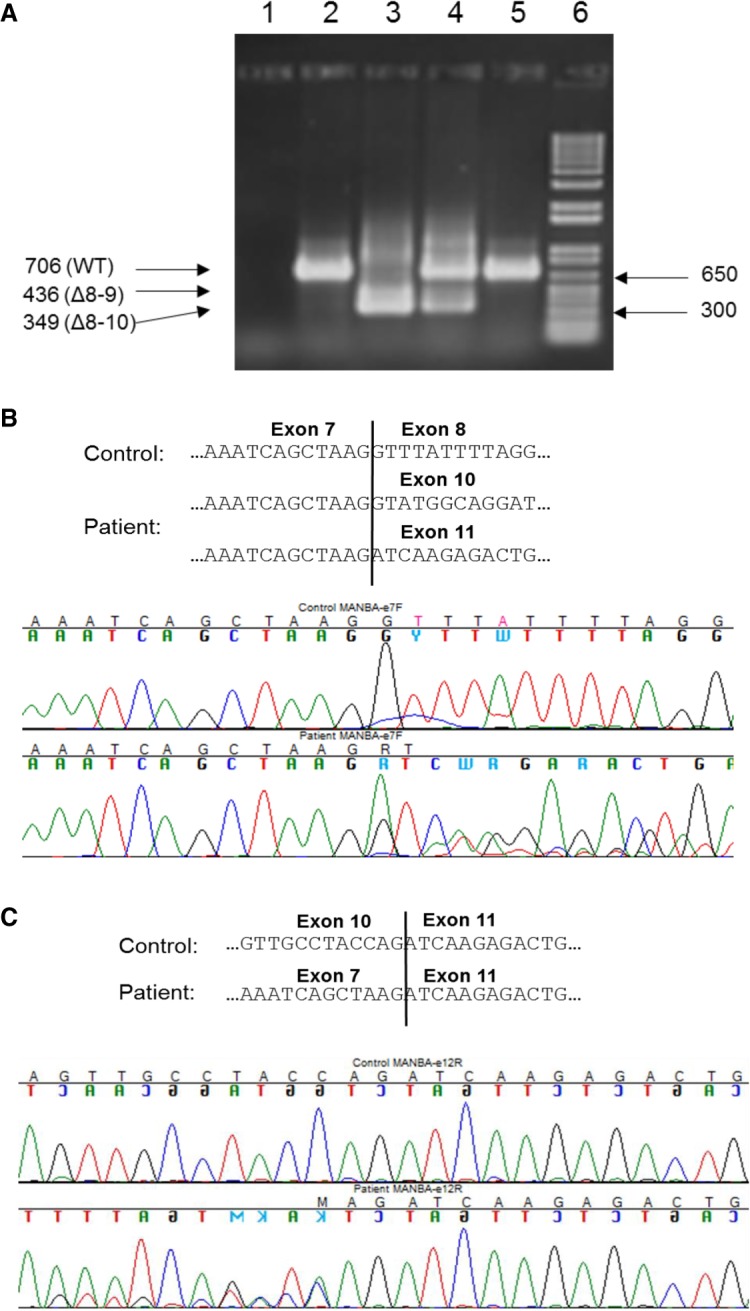
cDNA analysis reveals skipping of exons 8–9 and 8–10, respectively, in MANBA. (A) cDNA was prepared from RNA isolated from whole blood and PCR amplified using primers in exons 7 and 12. Agarose-gel electrophoresis of the PCR products. Lanes 1–6: PCR-mix (no cDNA); control 1; patient; mother; control 2; size standard (1 kb+ ladder, Sigma-Aldrich). (B) PCR products from control 1 and the patient were Sanger-sequenced in the forward direction with a primer in exon 7 and (C) reverse direction with a primer in exon 12.
