

Abstract
Variants in the mitochondrial alanyl-tRNA synthetase 2 gene AARS2 (OMIM 612035) are associated with infantile mitochondrial cardiomyopathy or later-onset leukoencephalopathy with premature ovarian insufficiency. Here, we report two newborn siblings who died soon after birth with primary pulmonary hypoplasia without evidence of cardiomyopathy. Whole-exome sequencing detected the same compound heterozygous AARS2 variants in both siblings (c.1774C>T, p.Arg592Trp and c.647dup, p.Cys218Leufs*6) that have previously been associated with infantile mitochondrial cardiomyopathy. Segregation analysis in the family confirmed carrier status of the parents and an unaffected sibling. To our knowledge, this is the first report of primary pulmonary hypoplasia in the absence of cardiomyopathy associated with recessive AARS2 variants and further defines the phenotypic spectrum associated with this gene.
Keywords: concentric hypertrophic cardiomyopathy, EEG with burst suppression, hydrops fetalis, mixed respiratory and metabolic acidosis, nonimmune hydrops fetalis, pulmonary hypoplasia, serous pericardial effusion, transudative pleural effusion
INTRODUCTION
The clinically and genetically heterogeneous mitochondrial respiratory chain conditions are an expanding group of disorders among the most common genetic human conditions with an estimated prevalence of at least 1 in 5000 (Khan et al. 2015). The spectrum and onset of features associated with mitochondrial respiratory chain defects is highly variable and often multisystemic, resulting from defective assembly and/or function of the oxidative phosphorylation system and reduced cellular energy production (McCormick et al. 2018).
Mitochondria have their own DNA (mtDNA), encoding important mitochondrial proteins, ribosomal RNAs, and transfer RNAs, and disorders caused by mtDNA variants are maternally inherited. However, replication, transcription, and translation of mtDNA rely on proteins encoded by nuclear genes, and mitochondrial disorders involving nuclear genes follow Mendelian inheritance (Frazier et al. 2017). Mitochondrial aminoacyl-tRNA synthetases are one such family of nuclear genes, synthesized in the cytosol and then imported into mitochondria. They play a crucial role in intramitochondrial protein synthesis by catalyzing the attachment of each amino acid to a cognate tRNA (Diodato et al. 2014). In recent years, an increasing number of variants in each of the 19 nuclear encoded mitochondrial aminoacyl-tRNA synthetases have been associated with widely variable clinical presentations, ranging from isolated manifestations such as hearing loss and intellectual disability to severe multisystemic clinical disorders often involving the central nervous system (Diodato et al. 2014; Sissler et al. 2017; González-Serrano et al. 2019).
The alanyl-tRNA synthetase 2 (AARS2) gene encodes the enzyme responsible for charging tRNA with alanine in the mitochondria. To date, at least 15 different variants in AARS2 have been described in patients. These variants are associated with two separate phenotypes: namely, an autosomal recessive lethal infantile cardiomyopathy (Götz et al. 2011; Taylor et al. 2014) and a later-onset autosomal recessive leukodystrophy with premature ovarian insufficiency (Dallabona et al. 2014; Lynch et al. 2017). Interestingly, a founder variant of European origin, c.1774C>T, p.Arg592Trp, has been described in a number of patients who have been reported to present with lethal infantile cardiomyopathy (Euro et al. 2015). We report here the case of two siblings possessing the p.Arg592Trp variant, in trans with a frameshift variant, c.647dup, p.Cys218Leufs*6, who presented with lethal pulmonary hypoplasia in the absence of cardiomyopathy, expanding the spectrum of recessive AARS2 disorders.
RESULTS
Clinical Presentations
Patient A was a female, the first child of a nonconsanguineous couple of European background with a history of hypogonadotropic hypogonadism in the mother. She was born after a natural pregnancy complicated by polyhydramnios and nonimmune hydrops, with small pulmonary effusions and significant ascites first detected at 35 wk of pregnancy. A prior anatomic ultrasound scan at 19 wk was normal. Upon detection of nonimmune hydrops, an amniocentesis with viral studies and cytogenetic studies was undertaken and returned normal results. She was delivered by cesarean section at 38 wk and 3 d because of breach position. At birth, she was not breathing and was intubated. Resuscitation attempts were unsuccessful and she died within the first hour. No laboratory or radiologic studies were obtained.
Patient B was the male sibling of Patient A and the parents’ third child, following the birth of a healthy sister. The pregnancy was closely monitored and pulmonary hypoplasia was not suspected until delivery by planned cesarean section at 38 wk + 5 d. At delivery, the child had no respiratory effort leading to a full resuscitation. Laboratory testing obtained during the resuscitation was from a single arterial puncture and revealed marked mixed respiratory and metabolic acidosis with pH 6.55, pCO2 107 mmHg, pO2 63 mmHg, base excess −26 mmol/L, bicarbonate 9 mmol/L, and lactate 18 mmol/L (Table 1). Full body radiograph demonstrated very poorly inflated lungs with only a small volume of aerated right lung and air bronchogram patterns on the left (Fig. 1A). Resuscitation was unsuccessful and the child died ∼90 min after birth.
