

Abstract
Gardner syndrome describes a variant phenotype of familial adenomatous polyposis (FAP), primarily characterized by extracolonic lesions including osteomas, dental abnormalities, epidermal cysts, and soft tissue tumors. We describe a 2-yr-old boy presenting with a 2-cm soft tissue mass of the forehead. Pathologic evaluation revealed a nuchal-type/Gardner-associated fibroma. Sequencing of the APC gene revealed a pathologic variant c.4666dupA. Parental sequencing of both blood and buccal tissue supported the de novo occurrence of this pathologic variant. Further imaging revealed a number of additional lesions including a large lumbar paraspinal desmoid, a 1-cm palpable lesion posterior to the left knee, firm lesions on bilateral heels, and multiple subdermal lesions. Colonoscopy was negative. This case illustrates a genetic variant of Gardner syndrome resulting in an aggressive early childhood phenotype and highlights the need for an individualized approach to treatment.
Keywords: desmoid tumors, facial neoplasm, intestinal polyp, neoplasm of the gastrointestinal tract, neoplasm of the large intestine
INTRODUCTION
Gardner syndrome is a known variant of familial adenomatous polyposis (FAP), a rare autosomal dominant condition arising from a germline variation in the adenomatous polyposis coli (APC) gene on Chromosome 5q21 (Galiatsatos and Foulkes 2006). FAP is typically clinically characterized by the development of multiple colorectal adenomatous polyps after the second decade of life, which predisposes to colorectal cancer. Gardner syndrome includes the intestinal characteristics of FAP in addition to extracolonic manifestations, including osteomas, dental abnormalities, epidermal cysts, and soft tissue tumors, particularly desmoid tumors (Galiatsatos and Foulkes 2006; Campos 2014). Gardner-associated fibromas may be seen in young patients as precursor lesions to the desmoid tumors of Gardner syndrome (Coffin et al. 2007). Here, we present an extreme presentation of Gardner syndrome with an aggressive early childhood phenotype in a child who presented with a soft tissue right temporal mass. This report is meant to provide a touchstone to document the severe early-onset phenotype and pain associated with c.4666dupA.
RESULTS
Clinical Presentation and Family History
The patient presented as an otherwise well appearing 2-yr-old boy with a progressively enlarging mass at the lateral edge of his right eyebrow over the course of 1 yr. The mass measured by ultrasound was 0.7 × 2.0 × 1.6 cm increasing over 3 mo to 0.7 × 2.3 × 1.8 cm. His past medical history was notable for a history of an enlarged atrial appendage noted in utero with no cardiac sequelae on echocardiogram performed after delivery. Additionally, this patient had dermatographia with frequent rashes. Although he met his motor milestones on time, his language was delayed. On exam, there was a nontender, smooth, ovoid mass at the lateral aspect of the right eyebrow without overlying skin changes (Fig. 1).
Figure 1.
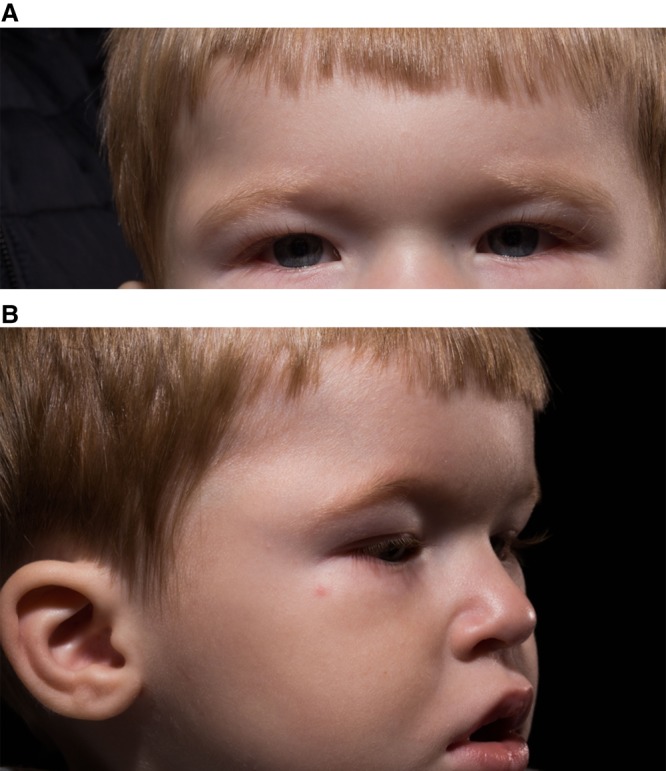
Facial aspects of the patient's clinical presentation.