WGS analysis showed that the increase in coverage found in the NGS panel analysis corresponded to an inverted duplication involving exons 8–11 (illustrated in Fig. 3A,B; see Table 1), which could also explain the exon skipping observed on cDNA analysis. Furthermore, WGS indicated the precise location of both junctions, which were confirmed by Sanger sequencing (Fig. 3C). The 13.1-kb duplicated fragment extends from position Chr 4:103,598,877 in intron 7 to position Chr 4:103,585,729 in intron 11 and was inserted in inverted orientation in positions Chr 4:103,590,920 and 103,590,854, creating a 67-base nonduplicated gap at the insertion point. Both junctions showed microhomology regions, as shown in Figure 3C. WGS did not detect any other pathogenic variants in the genes present in any of the six regions of homozygosity identified by SNP array.
Figure 3.

WGS analysis defines both junctions at the nucleotide level. (A) Overview of WGS data encompassing the duplicated region at MANBA. The coverage track clearly defines a duplicated region from introns 7–11. The drop in coverage observed in intron 9 (red arrow) corresponds to the 67-bp nonduplicated gap observed between the insertion points. (B) Paired reads with RR (right-right) and LL (left-left) orientation suggest the existence of an inversion, diagrammed in panel. (C) Definition of both junctions at the nucleotide level obtained from WGS data and confirmed by Sanger sequencing. The inserted sequence is marked in blue. Both junctions show areas of microhomology (yellow boxes).
Table 1.
Variant table
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect (substitution, deletion, etc.) | dbSNP/dbVar ID | Genotype (heterozygous/homozygous) |
|---|---|---|---|---|---|---|---|
| MANBA | 4q24 | g.103590853_103590921ins103585729_103598877inv (hg19) | Not available | Inverted duplication | Exon skipping (8–9 or 8–10) | - | Homozygous |
Retrospectively, the duplication was visible on the SNP array analysis from 2009 as a gain in only seven probes (Genome-Wide Human SNP array 6.0, Affymetrix). However, with standard settings still in use, it falls below the threshold for reporting of the clinical laboratory.
DISCUSSION
The combination of intellectual disability, sensorineural hearing loss, behavioral difficulties, autonomic features, and angiokeratomas raised in our patient the suspicion of a lysosomal storage disorder. The homozygosity area in 4q24 where MANBA is located specifically pointed to β-mannosidosis and prompted genetic investigation with an NGS panel for lysosomal disorders, in which copy-number variation analysis showed increased coverage in exons 8–11 of MANBA. Analysis of β-mannosidase in isolated lymphocytes showed very low activity, thereby confirming the diagnosis of β-mannosidosis. Close analysis of NGS panel data suggested that the observed increase in coverage could correspond to an inverted duplication involving exons 8–11, matching the exon skipping observed on cDNA analysis. WGS further supported this hypothesis and indicated the precise location of both junctions.
β-Mannosidosis is a very rare lysosomal storage disorder with an estimated incidence of 0.1 per 100,000 (Poorthuis et al. 1999; Poupětová et al. 2010). So far, to our knowledge, only 22 cases of β-mannosidosis in 18 families from a range of ethnic backgrounds have been reported since the disease was first described in 1986 (Cooper et al. 1986; Wenger et al. 1986). The disease has a highly variable clinical presentation (Bedilu et al. 2002; Riise Stensland et al. 2008) including intellectual disability/developmental delay, behavioral disturbance, recurrent infections, hearing loss, and severe neurological phenotypes such as epileptic encephalopathy, hydrocephalus, and spinocerebellar ataxia (Sedel et al. 2006; Labauge et al. 2009; Broomfield et al. 2013). Dysmorphic traits have been reported (Poenaru et al. 1992; Labauge et al. 2009). Angiokeratomas are also described in several cases of β-mannosidosis (Rodríguez-Serna et al. 1996; Suzuki et al. 2004; Molho-Pessach et al. 2007). Thus several features of our patient are in line with β-mannosidosis patients previously described, including the presence of angiokeratomas.