Table 1.
Laboratory results from the single arterial puncture performed on Patient B
| Arterial blood | Normal range | |
|---|---|---|
| pH | 6.55 | [7.26–7.49] |
| pCO2 | 107 | [27–40 mmHg] |
| pO2 | 63 | [55–80 mmHg] |
| Bicarbonate | 9 | [17–25 mmol/L] |
| Base excess | −26 | [−10–2 mmol/L] |
| Sodium | 139 | [132–147 mmol/L] |
| Potassium | 5.4 | [3.5–6.2 mmol/L] |
| Chloride | 117 | [98–115 mmol/L] |
| Anion gap | 18 | [7–17 mmol/L] |
| Calcium ionized | 1.42 | [1.00–1.35 mmol/L] |
| Glucose | 9.4 | [2.6–5.4 mmol/L] |
| Lactate | 18 | [1.2–4.0 mmol/L] |
| Hemoglobin | 170 | [135–195 g/L] |
| Methemoglobin | 1.7 | [<1.5%] |
| Carboxyhemoglobin | 1.4 | [<6.0%] |
Figure 1.

Clinical and postmortem images. (A) Body X ray from Patient B. Note the poor expansion of the thoracic space and minimal lung aeration. Postmortem examination views of the right chest for Patient A (B) and Patient B (C). White arrows indicate the hypoplastic lungs. Microscopy from postmortem examination of cardiac muscle from Patient A (D) and Patient B (E) showing normal tissue architecture.
Postmortem Examinations
Gross postmortem examination of Patient A was notable for a nondysmorphic, normally grown female infant with mild soft tissue edema, ascites, bilateral pleural effusions, and marked pulmonary hypoplasia with normal lung lobation (Fig. 1B). Growth parameters were consistent with full term gestation with a body weight of 3100 g. The lungs weighed 14.4 g, with an expected weight of 53 ± 15 g (Wigglesworth and Singer 1998). The lung to body weight ratio for Patient A was 0.004, which is ∼2% of the 50th percentile for term infants, 0.0179 (De Paepe et al. 2005). Microscopically, the lungs were at the appropriate developmental stage with normal broncho-vascular relationships and normal air space development. Pulmonary arteries and arterioles were markedly dilated, whereas intra-acinar arterioles showed normal muscularization, suggesting significant pulmonary hypertension after delivery. There was evidence of pericardial effusion. The heart itself was 23.4 g, with a normal range of 16.5–26.5 g (Wigglesworth and Singer 1998). The brain was of normal size and morphology.
Gross postmortem examination of Patient B showed a nondysmorphic male with normal growth parameters and marked pulmonary hypoplasia (Fig. 1C). Growth parameters were consistent with full term gestation with a body weight of 3340 g. The lungs weighed 10.0 g with an expected weight of 53 ± 15 g (Wigglesworth and Singer 1998). The lung/body weight ratio in Patient B was 0.003, which, similarly to Patient A, was ∼2% of the 50th percentile for term infants, 0.0179. Microscopically, the lungs were at the appropriate developmental stage with normal broncho-vascular relationships and normal air space development. Apart from the lungs, the weights of the other organs, including the heart, were normal. The heart was 25.1 g, with a normal range of 16.5–26.5 g (Wigglesworth and Singer 1998). There was thickening of the intraventricular septum but not the ventricular walls. The brain was of normal size and morphology.
Histologic examinations of both patients’ hearts were normal without features of cardiomyopathy, cardiomyocyte hypertrophy, or histologic changes suggestive of a mitochondriopathy (Fig. 1D,E). Neither heart showed endocardial fibroelastosis.
Genomic Analyses
Whole-exome sequencing (WES), which was performed for both children following the death of the second child, detected two previously reported pathogenic variants in the AARS2 gene (NM_020745.3). Both patients were compound heterozygous for AARS2 c.1774C>T, p.Arg592Trp and c.647dup, p.Cys218Leufs*6 (Table 2). They also shared rare compound heterozygous variants in USH2A, which is the gene associated with Usher Syndrome 2A, a well-described condition associated with only retinal disease and hearing loss, and not neonatal cardiopulmonary disease (Lentz and Keats 2016). Therefore, the USH2A variants were considered to be unrelated to the severe phenotype witnessed in the patients described here. No other rare (<0.5% population frequency) homozygous or compound heterozygous variants were detected. Patient A had two de novo coding variants (in ABCF3 and GRHL2), and Patient B had one de novo coding variant (in KLHL36), with no de novo variants in common. The AARS2 variants were confirmed by Sanger sequencing, and family studies confirmed parental and unaffected sibling's carrier status, consistent with the expected recessive inheritance (Fig. 2). Sequencing of mitochondrial DNA was not performed for either child.