Ultrasound demonstrated a heterogeneously hyperechoic subdermal mass with traversing internal vessels. Computed tomography showed a well-circumscribed mass located superficial to the temporalis muscle and zygomatic arch without bone or muscle invasion. The lesion demonstrated mild enhancement and was supplied by branches of the superficial temporal artery. Magnetic resonance imaging showed diffusely hypointense T1 and T2 signal with low-level enhancement, supporting a fibrous composition. The mass was completely surgically excised and sent to pathology, which revealed a nuchal-type/Gardner-associated fibroma (Fig. 2). Because of the known relationship between Gardner-associated fibromas and FAP, a family history was taken, and genetic testing was recommended to the family. The patient had some family history of colon polyps, but this history does not support the autosomal dominant inheritance pattern seen in FAP (Fig. 3).
Figure 2.

The histology shows thick haphazardly arranged collagen infiltrating adjacent structure with entrapped adipose tissue, consistent with nuchal-type/Gardner-associated fibroma. Both images are shown at 50× magnification.
Figure 3.

Family history obtained from the patient's parents.
The APC gene was sequenced using next-generation technology to test for the presence of a variant in all coding regions and intron/exon boundaries of the APC gene. All reported alterations detected by next-generation sequencing (NGS) were confirmed using Sanger sequencing. Testing result was significant for a heterozygous single-nucleotide insertion at position c.4666dupA (p.Thr1556AsnfsX3) NM_000038; build GRCh37 (hg19) of the APC gene resulting in a frameshift variant (see Table 1). This variant is not present in the Genome Aggregation Database (gnomAD), demonstrating the low frequency of this allele. This alteration was interpreted to be a pathogenic variant-based criterion by Richards et al. (2015). This variant is absent in population databases (PM2), a moderate indicator of pathogenicity; this variant is a predicted null variant in a gene in which loss of function is a known mechanism of disease (PVS1), a very strong indicator of pathogenicity; this variant is de novo, with paternity and maternity confirmed (PS2), a strong indicator of pathogenicity; and this patient's phenotype is highly specific for the gene (PP4), a supporting indicator of pathogenicity. This variant is reported as pathogenic in three out of four reports in ClinVar and was reported previously as a pathogenic variant in two studies (Aceto et al. 2005; Friedl and Aretz 2005). Array comparative genomic hybridization (CGH) was performed a year later for language delays, and no copy number variants were detected.
Table 1.
Genomic analysis
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | ClinGen/dbSNP | Genotype | ClinVar ID |
|---|---|---|---|---|---|---|---|---|
| APC | 5 | c.4666dupA | p.Thr1556AsnfsX3 | De novo | Frameshift | 428112/rs1114167560/ | Heterozygous | SCV000691753.1 |
Variant analysis in both parents from genomic DNA derived from both blood and buccal tissue did not reveal the APC variant found in the proband, thus reducing the probability of somatic mosaicism in the parent. Genomic DNA of his maternal half-brother was also negative. This, combined with the family history inconsistent for an autosomal dominant trait, supports that the genomic variant in APC most likely arose de novo in this patient.
Further investigation with thoracolumbar MRI was significant for fibrous appearing tumors in the right thoracolumbar paraspinal soft tissues and right posterior base of the neck (Fig. 4).
Figure 4.

Contrast-enhanced T1-weighted MRI with fat saturation at the level of the orbits (top left), cervical spine (top right), lumbar spine (lower left), and sacrum (lower right). There are multiple subcutaneous and intramuscular fibromas involving the right temporal scalp and paraspinal soft tissues (arrows), with hypointense signal and low-level enhancement.