Angiokeratoma corporis diffusum (ACD) is thought to be the cutaneous hallmark of Fabry disease (Orteu 2010) but also occurs in other lysosomal storage disorders as well as a few other conditions, like hereditary hemorrhagic telangiectasia. Lesions typically appear in the first or second decade and increase in number with age. Lesions are red to black pinpoint—10-mm macules or papules, sometimes described as hyperkeratotic. The typical distribution is on the trunk, including the genital area and proximal limbs, but may also involve distal extremities and lips. Histologically, dilated capillaries with thin walls are found in the dermis, underlying an epidermis with hyperkeratosis (Molho-Pessach et al. 2007). Electron microscopy reveals lysosomal storage vacuoles in the cytoplasm in β-mannosidosis, whereas the findings in Fabry disease are distinct with electron-dense lysosomal granules (Molho-Pessach et al. 2007). The macroscopic appearance appears to be similar, but with different ultrastructural appearances due to unique substrate depositions in each of the lysosomal disorders. The pathogenesis of ACD in β-mannosidosis is not yet clear but probably could be explained by a similar mechanism as in Fabry disease. Pathophysiological studies in Fabry disease demonstrate accumulation of globotriaosylceramide in dermal endothelial cells, ensuing capillary wall defects, and vessel ectasias (Lidove et al. 2006).
Behavioral difficulties are frequently seen in the previously described cases of β-mannosidosis (Bedilu et al. 2002; Sabourdy et al. 2009). Hyperactivity, Tourette syndrome, obsessive–compulsive disorder, aggressiveness, and self-mutilation are reported (Sedel et al. 2006). In our patient, hyperactivity was present early with a later formal diagnosis of ADHD. Severe behavioral disturbances emerged later, including self-mutilation, disinhibition, and aggressiveness. Psychiatric symptoms in adolescence or adulthood together with systemic, cognitive, and neurological signs, as in our patient, can reveal inborn errors of metabolism. The coexistence of (mild) intellectual disability and emerging psychiatric symptoms or behavioral changes is found in several lysosomal storage disorders (Sedel et al. 2007). This is, however, difficult to bear in mind as each of these disorders are rare but should be considered especially in presence of other typical signs and symptoms and in cases in which recessive disease is suspected. Symptoms of autonomic dysfunction like hypohidrosis, facial flushing, absent tear production, and high pain threshold, as in our patient, have not been previously described in β-mannosidosis, but are well known in Mb Fabry (Biegstraaten et al. 2010). Biegstraaten et al. (2010) have suggested that the autonomic dysfunction is not due to autonomic neuropathy, as previously described, but rather due to end-organ damage. Formal testing for autonomic dysfunction (like tilt-test, special sweat tests) were not undertaken, but could give more information and quantification of this feature. Extensive WGS analysis did not reveal other disease-associated variants, supporting that these features are symptoms of β-mannosidosis in our patient.
β-Mannosidosis in humans is, with a few exceptions, generally less severe than the disease found in goats and other kind of cattle in which severe neonatal neurological onset together with dysmorphism are particularly common (Jones and Dawson 1981). It has been suggested that the phenotypic differences among species are caused by differences in the size and nature of the storage compounds (Winchester 2005). Furthermore, two sequence variants found in ruminants are suggested to abolish the β-mannosidase enzyme activity, resulting in the severe neurodegenerative phenotype (Leipprandt et al. 1996, 1999). Comparing patients reported in the literature, no clear genotype–phenotype correlation could be shown in humans (Bedilu et al. 2002; Broomfield et al. 2013), and variability is suggested to be due to the involvement of other genetic and environmental factors (Gort et al. 2006; Riise Stensland et al. 2008; Lovell et al. 2014). However, comparative structural bioinformatics analyses of inherited mutations reported in MANBA until 2011 indicated the existence of genotype–phenotype correlation in β-mannosidosis (Huynh et al. 2011). In general, the authors suggest the proximity of mutations to the active site to be determinant of disease severity, and five mutational hotspots were identified. So far 18 disease-causing variants have been reported including both null mutations (splice, nonsense, small deletions/duplications) and missense mutations (The Human Gene Mutation Database Professional 2018.3; retrieved October 26, 2018 from http://www.hgmd.cf.ac.uk/ac/). Our patient shows a novel inverted duplication involving exons 8–11 in MANBA, resulting in exon skipping (exon 8–9 or 8–10), which in turn results in shortening of the protein by 90 or 119 amino acids, respectively. No western blot was performed to assess whether the abnormal transcripts gave stable protein products; however, despite the massive structural change involved, some residual β-mannosidase activity could be measured in lymphocytes, and thus we have to assume that some degree of translated protein is present.