Table 2.
Rare compound heterozygous variants that are common between the two affected siblings
| Gene | Name | Transcript | Chr:pos. (hg19) | Variation | dbSNP | Frequency (gnomAD) | CADD | SIFT | PolyPhen | ClinVar | Parent of origin |
|---|---|---|---|---|---|---|---|---|---|---|---|
| USH2A | Usherin | NM_206933.2 | 1:215820937 | c.14718G>T p.Lys4906Asn | rs144520489 | 4.1 × 10−06 | 8.46 | T | B | M | |
| 1:215960197 | c.10202T>C p.Ile3401Thr | rs755754255 | 1.2 × 10−05 | 0.096 | T | B | P | ||||
| AARS2 | Alanyl-tRNA synthetase 2, mitochondrial | NM_020745.3 | 6:44272249 | c.1774C>T p.Arg592Trp | rs138119149 | 2.0 × 10−04 | 27.4 | D | D | Pathogenic/likely pathogenic | P |
| 6:44278832 | c.647dupG p.Cys218Leufs*6 | rs746514660 | 7.2 × 10−05 | fs | fs | Pathogenic | M |
(T) Tolerated, (B) benign, (M) maternal, (P) paternal, (D) deleterious, (fs) frameshift.
Figure 2.
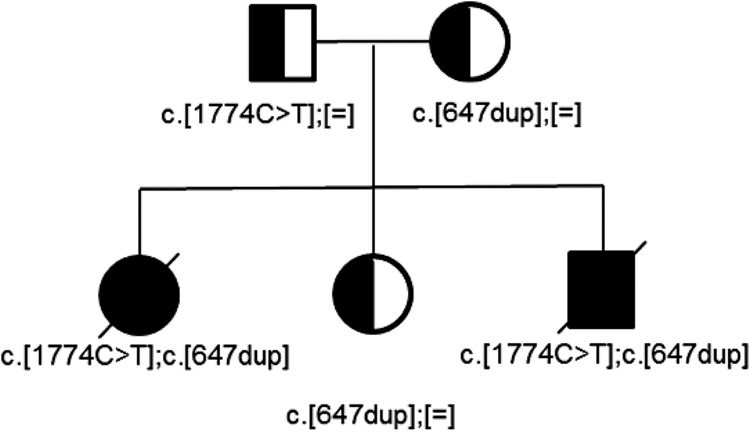
Family pedigree. Individuals with compound heterozygote variants are Patient A (bottom left) and Patient B (bottom right), both shaded black. Heterozygote carriers are shaded part black, part white. Exact variants are listed below each individual. [=] indicates reference sequence.
DISCUSSION
We present two siblings who died shortly after birth with severe, lethal primary pulmonary hypoplasia. The pulmonary hypoplasia in our two patients was marked, the lung to body weight ratio being 0.003–0.004, far less than even the 10th percentile for normal term infants (0.012) (De Paepe et al. 2005). No frozen tissue samples were taken before either child died, as the overall presentations were not immediately suspicious for a mitochondrial disorder. Ultimately, however, both children were found to be compound heterozygous for two previously reported AARS2 variants. The first, the European founder variant p.Arg592Trp, is located in the editing domain of AARS2. Molecular modeling suggests that this residue is important for securing target tRNA into the enzyme's aminoacylation site and that its mutation would cause impaired tRNA binding and markedly decreased aminoacylation (Euro et al. 2015). A recent study has also shown that this variant results in reduced protein stability (Sommerville et al. 2019). The second variant found in these siblings, p.Cys218Leufs*6, would be expected to result in nonsense-mediated decay, leaving our patients with only the less stable and poorly functioning p.Arg592Trp form of the protein. Consistent with this, Euro and colleagues have suggested that the combination of severe variants in AARS2 causes an early lethal clinical phenotype, whereas milder variants resulting in partial reduction of the synthetase function are associated with a later onset mostly neurological presentation with leukodystrophy and premature ovarian insufficiency in affected females (Euro et al. 2015).
