

Abstract
Adrenocortical carcinoma is a rare malignancy with a poor prognosis and few treatment options. Molecular characterization of this cancer remains limited. We present a case of an adrenocortical carcinoma (ACC) in a 37-yr-old female, with dual lung metastases identified 1 yr following commencement of adjuvant mitotane therapy. As standard therapeutic regimens are often unsuccessful in ACC, we undertook a comprehensive genomic study into this case to identify treatment options and monitor disease progress. We performed targeted and whole-genome sequencing of germline, primary tumor, and both metastatic tumors from this patient and monitored recurrence over 2 years using liquid biopsy for ctDNA and steroid hormone measurements. Sequencing revealed the primary and metastatic tumors were hyperhaploid, with extensive loss of heterozygosity but few structural rearrangements. Loss-of-function mutations were identified in MSH2, TP53, RB1, and PTEN, resulting in tumors with mismatch repair signatures and microsatellite instability. At the cellular level, tumors were populated by mitochondria-rich oncocytes. Longitudinal ctDNA mutation and hormone profiles were unable to detect micrometastatic disease, consistent with clinical indicators of disease remission. The molecular signatures in our ACC case suggested immunotherapy in the event of disease progression; however, the patient remains free of cancer. The extensive molecular analysis presented here could be applied to other rare and/or poorly stratified cancers to identify novel or repurpose existing therapeutic options, thereby broadly improving diagnoses, treatments, and prognoses.
Keywords: adrenocortical carcinoma
INTRODUCTION
Adrenocortical carcinoma (ACC) is an extremely rare malignancy, with an incidence of 0.7–2.0/million/year (Fassnacht et al. 2013). In 40%–60% of patients, ACC is diagnosed after the patient presents with clinical features of steroid excess (usually hypercortisolemia or Cushing's syndrome). Thirty percent of patients may present with an abdominal mass, and some 20%–30% of ACC may be incidental findings on unrelated imaging (Libé 2015). Surgery is the primary treatment modality, often followed by adjuvant radiotherapy to the adrenal bed, mitotane or cytotoxic drugs, or a combination thereof (Fassnacht et al. 2013). Pathology scoring relies heavily on the Weiss system, and the proliferation marker Ki67, where <10% or >10% Ki67 positivity, corresponds to low- and high-grade tumors respectively (Libé 2015). The Weiss system, assessed on a scale of 1–9, provides guidelines for differentiating adrenocortical adenoma from ACC. Studies have demonstrated an association between the Weiss score and survival (van't Sant et al. 2007). For instance, patients who score ≤3 have better prognoses (Lucon et al. 2002; Libé 2015). Stage I ACC are ≤5 cm, are strictly localized to the adrenal, and have a 5-yr survival of 81% (Fassnacht et al. 2013); however, just 3%–6% of patients present at this stage (Loncar et al. 2015). Stage II ACC are localized but >5 cm. In Stage III, tumor invades surrounding lymph nodes or the renal vein, whereas Stage IV represents metastatic disease (Fassnacht et al. 2013). Five-year survival declines dramatically with increasing stage—being 61% in Stage II, 50% in Stage III, and 13% in Stage IV (Fassnacht et al. 2013).
To date, mitotane is the only drug approved for adjuvant therapy of ACC, although there are no guidelines and poor consensus for its use (Atallah et al. 2017). It is a cytostatic and antineoplastic agent that works at the mitochondrial level to suppress steroidogenesis, but it is difficult to manage because of its long half-life, dose-limiting toxicity, and severe side effects (Berruti et al. 2012). Its adrenolytic activity means all patients require concomitant glucocorticoids (Berruti et al. 2012). Mitotane is recommended in patients with Ki67 of >10% and tumor size >8 cm, and those with potential residual disease following resection (Berruti et al. 2012). However, there is a low complete response rate (<10%) to treatment (Reidy-Lagunes et al. 2017). Patients with metastatic disease receive conventional chemotherapy, either a combination of streptozocin plus mitotane, or etoposide, doxorubicin, and cisplatin plus mitotane. Response rates are poor; 23% for the latter treatment versus 9% for the former, with little long-term benefit and significant side effects (Fassnacht et al. 2012).