The patient continued to be managed in accordance with the guidelines set by the National Comprehensive Cancer Network (2018) (Genetic/Familial High-Risk Assessment: Colorectal). Recommendations for further evaluation in a patient presenting at such a young age are not specified. Therefore, screening took place for the associated additional findings of congenital hypertrophy of retinal pigment epithelium (CHRPE), further desmoids, monitoring for the presence of colonic polyps, and evaluation for hepatoblastoma. A dilated retinal examination by an experienced pediatric ophthalmologist did not reveal any evidence of CHRPE. Throughout follow-up, the patient continued to develop multiple new painful fibromas on the scalp, face, back, knee, and foot. Treatment with gabapentin for pain management was unsuccessful. However, bilateral leg pain has improved since starting celecoxib and lidocaine patches. Multiple palpable lesions are currently being monitored, including three lesions on the head, a 3-cm lesion over the right abdomen, a 1-cm lesion in the epigastric area, a large firm lesion 3 cm to the right of midline on the back, a 1-cm palpable lesion at the left knee, and firm lesions on bilateral heels. α-Fetoprotein (AFP) has also been monitored quarterly and has remained within the normal range, and liver imaging has also been normal. Colonoscopy and capsule endoscopy took place, both of which were negative.
DISCUSSION
This case is significant because of the young age of the patient presenting with multiple desmoids and pain associated with Gardner syndrome due to a pathogenic c.4666dupA variant in APC. Although screening guidelines exist for children at risk of classic FAP, no unified approach exists for any of the extracolonic manifestations in pediatric patients with Gardner syndrome. Therefore, despite differing characteristics of disease presentation, screening practices for Gardner syndrome revert to those established for FAP.
There is currently no standard in the literature for when to start colonoscopies in patients with Gardner syndrome presenting this young. For children at risk of classic FAP, sigmoidoscopy every 1–2 yr beginning at 10–12 yr of age is the generally accepted colon screening guideline (Syngal et al. 2015). One report notes hundreds of polyps were found on colonoscopy of a 10-yr-old patient lacking a family history of FAP following 8 years of recurrent hematochezia, precipitating iron-deficiency anemia (Auricchio et al. 2005). Additionally, a recent report (Gutierrez Sanchez et al. 2018) details that esophagogastroduodenoscopy reveals clinically relevant lesions earlier than previously predicted in children with FAP, suggesting that earlier screening may be indicated. However, guidelines for the commencement of colonoscopy monitoring for children with FAP are not provided for patients presenting at such a young age (National Comprehensive Cancer Network 2018).
An association between hepatoblastoma and FAP has been confirmed, increasing risk and occurring primarily in boys (Giardiello et al. 1996; Spector and Birch 2012). Therefore, liver palpation along with AFP level screening and abdominal ultrasound every 2–3 mo is recommended until at least 5 yr old (Hirschman et al. 2005). Patients with FAP are also at increased risk of neoplasms of the thyroid and pancreas, and should therefore be subject to routine screenings in these areas as well (Giardiello et al. 1993). Children with FAP also have an increased risk of cancer in areas that are less easily monitored. Patients are at risk of brain tumors, especially medulloblastoma, as well as adrenal adenocarcinoma (Septer et al. 2016).
Early presentations of Gardner syndrome are rare and poorly documented in the literature. Beyond the report of the 10-yr-old patient with recurrent hematochezia from innumerable polyps mentioned previously (Auricchio et al. 2005), one report details a neonatal presentation of Gardner syndrome (Levesque et al. 2010). The patient presented with paraspinal masses at 3 wk and at 5 mo of age, the second of which was biopsied and found to be consistent with a Gardner-associated fibroma. Rectal bleeding at 47 mo prompted a colonoscopy that revealed nearly 100 colonic polyps, leading to a total proctocolectomy. The scarcity of early childhood Gardner syndrome cases in the literature highlights the need to document cases of early-onset Gardner syndrome.
Desmoid tumors in Gardner syndrome require a pragmatic approach, as complete excision may be technically challenging and may put important neurovascular structures at risk. Surgical resection has been reported to be associated with postsurgical growth of resected desmoids (Soravia et al. 2000). Thus, a watchful waiting approach is being taken with this patient. Although surgery is not curative, the painful nature of desmoids emphasizes the need to consider surgical excision for symptomatic relief on a case-by-case and lesion-by-lesion basis. Desmoid tumors may present in any location on the body. One study of 45 patients with 57 fibromas notes that lesion site occurrences were 61% in the back and paraspinal region, 14% in the head and neck, 14% on the extremities, and 11% in the chest and abdomen (Coffin et al. 2007). Desmoid tumors account for the greatest amount of postcolectomy morbidity and mortality in FAP patients (Septer et al. 2016), representing a significant chronic health burden even after dramatic surgical interventions. Not well documented in the literature is the pain associated with these tumors, which is a significant problem for this young patient and his family.