Pulmonary hypoplasia is typically secondary to one or more causative factors and is rarely found alone, or primary. Although precise pathophysiology connecting secondary pulmonary hypoplasia to associated pathologies often remains elusive, malformations of the thoracic cavity, impaired fetal breathing movements, and/or abnormal fluid volumes and pressures prevent normal fetal lung development (Wigglesworth and Singer 1998; Porter 1999; Hsu et al. 2012). Diaphragmatic abnormalities and congenital renal disease are among the most commonly associated anomalies with pulmonary hypoplasia (Porter 1999; Aghabiklooei et al. 2009). Severe fetal hydrops with pleural effusions or extreme cardiomegaly also may result in secondary pulmonary hypoplasia (Delgado-Peña et al. 2016), and, as noted above, previous authors have suggested that the pulmonary hypoplasia seen in some patients with early-onset disease from variants in AARS2 is secondary to cardiomegaly. Neither hydrops fetalis nor polyhydramnios was a reported feature for other patients with the European founder variant, and both features were only identified in Patient A, not B. Furthermore, neither Patient A nor Patient B showed clinical or postmortem evidence of cardiomegaly or cardiomyopathy, either grossly or microscopically. Therefore, with no clear underlying abnormality to explain the extent of severe lung hypoplasia observed in these siblings we conclude that they likely had primary pulmonary hypoplasia.
In contrast to the patients described here, almost all previous patients with the p.Arg592Trp variant presented with severe cardiomyopathy (Table 3). Seven of these patients (including one set of twins) were homozygous for p.Arg592Trp, five were in trans with a second AARS2 missense variant, and four were in trans with a nonsense AARS2 variant. The one child without a clearly described cardiomyopathy was a 32-wk stillborn delivered after severe fetal distress with postmortem findings showing prominent endocardial fibroelastosis involving the right ventricle and hypoplastic lungs, in addition to mitochondrial myopathy and bilateral fracture of the femora without evidence of a bone dysplasia (Table 3, Patient 4, from D. Thorburn, pers. comm.). For all these patients, age of presentation ranged from antenatal to 6 mo of age, with neonatal diagnosis in most patients and fatal outcome in all. Lactic acidosis was always present, and hypotonia, with or without evidence of muscular mitochondriopathy, was present in the majority. Neurological features including abnormal EEG were present in some patients.
Table 3.
Reported patients with the European founder c.1774C>T (p.Arg592Trp) variant in AARS2
| Reference | Ethnicity | Variant 1 | Variant 2 | Presentation | Death | HCM | Lactic acidosis | Pulmonary hypoplasia | Other findings, comments | |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 (Sib 1) | Götz et al. 2011 | European (Finnish) | c.1774C>T; p.Arg592Trp | c.464T>G; p.Leu155Arg | Birth | 3 d | + | + | + | Mitochondrial myopathy; abnormal EEG; normal brain postmortem; mechanical ventilation |
| Patient 2 (Sib 2) | Götz et al. 2011 | European (Finnish) | c.1774 C>T; p.Arg592Trp | c.464T>G; p.Leu155Arg | In utero | In utero | + | N/A | + | Stillborn at 40 wk |
| Patient 3 | Götz et al. 2011 | European (Finnish) | c.1774 C>T; p.Arg592Trp | c.1774 C>T; p.Arg592Trp | 3½ mo | 10 mo | + | + | +(mild) | Mitochondrial myopathy; normal CK; EEG abnormality; failure to thrive; mild and nonspecific neuropathology |
| Patient 4 | Calvo et al. 2012 | Australian | c.1774 C>T; p.Arg592Trp | c.986G>A; p.Arg329His | In utero | In utero | NR | N/A | + | Stillborn; multiple fractures; mtDNA depletion in muscle; prominent endocardiac fibroelastosisa |
| Patient 5 | Taylor et al. 2014 | European (UK) | c.1774 C>T; p.Arg592Trp | c.2882 C>T ; p.Ala961Val | Birth | 6 wk | + | + | NR | Muscle involvement, CNS involvement |
| Patient 6 | Taylor et al. 2014 | European (Germany) | c.1774 C>T; p.Arg592Trp | c.1616A>G; p.Tyr539Cys | Birth | 1 mo | + | + | NR | Muscle involvement, CNS involvement |
| Patient 7 | Taylor et al. 2014 | European (Germany) | c.1774 C>T; p.Arg592Trp | c.1774 C>T; p.Arg592Trp | 3 wk | 2 mo | + | + | NR | Muscle involvement |
| Patient 8 | Taylor et al. 2014 | European (UK) | c.1774 C>T; p.Arg592Trp | c.647dupG; p. Cys218Leufs*6 | Birth | 3 mo | + | + | NR | |