WGS is a very efficient tool for identification and characterization of structural variants. Providing nucleotide-level resolution at the breakpoints, WGS gives us valuable information concerning the mechanism causing the observed inverted duplication. The insertion point of this duplication is located in an Alu element [AluY (+)] as well as the end of the duplication contributing to the telomeric junction [AluYc (−)]. Moreover, areas of microhomology can be observed in both junctions. Taken together, we propose that the duplication presented here is caused by a microhomology-mediated, replication-based template switching between Alu elements. The fact that both elements are in opposite orientations probably explains the inverted nature of the duplication (Fig. 4A). This is a common mechanism underlying nonrecurrent copy-number variations (Verdin et al. 2013; Carvalho and Lupski 2016). Analysis of cDNA revealed exon skipping as a result of the presence of the inverted duplication. Alternative splicing caused by insertion of inverted repeats is a well-known phenomenon and has been proposed to be caused by hairpin formation at the level of precursor mRNAs (Solnick 1985). Based on this, we propose that the presence of the inverted duplication causes a large hairpin at the pre-mRNA level that compromises correct splicing of exons 8–10 (Fig. 4B).
Figure 4.

Proposed mechanisms underlying the observed inverted duplication and exon skipping. (A) We propose the following sequence of events causing the inverted duplication: (1) replication stalls at AluY (+), possibly facilitated by the interaction with a second Alu element, AluYc, in opposite orientation; (2) template switching between both Alu elements takes place, which is mediated by a 20-base-long microhomology region and causes the large inverted duplication; and (3) replication switches back to the original template directly downstream from the highly homologous regions between the Alu elements, leaving a 67-base-long nonreplicated gap. This template switching is mediated by a 3-base-long area of microhomology. (B) The inverted duplication creates a large area of full complementarity within the pre-mRNA that induces the formation of a hairpin, which in turn forces the spliceosome to skip exons 8 and 9. Even proper splicing of exon 10 is compromised.
In conclusion, this study reports a new case of the ultra-rare disease β-mannosidosis with a novel pathogenic variant in MANBA—namely, a homozygous intragenic inverted duplication. Diagnostic clues to β-mannosidosis were tiny angiokeratomas, behavioral problems, intellectual disability, sensorineural hearing loss, and homozygosity of 4q24. Other symptoms such as hypohidrosis, facial flushing, no tears, and high pain threshold have not previously been described in β-mannosidosis but are well known in Fabry disease, another lysosomal storage disorder. Our case clearly shows the importance of reevaluation of undiagnosed patients, searching for tiny clues and special features and correct phenotyping. This report further highlights the importance of copy-number variation analysis of NGS data, and, in particular, the power of WGS in the identification and characterization of the inverted duplication in MANBA.
METHODS
NGS Panel for Lysosomal Storage Disorders
Mutation analysis was performed by using a custom-made NGS gene panel (SureSelectQXT, Agilent Technologies). The panel targets coding exons of 61 genes described to be involved in lysosomal disorders (±25 bases, according to the RefSeq database and assembly February 2009 [GRCh37/hg19]). The list of genes is available upon request. Prepared libraries were sequenced in a paired-end run (2 × 150 bp) on the MiSeq instrument (Illumina). Demultiplexing, adaptor trimming, and mapping of sequencing reads to the human reference sequence hg19, as well as subsequent indel realigning and variant calling, was performed using the MiSeq Reporter Software (Illumina). Annotation and filtering of variants were addressed with Variant Studio Analysis Software (Illumina). Variants of interest were filtered according to allele frequency, exonic/splice site location, and autosomal recessive or X-linked pattern of inheritance. Copy-number variation analysis, executed with CLC Cancer Research Workbench 2.0 (CLC Bio, QIAGEN), was used to detect larger deletions (>1 kb). Quality assessment was performed by using Q30 as pass filter cutoff for a Phred score of called variants, together with a minimum sequencing read depth of 20× for target regions. Statistics for the NGS panel are summarized in Supplemental Table 1.