Gardner syndrome is not typically diagnosed by a sentinel facial lesion, as seen in this patient's case. However, providers must be able to recognize the distinct fibrous lesions of this disease regardless of presentation. Lesion identification leads to diagnosis of Gardner syndrome in pediatric patients in whom this condition would not be expected based on family history, highlighting the importance of recognizing nuchal-type fibromas and their diagnostic potential (Wehrli et al. 2001). FAP must be excluded in a child with fibromas suggestive of Gardner syndrome because de novo variants can occur in patients with a negative family history, as seen in this patient and other cases (Levesque et al. 2010). However, diagnosis may also identify previously unrecognized FAP families (Vieira et al. 2015), so genetic testing of parents and siblings is a critical part of the evaluation process.
This patient's genetic variation of c.4666dupA (p.Thr1556AsnfsX3) is a known pathogenic variant, but the associated clinical phenotype is not well documented in the literature. Although this patient's variant is documented as pathogenic in two studies (Aceto et al. 2005; Friedl and Aretz 2005), no phenotype specific to the patient was provided in the first study. In the second study, the patient with this variant, known as “GD23,” presented with FAP, without desmoid tumors. However, five unrelated patients from this study, including GD23, carried germline variants within codons 1445–1580 of the APC gene, and two of these patients (GD14 and GD34) presented with desmoids. GD23 was also noted to have a positive family history, with vertical transmission.
Several studies state that this patient's variation is within a region of the APC gene associated with a greater incidence of desmoid tumors (Caspari et al. 1995; Friedl et al. 2001; Slowik et al. 2015), but this specific variant has not been previously implicated in such an aggressive early childhood phenotype. In addition to desmoid tumors, the variant is also within a region of the APC gene associated with a lack of CHRPE and a greater incidence of dental abnormalities (Davies et al. 1995). Variants near base pair 1309 in the APC gene are correlated to hepatoblastoma (Hirschman et al. 2005), and although this patient's variant does not fall within this proximity, he is being screened regularly.
Interestingly, dysfunction of the Wnt-β catenin pathway and abnormalities of cilia development have been hypothesized to be the underlying mechanisms of pathogenic extra-intestinal manifestations in FAP patients (Gómez and Knoers 2009; Nelson and Näthke 2013). This connection is supported by the presence of symptoms similar to Gardner syndrome in cilia-related disorders, and by the fact that APC is critical for the degradation of β-catenin in the Wnt-signaling pathway. Therefore, this pathway-specific function of APC may create a new context for therapeutic options for patients with Gardner syndrome.
This patient provides a compelling case for the utilization of genetic testing to direct care of patients with Gardner syndrome. Genetic testing, if positive, provides a definitive diagnosis, as well as a road map for providing a care plan to the patient for his health. Genetic testing, considered with the pathology findings and clinical presentation of this patient, led to a diagnosis of Gardner syndrome, directing the care and screening processes for this patient. Even when screening processes are not standardized for Gardner syndrome, the knowledge of this patient's genetic variation allows providers to at least consider analogous forms of care, seen here in comparison to FAP.
This case supports that the de novo c.4666dup (p.Thr1556AsnfsX3) variation of the APC gene should be documented in the literature as resulting in Gardner syndrome phenotype with multiple extracolonic tumors occurring early in life. Variations in this specific portion of the APC gene are known to cause classical FAP and may increase the risk of desmoid tumors (Delaini et al. 2009). Until now, however, this variation had not been documented as causing the early, aggressive phenotype seen in this patient. Clearly, Gardner syndrome demonstrates a variable expression and severity, depending on the location of the genetic variation within the APC gene. Dissemination of the information gathered from this case and other similar instances of pediatric patients with FAP and Gardner syndrome will better anticipate their specific needs and help establish a standard of care.