| Patient 9 | Taylor et al. 2014 | European (UK) | c.1774 C>T; p.Arg592Trp | c.1774 C>T; p.Arg592Trp | 6 mo | 11 mo | + | - | NR | |
| Patient 10 (Twin 1) | Euro et al. 2015 | European (Germany) | c.1774 C>T; p.Arg592Trp | c.1774 C>T; p.Arg592Trp | Birth | 16 wk | + | + | NR | Mitochondrial myopathy; respiratory distress |
| Patient 11 (Twin 2) | Euro et al. 2015 | European (Germany) | c.1774 C>T; p.Arg592Trp | c.1774 C>T; p.Arg592Trp | Few weeks | 20 wk | + | + | NR | Mitochondrial myopathy; respiratory distress |
| Patient 12 | Euro et al. 2015 | European (Swedish) | c.1774 C>T; p.Arg592Trp | c.1774 C>T; p.Arg592Trp | Birth | 7 wk | + | + | NR | Mitochondrial myopathy; respiratory distress |
| Patient 13 | Mazurova et al. 2017 | NR | c.1774 C>T; p.Arg592Trp | c.1774 C>T; p.Arg592Trp | Birth | 9 wk | + | + | NR | Severe hypotonia; WPW; respiratory distress; poor feeding; poor glycemic control; elevated CK |
| Patient 14 (Twin 1) | Kamps et al. 2018 | European (Dutch) | c.1774 C>T; p.Arg592Trp | c.2872 C>T ; p.Arg958* | Birth | 4 d | + | + | + | Hypotonia, encephalomyopathy, severe HCM |
| Patient 15 (Twin 2) | Kamps et al. 2018 | European (Dutch) | c.1774 C>T; p.Arg592Trp | c.2872 C>T ; p.Arg958* | Birth | 2 h | +/- | NR | + | Presented similar to Patient 14; signs of lung hypoplasia at birth; no autopsy |
| Patient 16 (Sib 3) | Kamps et al. 2018 | European (Dutch) | c.1774 C>T; p.Arg592Trp | c.2872 C>T ; p.Arg958* | Birth | 16 d | +/- | NR | NR | Presented similarly to Patient 14 and 15, no autopsy |
| Patient 17 | This report (Patient A) | European (UK) | c.1774C>T; p.Arg592Trp | c.647dupG; p.Cys218Leufs*6 | Birth | 30 min | - | NR | +++ | Hydrops, ascites, pleural effusions (first detected at 35 wk gestation) |
| Patient 18 | This report (Patient B) | European (UK) | c.1774C>T; p.Arg592Trp | c.647dupG; p.Cys218Leufs*6 | Birth | 90 min | - | + | +++ | Thickened intraventricular septum |
(CK) Creatine kinase, (HCM) hypertrophic cardiomyopathy, (EEG) electroencephalogram, (WPW) Wolf–Parkinson–White, (+) finding reported to be present, (-) finding reported to be absent, (+/-) presence of finding unclear from report, (NR) not reported.
aPersonal communication with D. Thorburn.
Interestingly, Taylor and colleagues reported a British girl (Table 3, Patient 8) having the exact same allele combination as our siblings (p.Arg592Trp and p.Cys218Leufs*6 variants in AARS2) who presented at birth and died at 3 mo of age with cardiomyopathy, but without pulmonary hypoplasia (Taylor et al. 2014). Pulmonary hypoplasia has been reported, however, in at least four other families with AARS2 variants, although typically in association with cardiomyopathy. One child, a female homozygous for p.Arg592Trp, had poor growth, lactic acidosis, and a hypertrophic, dilated left ventricle (Götz et al. 2011). She died at 10 mo of age and autopsy revealed mild pulmonary hypoplasia that was interpreted as secondary to compression of the lung by the severely enlarged heart. Two other siblings in a second family died, one at 3 d of life and the other in utero, with hypertrophic cardiomyopathy and pulmonary hypoplasia (Götz et al. 2011). Sequencing showed the p.Arg592Trp variant along with a second missense variant, c.464T>G (p.Leu155Arg). Three siblings in a third family were described to have cardiomyopathy, pulmonary hypoplasia, lactic acidosis, and hypotonia, with each dying within 2 wk (Kamps et al. 2018). All three were compound heterozygous in AARS2 for p.Arg592Trp and a second truncating variant, p.Arg958*. A child in a fourth family was recently described with respiratory distress, cardiomyopathy, and lactic acidosis and had a novel c.1738C>T (p.Arg580Trp) variant in trans with c.1008dupT (p.Asp337*) (Sommerville et al. 2019). Although hypoplastic lungs were not reported in this individual, an ungenotyped sibling who previously died with lactic acidosis and coagulopathy was found on autopsy to have pulmonary hypoplasia without report of cardiomegaly. Finally, a novel AARS2 homozygous missense variant was detected in five fetuses with nonimmune hydrops from consanguineous Omani families (Bruwer et al. 2017). Of these, one stillborn baby had normal echocardiography. However, postmortem examination was not available on any of the children and it is not known if pulmonary hypoplasia or cardiomyopathy were present.