RNA Isolation and cDNA Analysis
Blood was collected in Tempus Blood RNA Tubes (Thermo Fisher Scientific), and RNA was isolated using the Tempus Spin RNA Isolation Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. The RNA concentrations were measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized using the SuperScript VILO cDNA Synthesis Kit (Invitrogen) according to the manufacturer's protocol. Primers APRT-e3F (5′-GGGGAAGCTGCCAGGCCCCACT-3′) and APRT-e5R (5′-GCGAGGTCAGCTCCACCAGGCT-3′), positioned in exons 3 and 5, respectively, of the housekeeping gene APRT (NM_000485.2), were used to monitor the cDNA synthesis and to identify possible contamination of genomic DNA (presence of a 721-bp PCR product in addition to the expected 218-bp PCR cDNA product). Primers MANBA-e7F (5′-GAAACTTGGTGGCCTCATGG-3′) and MANBA-e12R (5′-GGTTTTGAGAGACCCAGGCT-3′), positioned in exons 7 and 12, respectively, of the MANBA gene (NM_005908.3), were used for PCR amplification of MANBA exons 8–11 (PCR product 706 bp) using the JumpStart REDTaq ReadyMix Reaction Mix (Sigma-Aldrich) (PCR conditions available on request). PCR products were visualized on agarose gels, purified using the A'SAP PCR Purification Kit (ArcticZymes), and Sanger-sequenced in both directions using PCR-primers and BigDye v.3.1 (Life Technology). Fragments were separated on a 3500xL Genetic Analyzer (Applied Biosystems), and the sequences were analyzed in Sequencher version 5.3 (Gene Codes Corporation). Primers were designed using the Primer3 software (Primer3 v0.4.0/).
Whole-Genome Sequencing
DNA extraction from blood, WGS, secondary analysis, and variant analysis was carried out essentially as described in Darin et al. (2018). Sequencing returned 900 million paired sequences, corresponding to a coverage of 40× across the human genome. Data was then trimmed and mapped, and variants were called using CLC Genomics Server (QIAGEN). Trimming was performed using an ambiguous limit of 2, minimum Phred score of 17, and minimal length of 30. Reads were mapped using default settings and three passes of local realignment. Variants were called with a 90% probability cutoff, minimum count of 2%, minimum coverage of 7%, and 15% minimum frequency. Long indels were called with Manta (Chen et al. 2016) and CNVs with Canvas (Roller et al. 2016). Variants were then uploaded into Ingenuity (QIAGEN) for analysis. Statistics for the WGS are summarized in Supplemental Table 2.
Sanger Verification of Duplication Junctions
DNA was extracted from EDTA blood using the DSP DNA Midi Kit 96 and the QIAsymphony extractor (both QIAGEN). DNA concentrations were measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Primers MANBA-i10R (5′-AATCTGGGAGTCATCAGCCAA-3′ in intron 10) and MANBA-i7R (5′-AGCAGTATTTGTGGCCAAGGA-3′ in intron 7) were used for amplification of the centromeric junction, and primers MANBA-i10F (5′-GCAGAAATGCAAGGGCTTTA-3′ in intron 10) and MANBA-i8F (5′-AACCATCATGATTTTAGAGTGTGTTT-3′ in intron 8) were used for PCR amplification of the telomeric junction. PCR conditions are available on request. PCR products were visualized on agarose gels, purified using the A'SAP PCR Purification Kit (ArcticZymes), and Sanger-sequenced in both directions using PCR primers and BigDye v.3.1 (Life Technology). Fragments were separated on a 3500xL Genetic Analyzer (Applied Biosystems), and the sequences were analyzed in Sequencher version 5.3 (Gene Codes Corporation).
β-Mannosidase Activity in Lymphocytes
EDTA blood was collected and lymphocytes were isolated by Ficoll-Isopaque density centrifugation (Lymphoprep, Axis-Shield PoC AS). β-Mannosidase activity in isolated lymphocytes was measured using a fluorometric assay utilizing 4-metylumbelliferyl-β-D-mannopyranosid (Sigma-Aldrich) as substrate according to Panday et al. (1984). β-Galactosidase activity was measured as a control for sample quality, using the 4-metylumbelliferyl-β-D-galactopyranosid (TRC) as substrate. Protein concentration was assayed using the bicinchoninic protein assay (BCA, Pierce, Thermo Fisher Scientific).