The lack of adjuvant therapies and poor response rates to targeted therapies such as linsitinib (Fassnacht et al. 2015), sunitinib (Kroiss et al. 2013), and bevacizumab plus capecitabine (Wortmann et al. 2010) are exacerbated by the dearth of preclinical models, as well as limited molecular stratification of these malignancies (Hantel et al. 2016). Genomic stratification of common cancers has helped inform diagnoses (predominantly from a risk stratification perspective) and inform novel treatment options that have led to a marked improvement in prognoses or the quality and duration of life in some patients. In landmark studies, next-generation sequencing (NGS) identified therapeutically actionable mutations in 76% of 2221 tumor samples of various types studied and has also facilitated successful repurposing of existing therapeutics (Bang et al. 2010; Frampton et al. 2013). Over time, however, many of these targeted therapies have become ineffective because of ongoing tumor evolution leading to the eventual acquisition of tumor resistance. For instance, 20% of melanoma patients sustained a response to MAPK-targeting treatments, whereas another subset of patients had life extended by 10 yr with the checkpoint inhibitor ipilimumab (Lim et al. 2018), but after 12 mo resistance to the therapies emerged in most patients. Targetable genetic changes are highly likely to be detected in other less common malignancies also, but like their common-cancer counterparts, the buildup of resistance needs to be countered.
This may potentially be mediated through liquid biopsy for circulating tumor DNA (ctDNA), which is also emerging as a powerful tool for the real-time monitoring of tumor burden or for early detection of cancer initiation or recurrence (Dawson et al. 2013; Wang and Bettegowda 2017 Lim et al. 2018; O'Leary et al. 2018). Small surgical core biopsies are spatiotemporally constrained, informing a limited choice of targeted therapeutic options and allowing missed tumoral subclones to develop, leading to drug resistance. Liquid biopsy potentially negates this, by providing a global picture of all mutations in real time and informing broader treatment plans (Lim et al. 2018). Recent studies have applied NGS and liquid biopsy to improve the molecular stratification of ACC, as well as monitor tumor burden and its response to treatment (Creemers et al. 2017; Armignacco et al. 2018; Garinet et al. 2018).
At the germline level, mutations in TP53 are found in 7.5% of ACC cases generally, but much more frequently (50%–97%) in childhood ACC (Raymond et al. 2013). Further germline mutations have been identified in several genes on Chromosome 11p15 (Else 2012) and BRCA2 (El Ghorayeb et al. 2016). Somatically, dysregulation of protein kinase A and mutations in ADIPOR1, ADIPOR2, APC, ARMC5, SDHA, SDHC, SF1, and IGF2, among others, have also been implicated as mediators of ACC tumorigenesis, along with mutations in RB1 and APC (Babińska et al. 2017; Else et al. 2017). Recent expression studies have identified sonic hedgehog and somatostatin receptor pathways activated in ACC, along with markedly increased expression in cytosolic protein Stathmin1, SLC12A7 and phospho-mTOR, EGFR, and WNT/β-catenin signaling pathways (Germano et al. 2017; Aronova et al. 2018; Brown et al. 2018a,b; Hasanovic et al. 2018). At the miRNA level, miR-483-3p and miR-139-5p promote ACC aggressiveness (Agosta et al. 2018). These studies have led to the emergence of potential new drug targets or targeted therapies, such as VAV2, Nutlin-3a, and the GRP78/BiP inhibitor HA15 (Ruggiero and Lalli 2017; Hui et al. 2018; Ruggiero et al. 2018).
We hypothesized that a genomics-based strategy could identify optimal treatment options when presented with metastatic ACC. We aimed to perform this using whole-genome or targeted sequencing on primary and metastatic tissue, as well as monitor for early relapse by serial liquid biopsy for ctDNA analysis.
RESULTS
Clinical Presentation
A 37-yr-old female Caucasian patient was diagnosed with stage II, high-grade ACC in April 2014, after presenting with abdominal pain and symptoms of cortisol and androgen excess. Abdominal imaging revealed a large heterogeneous, right-sided adrenal mass, measuring 11 × 14 × 17 cm (Fig. 1A–D). Hormone measurements confirmed Cushing's syndrome and hyperandrogenism, consistent with a glucocorticoid and androgen-producing tumor (Table 1). The patient was a fragile X carrier and has a son with fragile X syndrome. There was no personal or family history of malignancy.
Figure 1.

A case with metastatic ACC. Abdominal (A) and whole-body computed tomography (CT) (C) and positron emission tomography (PET) (B) scanning in March 2014, of a large, right-sided adrenal mass (red arrow and red asterisks), which underwent immediate adrenalectomy with nephrectomy (D, white asterisk). Grading of the tumor was assessed by Ki67 (E) and Weiss scoring. The Ki67 image is presented alongside H&E (F) and programmed cell death ligand-1 (PD-L1) (G) staining. An enlarged inset image is provided for PD-L1, with the dashed line separating positively staining cells from negative cells. Follow-up (May 2015) PET (H) and CT (I) scan imaging demonstrating bilateral lung nodules (red circles).