As observed with AARS2, distinct variants in other aminoacyl-tRNA synthetase genes can result in severe early-onset clinical presentations. In particular, two sisters with compound heterozygous variants in the arginine-tRNA synthetase 2 gene (RARS2) were reported to have severe pulmonary hypoplasia (Lax et al. 2015). One sister died soon after birth with pulmonary hypoplasia and a grossly normal-appearing heart without cardiomegaly, although microscopy revealed central myocyte clearing and evidence of abundant mitochondria and was interpreted as consistent with a mitochondrial cardiomyopathy. The second sister had biventricular hypertrophy, lactic acidosis, and EEG with burst suppression and died after support was withdrawn on day 14 of life. Her autopsy revealed pulmonary hypoplasia with biventricular free wall thickening in the heart.
The lack of evidence of extrinsic factors in our patients, aside from hydrops in Patient A, would suggest that there is an independent as-yet undefined mechanism relating to their AARS2 variants that explains the severe primary pulmonary hypoplasia. The validity of this hypothesis is indirectly supported by the patients with RARS2 variants who also displayed pulmonary hypoplasia in the absence of cardiomegaly and hydrops (Lax et al. 2015) and directly by the sibling of an AARS2 variant–affected proband lacking cardiac features but having noted pulmonary hypoplasia (Sommerville et al. 2019). Furthermore, the phenotypic heterogeneity seen with AARS2 variants is of great interest, exemplified by the differences between siblings reported in this and prior reports, as well as differences between patients with identical variants. This suggests that although variants in specific residues may largely determine phenotype, such as early- or late-onset diseases, there are likely other modifier genes, epigenetic factors, or environmental modifiers that further define the precise clinical presentation (Sissler et al. 2017; González-Serrano et al. 2019). What these are remains to be discovered.
In summary, we suggest that severe, lethal primary pulmonary hypoplasia in the absence of cardiomyopathy, with or without hydrops, is a potential presentation of recessive variants in aminoacyl-tRNA synthetases, particularly but not limited to AARS2. These disorders should be considered with presentations of isolated pulmonary hypoplasia and even for unexplained hydrops fetalis, which has important implications for genetic investigations for the affected child and for subsequent parental counseling.
METHODS
WES was performed on germline DNA under a research protocol at the Yale Center for Genome Analysis using IDT xGen exome capture kit followed by Illumina DNA sequencing (HiSeq 4000). Paired-end sequence reads were converted to FASTQ format and were aligned to the reference human genome (hg19). GATK best practices were applied to identify genetic variants, and variants were annotated by ANNOVAR. The two siblings and their parents were sequenced to a mean depth of 37.3–45.2 independent reads per targeted base across all the samples. There were greater than eight independent reads in an average of 93.4%–94.0% of targeted bases in all of the samples. Data for sequencing coverage are shown in Table 4.
Table 4.
Metrics of sequencing data
| Patient A | Patient B | Father | Mother | |
|---|---|---|---|---|
| Paired-end; read length | 99 | 99 | 99 | 99 |
| Number reads (M) | 46.4 | 35.9 | 43.4 | 48.4 |
| Number bases (G) | 4.6 | 3.6 | 4.3 | 4.8 |
| Mean coverage | 45.2 | 37.3 | 42.8 | 42.4 |
| Median coverage | 42 | 35 | 40 | 39 |
| PCR duplicates | 21.40% | 19.92% | 20.38% | 22.93% |
| Unmapped | 0.03% | 0.02% | 0.03% | 0.04% |
| Reads on-target | 56.47% | 58.79% | 56.40% | 51.59% |
| Bases on-target | 45.77% | 47.99% | 45.45% | 41.12% |
| Mean error rate | 0.26% | 0.24% | 0.27% | 0.30% |
| 8× target base coverage | 93.80% | 93.40% | 93.90% | 94.00% |
| 10× target base coverage | 93.30% | 92.60% | 93.40% | 93.40% |
| 15× target base coverage | 90.80% | 88.90% | 90.70% | 90.20% |
| 20× target base coverage | 86.00% | 82.20% | 85.50% | 84.40% |
| 30× target base coverage | 71.20% | 62.60% | 69.50% | 67.80% |
| 50× target base coverage | 38.00% | 23.70% | 34.00% | 33.10% |
| Variant allele #1 in AARS2 (NM_020745) | c.1774C>T, p.Arg592Trp | c.1774C>T, p.Arg592Trp | c.1774C>T, p.Arg592Trp | — |
| Variant #1 reads/total reads | 33/59 | 18/38 | 25/50 | 0/52 |
| Variant allele #2 in AARS2 (NM_020745) | c.647dupG, p.Cys218Leufs*6 | c.647dupG, p.Cys218Leufs*6 | — | c.647dupG, p.Cys218Leufs*6 |
| Variant #2 reads/total reads | 16/38 | 13/27 | 0/44 | 18/40 |
| Compound heterozygosity in USH2A (NM_206933) | c.14718G>T, p.Lys4906Asn | c.14718G>T, p.Lys4906Asn | — | c.14718G>T, p.Lys4906Asn |
| c.10202T>C, p.Ile3401Thr | c.10202T>C, p.Ile3401Thr | c.10202T>C, p.Ile3401Thr | ||
| De novo nonsynonymous Variant 1 | ABCF3:NM_018358, c.496C>T, p.Arg166Trp | KLHL36:NM_024731, c.916G>A, p.Glu306Lys | — | — |
| De novo nonsynonymous Variant 2 | GRHL2:NM_024915,c.1400A>G, p.Tyr467Cys | — | — | — |
Bold rows refer to the AARS2 gene.