ADDITIONAL INFORMATION
Data Deposition and Access
The inverted duplication reported in this manuscript has been submitted to the “Global Variome shared LOVD” (databases.lovd.nl/shared) as individual 00208206, variant 0000439394. Patient consent was not obtained to deposit raw sequencing data.
Ethics Statement
The patient was evaluated in a clinical diagnostic setting. Written informed consent for NGS in a diagnostic setting was obtained from the patient's parents. Written informed consent for publication of results and any accompanying images was similarly obtained. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with Helsinki Declaration of 1975, as revised in 2000.
Acknowledgments
We wish to thank the patient and her parents for consenting to the publication of this case report. Warm thanks also to professor Øivind Nilssen for sharing knowledge from decades of research on mannosidosis.
Author Contributions
M.B. contributed to conception and design, data acquisition, interpretation of data, and drafting of the paper as well as final approval of the manuscript. H.M.F.R.S. performed the cDNA analysis and the Sanger verification of duplication junctions and contributed to the drafting and correction of the manuscript. J.L. coordinated the NGS-panel analysis, performed the data interpretation, and contributed to the drafting and correction of the manuscript. M.F.S. was physician in charge of the patient and contributed to manuscript drafting. J.A.-C. coordinated the WGS and performed the data interpretation, and contributed to the interpretation of NGS data and to the drafting and correction of the manuscript. P.S. performed the WGS data processing and analysis and contributed to the drafting and correction of the manuscript.
Funding
This work was partially financed with Laboratory Medicine Research and Development funds, Sahlgrenska University Hospital, Gothenburg, Sweden.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Gholson Lyon
Neal Sondheimer
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Alkhayat AH, Kraemer SA, Leipprandt JR, Macek M, Kleijer WJ, Friderici KH. 1998. Human β-mannosidase cDNA characterization and first identification of a mutation associated with human β-mannosidosis. Hum Mol Genet 7: 75–83. 10.1093/hmg/7.1.75 [DOI] [PubMed] [Google Scholar]
- Bedilu R, Nummy KA, Cooper A, Wevers R, Smeitink J, Kleijer WJ, Friderici KH. 2002. Variable clinical presentation of lysosomal β-mannosidosis in patients with null mutations. Mol Genet Metab 77: 282–290. 10.1016/S1096-7192(02)00172-5 [DOI] [PubMed] [Google Scholar]
- Biegstraaten M, van Schaik IN, Wieling W, Wijburg FA, Hollak CE. 2010. Autonomic neuropathy in Fabry disease: a prospective study using the Autonomic Symptom Profile and cardiovascular autonomic function tests. BMC Neurol 10: 38 10.1186/1471-2377-10-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broomfield A, Gunny R, Ali I, Vellodi A, Prabhakar P. 2013. A clinically severe variant of β-mannosidosis, presenting with neonatal onset epilepsy with subsequent evolution of hydrocephalus. JIMD Rep 11: 93–97. 10.1007/8904_2013_227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. 10.1038/nrg.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. 10.1093/bioinformatics/btv710 [DOI] [PubMed] [Google Scholar]
- Cherian MP. 2004. β-Mannosidae deficiency in two mentally retarded girls with intractable seizures. Ann Saudi Med 24: 393–395. 10.5144/0256-4947.2004.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A, Sardharwalla IB, Roberts MM. 1986. Human γ-mannosidase deficiency. N Engl J Med 315: 1231 10.1056/NEJM198611063151918 [DOI] [PubMed] [Google Scholar]