Table 1.
ACC patient biochemistry
| Hormone | March 2014 preoperative | April 2014 postoperative | April 2015 | February 2016 | June 2017 | November 2017 | May 2018 |
|---|---|---|---|---|---|---|---|
| Cortisol (150–520 nmol/L) | 748 | 33 | 464 | 445 | 309 | 397 | |
| ACTH (0–12 pmol/L) | <2.2 | 24.9 | 14.0 | 13.7 | 17.5 | ||
| Midnight salivary cortisol (0–10 nmol/L) | 19 | ||||||
| 24 UFC (0–250 nmol/d) | 414 | ||||||
| 1 mg overnight DST | 680 | ||||||
| Low-dose DST | 505 | ||||||
| DHEAS (premenopausal 2.2–9.1 µmol/L) | 6.9 | <0.9 | <0.9 | <0.9 | 0.4 | <0.4 | <0.4 |
| Androstenedione (premenopausal 2.5–10 nmol/L) | 30 | 0.3 | 1.6 | 1.6 | 1.6 | 2.1 | 2.6 |
| Testosterone (0–1.8 nmol/L) | 4.9 | <0.3 | <0.3 | <0.3 | <0.3 | <0.3 | <0.3 |
| Free testosterone (0–24 pmol/L) | 71 | <6 | |||||
| SHBG (20–110 nmol/L) | 61 | ||||||
| 17 OH Progesterone (1.2–17 nmol/L luteal phase) | 55.9 | ||||||
| Plasma catecholamines | Normal | ||||||
| Urine catecholamines | Normal | ||||||
| Aldosterone/renin ratio | 0.5 |
Bold type indicates numbers that exceed the normal range provided in the first column. (ACTH) Adrenocorticotropic hormone, (DHEAS) dehydroepiandrosterone, (DST) dexamethasone suppression test, (SHBG) sex hormone-binding globulin, (UFC) urine-free cortisol.
A right adrenalectomy/nephrectomy was performed in April 2014, with no lymphadenopathy or distant metastases identified. Histopathology revealed high-grade ACC with lymphovascular invasion but clear margins. Ki67 index was elevated at 60%–75% and PD-L1 staining of 1% (Fig. 1E–G). PD-L1-positive cells exhibited diffuse cytoplasmic staining that was sharper at the membrane, and were larger than surrounding, PD-L1-negative cells (Fig. 1G, inset). The tumor had a Weiss score of 5. Following resection, blood and urine steroid profiling revealed reductions in the previously elevated metabolites and precursors (Tables 1 and 2), except urine TH cortisone/cortisol postoperatively, because of the patient's Hysone (hydrocortisone) treatment. The patient completed adjuvant fractionated adrenal bed radiotherapy and commenced mitotane in June 2014. Therapy was complicated by reversible hepatotoxicity and central hypothyroidism.
Table 2.
ACC patient 24-h urine steroid profile
| Hormone | March 2014 preoperative | April 2014 postoperative | June 2015 | February 2016 | October 2016 | June 2017 | November 2017 |
|---|---|---|---|---|---|---|---|
| Androsterone (1.5–12 µmol/24 h) | 13.6 | 0.3 | 0.3 | 0.8 | 0.2 | 0.1 | 0.1 |
| Etiocholanolone (1.5–12 µmol/24 h) | 37.2 | 0.7 | 0.3 | 1.1 | 2.0 | 0.6 | 0.8 |
| 5b 17A OHPNL (<1 µmol/24 h) | 14.8 | <0.1 | 0.2 | 2.1 | 0.6 | 0.1 | 0.1 |
| Pregnanetriol (0.5–3.5 µmol/24 h) | 9.1 | 0.4 | 0.1 | 2.2 | 6.2 | 0.5 | 0.7 |
| TH-11 deoxycortisol (<0.5 µmol/24 h) | 5.5 | <0.1 | 0.2 | 0.3 | 0.3 | 0.1 | 0.3 |
| TH cortisone (2.5–12 µmol/24 h) | 9.6 | 22.4 | 3.1 | 8.5 | 8.3 | 5.7 | 6.2 |
| TH cortisol (0.7–6.0 µmol/24 h) | 11.8 | 32.3 | 5.8 | 7.6 | 3.6 | 4.6 | 3.9 |
The bold type indicates numbers that exceed the normal range provided in the first column. (OHPNL) Hydroxypregnenolone, (TH) tetrahydro.