Additional Information
Data Deposition and Access
Both variants described in these two siblings were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), with the accession numbers SCV000882690 (c.647dupG; p.Cys218Leufs*6) and SCV000882678 (c.1774C>T; p.Arg592Trp). This variant was also deposited in MSeqDR (https://mseqdr.org/). Raw data were not deposited to public access databases in the absence of patient consent but may be available through correspondence with the authors.
Ethics Statement
This study was approved by the Institutional Review Board (IRB) at Yale University. Written informed consent for the study and publication was obtained from the parents of the two siblings.
Acknowledgments
We gratefully acknowledge the generous support for the Pediatric Genomics Discovery Program by Yale New Haven Hospital and Sara and Jeffery Buell.
Author Contributions
C.K.-B. provided clinical data and wrote the manuscript. G.J. analyzed pathology specimens and critically reviewed the manuscript. W.J. analyzed sequencing data and critically reviewed the manuscript. L.J. analyzed clinical data and contributed to writing the manuscript. J.-L.R. analyzed clinical data and critically reviewed the manuscript. M.K. gathered clinical data and specimens and critically reviewed the manuscript. K.G.A. contributed to experimental design and critically reviewed the manuscript. S.A.L. guided experimental design and data analysis and contributed to writing the manuscript.
Funding
This work was not supported by any external funding sources.
Competing Interest Statement
Dr. Lakhani is part owner of Qiyas Higher Health, a start-up company unrelated to this work. No other authors have any disclosures to report.
Referees
David Thorburn
Anonymous
REFERENCES
- Aghabiklooei A, Goodarzi P, Kariminejad MH. 2009. Lung hypoplasia and its associated major congenital abnormalities in perinatal death: an autopsy study of 850 cases. Indian J Pediatr 76: 1137–1140. 10.1007/s12098-009-0229-4 [DOI] [PubMed] [Google Scholar]
- Bruwer Z, Al Riyami N, Al Dughaishi T, Al Murshedi F, Al Sayegh A, Al Kindy A, Meftah D, Al Kharusi K, Al Foori A, Al Yarubi N, et al. 2017. Inborn errors of metabolism in a cohort of pregnancies with nonimmune hydrops fetalis: a single center experience. J. Perinat Med 46: 968–974. 10.1515/jpm-2017-0124 [DOI] [PubMed] [Google Scholar]
- Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, et al. 2012. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 4: 118ra110 10.1126/scitranslmed.3003310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallabona C, Diodato D, Kevelam SH, Haack TB, Wong LJ, Salomons GS, Baruffini E, Melchionda L, Mariotti C, Strom TM, et al. 2014. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 82: 2063–2071. 10.1212/WNL.0000000000000497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Peña YP, Torrent-Vernetta A, Sacoto G, de Mir-Messa I, Rovira-Amigo S, Gartner S, Moreno-Galdó A, Molino-Gahete JA, Castillo-Salinas F. 2016. [Pulmonary hypoplasia: an analysis of cases over a 20-year period]. An Pediatr (Barc) 85: 70–76. 10.1016/j.anpedi.2015.10.008 [DOI] [PubMed] [Google Scholar]
- De Paepe ME, Friedman RM, Gundogan F, Pinar H. 2005. Postmortem lung weight/body weight standards for term and preterm infants. Pediatr Pulmonol 40: 445–448. 10.1002/ppul.20218 [DOI] [PubMed] [Google Scholar]
- Diodato D, Ghezzi D, Tiranti V. 2014. The mitochondrial aminoacyl tRNA synthetases: genes and syndromes. Int J Cell Biol 2014: 787956 10.1155/2014/787956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euro L, Konovalova S, Asin-Cayuela J, Tulinius M, Griffin H, Horvath R, Taylor RW, Chinnery PF, Schara U, Thorburn DR, et al. 2015. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front Genet 6: 21 10.3389/fgene.2015.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier AE, Thorburn DR, Compton AG. 2017. Mitochondrial energy generation disorders: genes, mechanisms and clues to pathology. J Biol Chem 10.1074/jbc.R117.809194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Serrano LE, Chihade JW, Sissler M. 2019. When a common biological role does not imply common disease outcomes: disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J Biol Chem 10.1074/jbc.REV118.002953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz A, Tyynismaa H, Euro L, Ellonen P, Hyötyläinen T, Ojala T, Hämäläinen RH, Tommiska J, Raivio T, Oresic M, et al. 2011. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 88: 635–642. 10.1016/j.ajhg.2011.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JS, Lee YS, Lin CH, Li FY, Jeng MJ, Soong WJ, Chen SJ, Tang RB. 2012. Primary congenital pulmonary hypoplasia of a neonate. J Chin Med Assoc 75: 87–90. 10.1016/j.jcma.2011.12.004 [DOI] [PubMed] [Google Scholar]