METHODS
Targeted NGS was performed on a genomic DNA sample extracted from whole blood from the proband in the Genomics Laboratory at Mayo Clinic (Test code: APCZ). Briefly, genomic DNA was sheared by sonication and captured using SureSelect probes (Agilent). Then, paired-end sequencing (2 × 101 bp) was performed on Illumina HiSeq 2500 platform (Illumina). NGS data were analyzed using CLC Bio (QIAGEN) and in-house-developed bioinformatic tools. APC coding exons and flanking noncoding region (transcript NM_ 000038) were analyzed with average coverage at ∼4000× in this sample. The reportable variant was verified by Sanger sequencing. Targeted Sanger sequencing analyses were performed on parental samples received (Test code: FMTT). (See Table 2.)
Table 2.
NGS sequencing coverage
ADDITIONAL INFORMATION
Data Deposition and Access
This variant was submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) NM_000038.5 (APC):c.4666dup (p.Thr1556Asnfs) by Mayo Medical Laboratories submission access SCV000691753.1.
Ethics Statement
This patient report was determined to be IRB exempt, and the patient's parents provided written informed consent to allow publication of his image. Existing patient data in the electronic medical record were reviewed, and no new information was collected during the report's construction.
Acknowledgments
We thank our patient and their family for their kindness and willingness to allow us to tell their story.
Author Contributions
P.K. provided manuscript composition, figure and legend composition, editing, and consolidation of revisions. E.D. provided results and discussion section contributions and manuscript editing. Y.H. provided figure and legend composition and manuscript editing. M.L.H. provided figure and legend composition and manuscript editing. K.B. provided results section composition and manuscript editing. W.E.H. provided genomic analysis. Z.N. provided genomic analysis and methods section composition. B.J.W. provided manuscript editing. L.A.S. was the principal investigator, ensuring communication with the patient's family, manuscript composition, editing, and consolidation of revisions.
Funding
We gratefully acknowledge support from the Mayo Clinic Center for Individualized Medicine, Nelson Fund, and the Mayo Clinic Department of Otorhinolaryngology, Head and Neck Surgery.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Raymond Kim
Anonymous
REFERENCES
- Aceto G, Curia MC, Veschi S, De Lellis L, Mammarella S, Catalano T, Stuppia L, Palka G, Valanzano R, Tonelli F, et al. 2005. Mutations of APC and MYH in unrelated Italian patients with adenomatous polyposis coli. Hum Mutat 26: 394 10.1002/humu.9370 [DOI] [PubMed] [Google Scholar]
- Auricchio R, De Rosa M, Quaglietta L, Miele E, Boccia G, Staiano A, Izzo P. 2005. A dramatic case of early-onset familial adenomatous polyposis. Clin Genet 67: 104–106. 10.1111/j.1399-0004.2005.00381.x [DOI] [PubMed] [Google Scholar]
- Campos FG. 2014. Surgical treatment of familial adenomatous polyposis: dilemmas and current recommendations. World J Gastroenterol 20: 16620–16629. 10.3748/wjg.v20.i44.16620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari R, Olschwang S, Friedl W, Mandl M, Boisson C, Böker T, Augustin A, Kadmon M, Möslein G, Thomas G, et al. 1995. Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet 4: 337–340. 10.1093/hmg/4.3.337 [DOI] [PubMed] [Google Scholar]
- Coffin CM, Hornick JL, Zhou H, Fletcher CD. 2007. Gardner fibroma: a clinicopathologic and immunohistochemical analysis of 45 patients with 57 fibromas. Am J Surg Pathol 31: 410–416. 10.1097/01.pas.0000213348.65014.0a [DOI] [PubMed] [Google Scholar]
- Davies DR, Armstrong JG, Thakker N, Horner K, Guy SP, Clancy T, Sloan P, Blair V, Dodd C, Warnes TW, et al. 1995. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet 57: 1151–1158. [PMC free article] [PubMed] [Google Scholar]
- Delaini GG, Skricka T, Colucci G. 2009. Intestinal polyps and polyposis: from genetics to treatment and follow-up. Springer, Milan. [Google Scholar]