- Cooper A, Hatton C, Thornley M, Sardharwalla IB. 1988. Human β-mannosidase deficiency: biochemical findings in plasma, fibroblasts, white cells and urine. J Inherit Metab Dis 11: 17–29. 10.1007/BF01800054 [DOI] [PubMed] [Google Scholar]
- Cooper A, Wraith JE, Savage WJ, Thornley M, Noronha MJ. 1991. β-Mannosidase deficiency in a female infant with epileptic encephalopathy. J Inherit Metab Dis 14: 18–22. 10.1007/BF01804383 [DOI] [PubMed] [Google Scholar]
- Darin N, Leckström K, Sikora P, Lindgren J, Almén G, Asin-Cayuela J. 2018. γ-Glutamyl transpeptidase deficiency caused by a large homozygous intragenic deletion in GGT1. Eur J Hum Genet 26: 808–817. 10.1038/s41431-41018-40122-41436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gort L, Duque J, Fabeiro JM, Zulaica A, Coll MJ, Chabás A. 2006. Molecular analysis in two β-mannosidosis patients: description of a new adult case. Mol Genet Metab 89: 398–400. 10.1016/j.ymgme.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Huynh T, Khan JM, Ranganathan S. 2011. A comparative structural bioinformatics analysis of inherited mutations in β-D-mannosidase across multiple species reveals a genotype–phenotype correlation. BMC Genomics 12: S22 10.1186/1471-2164-12-S3-S22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MZ, Dawson G. 1981. Caprine β-mannosidosis. Inherited deficiency of β-D-mannosidase. J Biol Chem 256: 5185–5188. [PubMed] [Google Scholar]
- Labauge P, Renard D, Castelnovo G, Sabourdy F, de Champfleur N, Levade T. 2009. β-Mannosidosis: a new cause of spinocerebellar ataxia. Clin Neurol Neurosurg 111: 109–110. 10.1016/j.clineuro.2008.09.007 [DOI] [PubMed] [Google Scholar]
- Leipprandt JR, Kraemer SA, Haithcock BE, Chen H, Dyme JL, Cavanagh KT, Friderici KH, Jones MZ. 1996. Caprine β-mannosidase: sequencing and characterization of the cDNA and identification of the molecular defect of caprine β-mannosidosis. Genomics 37: 51–56. 10.1006/geno.1996.0519 [DOI] [PubMed] [Google Scholar]
- Leipprandt JR, Chen H, Horvath JE, Qiao XT, Jones MZ, Friderici KH. 1999. Identification of a bovine β-mannosidosis mutation and detection of two β-mannosidase pseudogenes. Mamm Genome 10: 1137–1141. 10.1007/s003359901179 [DOI] [PubMed] [Google Scholar]
- Lidove O, Jaussaud R, Aractingi S. 2006. Dermatological and soft-tissue manifestations of Fabry disease: characteristics and response to enzyme replacement therapy. In Fabry disease: perspectives from 5 years of FOS (ed. Mehta A, et al. ), Chap. 24, pp. 315–322. Oxford PharmaGenesis, Oxford. [PubMed] [Google Scholar]
- Lovell KL, Zhu M, Drummond MC, Switzer RC III, Friderici KH. 2014. Distribution and severity of neuropathology in β-mannosidase-deficient mice is strain dependent. JIMD Rep 13: 73–81. 10.1007/8904_2013_258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molho-Pessach V, Bargal R, Abramowitz Y, Doviner V, Ingber A, Raas-Rothschild A, Ne'eman Z, Zeigler M, Zlotogorski A. 2007. Angiokeratoma corporis diffusum in human β-mannosidosis: report of a new case and a novel mutation. J Am Acad Dermatol 57: 407–412. 10.1016/j.jaad.2007.01.037 [DOI] [PubMed] [Google Scholar]
- Orteu CH. 2010. Dermatological manifestations of Fabry Disease. In Fabry disease (ed. Elstein D, et al. ), pp. 259–274. Springer, Netherlands. [Google Scholar]
- Panday RS, van Diggelen OP, Kleijer WJ, Niermeijer MF. 1984. β-Mannosidase in human leukocytes and fibroblasts. J Inherit Metab Dis 7: 155–156. 10.1007/BF01805598 [DOI] [PubMed] [Google Scholar]
- Poenaru L, Akli S, Rocchiccioli F, Eydoux P, Zamet P. 1992. Human β-mannosidosis: a 3-year-old boy with speech impairment and emotional instability. Clin Genet 41: 331–334. 10.1111/j.1399-0004.1992.tb03408.x [DOI] [PubMed] [Google Scholar]
- Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, Niezen-Koning KE, van Diggelen OP. 1999. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet 105: 151–156. 10.1007/s004399900075 [DOI] [PubMed] [Google Scholar]
- Poupětová H, Ledvinová J, Berná L, Dvořáková L, Kožich V, Elleder M. 2010. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 33: 387–396. 10.1007/s10545-010-9093-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riise Stensland HM, Persichetti E, Sorriso C, Hansen GM, Bibi L, Paciotti S, Balducci C, Beccari T. 2008. Identification of two novel β-mannosidosis-associated sequence variants: biochemical analysis of β-mannosidase (MANBA) missense mutations. Mol Genet Metab 94: 476–480. 10.1016/j.ymgme.2008.04.010 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Serna M, Botella-Estrada R, Chabas A, Coll MJ, Oliver V, Febrer MI, Aliaga A. 1996. Angiokeratoma corporis diffusum associated with β-mannosidase deficiency. Arch Dermatol 132: 1219–1222. 10.1001/archderm.1996.03890340083013 [DOI] [PubMed] [Google Scholar]
- Roller E, Ivakhno S, Lee S, Royce T, Tanner S. 2016. Canvas: versatile and scalable detection of copy number variants. Bioinformatics 32: 2375–2377. 10.1093/bioinformatics/btw163 [DOI] [PubMed] [Google Scholar]
- Sabourdy F, Labauge P, Stensland HM, Nieto M, Garcés VL, Renard D, Castelnovo G, de Champfleur N, Levade T. 2009. A MANBA mutation resulting in residual β-mannosidase activity associated with severe leukoencephalopathy: a possible pseudodeficiency variant. BMC Med Genet 10: 84 10.1186/1471-2350-10-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samra ZQ, Athar MA. 2008. Cloning, sequence, expression and characterization of human β-mannosidase. Acta Biochim Pol 55: 479–490. [PubMed] [Google Scholar]
- Sedel F, Friderici K, Nummy K, Caillaud C, Chabli A, Dürr A, Lubetzki C, Agid Y. 2006. Atypical Gilles de la Tourette Syndrome with β-mannosidase deficiency. Arch Neurol 63: 129–131. 10.1001/archneur.63.1.129 [DOI] [PubMed] [Google Scholar]
- Sedel F, Baumann N, Turpin JC, Lyon-Caen O, Saudubray JM, Cohen D. 2007. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J Inherit Metab Dis 30: 631–641. 10.1007/s10545-007-0661-4 [DOI] [PubMed] [Google Scholar]
- Solnick D. 1985. Alternative splicing caused by RNA secondary structure. Cell 43: 667–676. 10.1016/0092-8674(85)90239-9 [DOI] [PubMed] [Google Scholar]
- Suzuki N, Konohana I, Fukushige T, Kanzaki T. 2004. β-Mannosidosis with angiokeratoma corporis diffusum. J Dermatol 31: 931–935. 10.1111/j.1346-8138.2004.tb00630.x [DOI] [PubMed] [Google Scholar]
- van Pelt J, Hokke CH, Dorland L, Duran M, Kamerling JP, Vliegenthart JF. 1990. Accumulation of mannosyl-β(1→4)-N-acetylglucosamine in fibroblasts and leukocytes of patients with a deficiency of β-mannosidase. Clin Chim Acta 187: 55–60. 10.1016/0009-8981(90)90261-P [DOI] [PubMed] [Google Scholar]
- Verdin H, D'haene B, Beysen D, Novikova Y, Menten B, Sante T, Lapunzina P, Nevado J, Carvalho CM, Lupski JR, et al. 2013. Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet 9: e1003358 10.1371/journal.pgen.1003358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Sujansky E, Fennessey PV, Thompson JN. 1986. Human β-mannosidase deficiency. N Engl J Med 315: 1201–1205. 10.1056/NEJM198611063151906 [DOI] [PubMed] [Google Scholar]
- Winchester B. 2005. Lysosomal metabolism of glycoproteins. Glycobiology 15: 1–15. 10.1093/glycob/cwi041 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The inverted duplication reported in this manuscript has been submitted to the “Global Variome shared LOVD” (databases.lovd.nl/shared) as individual 00208206, variant 0000439394. Patient consent was not obtained to deposit raw sequencing data.