- Kamps R, Szklarczyk R, Theunissen TE, Hellebrekers D, Sallevelt S, Boesten IB, de Koning B, van den Bosch BJ, Salomons GS, Simas-Mendes M, et al. 2018. Genetic defects in mtDNA-encoded protein translation cause pediatric, mitochondrial cardiomyopathy with early-onset brain disease. Eur J Hum Genet 26: 537–551. 10.1038/s41431-017-0058-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Govindaraj P, Meena AK, Thangaraj K. 2015. Mitochondrial disorders: challenges in diagnosis & treatment. Indian J Med Res 141: 13–26. 10.4103/0971-5916.154489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax NZ, Alston CL, Schon K, Park SM, Krishnakumar D, He L, Falkous G, Ogilvy-Stuart A, Lees C, King RH, et al. 2015. Neuropathologic characterization of pontocerebellar hypoplasia type 6 associated with cardiomyopathy and hydrops fetalis and severe multisystem respiratory chain deficiency due to novel RARS2 mutations. J Neuropathol Exp Neurol 74: 688–703. 10.1097/NEN.0000000000000209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz J, Keats B. 2016. Usher syndrome type II. In GeneReviews® (ed. Adam MP, et al. ). University of Washington, Seattle, WA. [PubMed] [Google Scholar]
- Lynch DS, Rodrigues Brandão de Paiva A, Zhang WJ, Bugiardini E, Freua F, Tavares Lucato L, Macedo-Souza LI, Lakshmanan R, Kinsella JA, Merwick A, et al. 2017. Clinical and genetic characterization of leukoencephalopathies in adults. Brain 140: 1204–1211. 10.1093/brain/awx045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurova S, Magner M, Kucerova-Vidrova V, Vondrackova A, Stranecky V, Pristoupilova A, Zamecnik J, Hansikova H, Zeman J, Tesarova M, et al. 2017. Thymidine kinase 2 and alanyl-tRNA synthetase 2 deficiencies cause lethal mitochondrial cardiomyopathy: case reports and review of the literature. Cardiol Young 27: 936–944. 10.1017/S1047951116001876 [DOI] [PubMed] [Google Scholar]
- McCormick EM, Zolkipli-Cunningham Z, Falk MJ. 2018. Mitochondrial disease genetics update: recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr Opin Pediatr 30: 714–724. 10.1097/MOP.0000000000000686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter HJ. 1999. Pulmonary hypoplasia. Arch Dis Child Fetal Neonatal Ed 81: F81–F83. 10.1136/fn.81.2.F81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissler M, González-Serrano LE, Westhof E. 2017. Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol Med 23: 693–708. 10.1016/j.molmed.2017.06.002 [DOI] [PubMed] [Google Scholar]
- Sommerville EW, Zhou XL, Oláhová M, Jenkins J, Euro L, Konovalova S, Hilander T, Pyle A, He L, Habeebu S, et al. 2019. Instability of the mitochondrial alanyl-tRNA synthetase underlies fatal infantile-onset cardiomyopathy. Hum Mol Genet 28: 258–268. 10.1093/hmg/ddy294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, Smertenko T, Alston CL, Neeve VC, Best A, et al. 2014. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312: 68–77. 10.1001/jama.2014.7184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigglesworth JS, Singer DB. 1998. Textbook of fetal and perinatal pathology, 2nd ed Blackwell Science, Oxford. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Both variants described in these two siblings were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), with the accession numbers SCV000882690 (c.647dupG; p.Cys218Leufs*6) and SCV000882678 (c.1774C>T; p.Arg592Trp). This variant was also deposited in MSeqDR (https://mseqdr.org/). Raw data were not deposited to public access databases in the absence of patient consent but may be available through correspondence with the authors.