- Friedl W, Aretz S. 2005. Familial adenomatous polyposis: experience from a study of 1164 unrelated German polyposis patients. Hered Cancer Clin Pract 3: 95–114. 10.1186/1897-4287-3-3-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl W, Caspari R, Sengteller M, Uhlhaas S, Lamberti C, Jungck M, Kadmon M, Wolf M, Fahnenstich J, Gebert J, et al. 2001. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 48: 515–521. 10.1136/gut.48.4.515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiatsatos P, Foulkes WD. 2006. Familial adenomatous polyposis. Am J Gastroenterol 101: 385–398. 10.1111/j.1572-0241.2006.00375.x [DOI] [PubMed] [Google Scholar]
- Giardiello FM, Offerhaus GJ, Lee DH, Krush AJ, Tersmette AC, Booker SV, Kelley NC, Hamilton SR. 1993. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut 34: 1394–1396. 10.1136/gut.34.10.1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardiello FM, Petersen GM, Brensinger JD, Luce MC, Cayouette MC, Bacon J, Booker SV, Hamilton SR. 1996. Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 39: 867–869. 10.1136/gut.39.6.867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez García EB, Knoers NV. 2009. Gardner's syndrome (familial adenomatous polyposis): a cilia-related disorder. Lancet Oncol 10: 727–735. 10.1016/S1470-2045(09)70167-6 [DOI] [PubMed] [Google Scholar]
- Gutierrez Sanchez LH, Alsawas M, Stephens M, Murad MH, Absah I. 2018. Upper GI involvement in children with familial adenomatous polyposis syndrome: single center experience and meta-analysis of the literature. Gastrointest Endosc 87: 648–656.e3. 10.1016/j.gie.2017.10.043 [DOI] [PubMed] [Google Scholar]
- Hirschman BA, Pollock BH, Tomlinson GE. 2005. The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 147: 263–266. 10.1016/j.jpeds.2005.04.019 [DOI] [PubMed] [Google Scholar]
- Levesque S, Ahmed N, Nguyen VH, Nahal A, Blumenkrantz M, Puligandla P, Chong G, Foulkes WD. 2010. Neonatal Gardner fibroma: a sentinel presentation of severe familial adenomatous polyposis. Pediatrics 126: e1599–e1602. 10.1542/peds.2010-1045 [DOI] [PubMed] [Google Scholar]
- National Comprehensive Cancer Network (NCCN). 2018. Genetic/familial high-risk assessment: colorectal. Retrieved December 15, 2018, from https://www.nccn.org/professionals/physician_gls/default.aspx
- Nelson S, Näthke IS. 2013. Interactions and function of the adenomatous polyposis coli protein at a glance. J Cell Sci 126: 873–877. 10.1242/jcs.100479 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Gene Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Septer S, Lawson CE, Anant S, Attard T. 2016. Familial adenomatous polyposis in pediatrics: natural history, emerging surveillance and management protocols, chemopreventive strategies, and areas of ongoing debate. Fam Cancer 15: 477–485. 10.1007/s10689-016-9905-5 [DOI] [PubMed] [Google Scholar]
- Slowik V, Attard T, Dai H, Shah R, Septer S. 2015. Desmoid tumors complicating familial adenomatous polyposis: a meta-analysis mutation spectrum of affected individuals. BMC Gastroenterol 15: 84 10.1186/s12876-015-0306-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soravia C, Berk T, McLeod RS, Cohen Z. 2000. Desmoid disease in patients with familial adenomatous polyposis. Dis Colon Rectum 43: 363–369. 10.1007/BF02258303 [DOI] [PubMed] [Google Scholar]
- Spector LG, Birch J. 2012. The epidemiology of hepatoblastoma. Pediatr Blood Cancer 59: 776–779. 10.1002/pbc.24215 [DOI] [PubMed] [Google Scholar]
- Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW, American College of Gastroenterology. 2015. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 110: 223–262. 10.1038/ajg.2014.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, Pinto C, Afonso M, do Bom Sucesso M, Lopes P, Pinheiro M, Veiga I, Henrique R, Teixeira MR. 2015. Identification of previously unrecognized FAP in children with Gardner fibroma. Eur J Hum Genet 23: 715–718. 10.1038/ejhg.2014.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrli BM, Weiss SW, Yandow S, Coffin CM. 2001. Gardner-associated fibromas (GAF) in young patients: a distinct fibrous lesion that identifies unsuspected Gardner syndrome and risk for fibromatosis. Am J Surg Pathol 25: 645–651. 10.1097/00000478-200105000-00012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This variant was submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) NM_000038.5 (APC):c.4666dup (p.Thr1556Asnfs) by Mayo Medical Laboratories submission access SCV000691753.1.