Despite maintaining low circulating and urine adrenal steroids in the year following surgery (Tables 1 and 2), single bilateral lung ACC metastases (10 mm, right middle lobe; 13 mm, left lower lobe) were identified on routine surveillance imaging in May 2015 (Fig. 1H,I), with subsequent wedge resections in June (right lung metastasis [RLM]) and July (left lung metastasis [LLM]). Following the development of metastatic disease, mitotane therapy was ceased. The patient has received no additional radiotherapy or systemic therapy. Now 3.5 yr later, urine steroid profiling and serial imaging (PET or CT) performed every 3–6 mo, have not suggested further recurrence.
Genomic Analyses and Fluorescence In Situ Hybridization (FISH)
Targeted sequencing of the primary tumor to a mean depth of 512× revealed a tumor of high cellularity/purity (85%), with a large proportion of haploid cells suggestive of a mass loss-of-heterozygosity (LOH)/haploidization event. This was confirmed by FISH, which identified 47% of cells as haploid (Supplemental Fig. S1). Only chromosomes 5, 7, 9, 19, and 21 remained diploid, and the tumor exhibited a tumor mutation burden (TMB) of 15.8 mutations/Mb.
As metastatic tissue had been derived from fresh-frozen sources, we could conduct a more extensive, WGS characterization compared to the primary tumor. As per the primary tumor, hyperhaploidy was detected in both metastases: in 77% of cells in the LLM (Fig. 2A,C; 71% pure) and 88% of cells in the RLM (Fig. 2B,D; 82% pure). Both metastatic lesions were likely derived from the same clone carrying the hyperhaploidization event seen in the primary tumor. The most apparent difference between metastases was in Chromosome 18, which was diploid in the LLM (Fig. 2A), and the RLM had a subclonal deletion (Fig. 2B). There were very few structural variants observed in either metastasis, but high levels of indels and single-nucleotide variants (SNVs), with a preponderance of C>T SNVs occurring in both (Fig. 2A,B).
Figure 2.

Both metastases are hyperhaploid. Circos plots generated following WGS of the patient's left lung metastasis (A) and right lung metastasis (B). From the outside of the Circos plots toward the center, chromosomes are first numbered from 1 to 22 and X. SNVs, C>A (blue), C>G (black), C>T (red), T>A (grey), T>C (green), and T>G (pink), are then represented as dots and ordered by their genomic location and variant allele frequency (VAF). The next ring shows the locations of small indels (orange). Next, purity-adjusted chromosomal copy number levels are represented in green (amplifications) and red (deletions) (scale 0–8). The innermost ring shows the copy number of the minor allele, highlighting that most chromosomes exhibit loss of heterozygosity (orange). The center of the plots represent structural variants including deletions (red), duplications (green), inversions (black), and translocations (blue). The hyperhaploid tumors were confirmed with fluorescence in situ hybridization probing Chromosomes 9 (red) and 22 (green). (C,D) White circles represent example haploid cells; cell nuclei are highlighted in blue.
The TMBs of the LLM and RLM were similar to the primary, at 15.1 and 17.5 mutations/Mb respectively, which are considered to be high (Fig. 3A). By comparing the mutations observed in both metastases, we identified a total of 50,289 somatic variants, of which 24,735 were shared, and 8237 and 17,317 were private to the LLM and RLM respectively (Fig. 3B). Mutational signature analysis revealed a strong signal for defective DNA mismatch repair (MMR), along with strong signals for deamination of 5-methylcytosine and polymerase η; all attributable to C>T mutations (Fig. 3B). Filtering and prioritization of the somatic variant data, revealed a 24-bp deletion, overlapping a splice acceptor in the MMR gene MSH2, as well as six functionally deleterious mutations, in IGF2R, PTPRD, PTEN, MEN1, RB1, and TP53 (Table 3). All were validated by deep targeted sequencing (data not shown). The MSH2 deletion, and RB1 and TP53 mutations were also detected in the primary tumor. The PTEN nonsense mutation detected in the metastases was not detected in the primary, whereas IGF2R, PTRPD, and MEN1, were not targeted for sequencing in the primary. A functionally deleterious missense variant, c.793C>T, p.Arg265Cys was detected in MMR gene MLH1 in the primary, but not in the metastases.
Figure 3.

Both metastases have microsatellite instability. Plot of tumor mutation burden (TMB) mutation counts/Mb for primary tumor and each metastasis (A). Phylogenetic analysis of variants shared between, or private to either metastasis, plus somatic mutation signature analysis indicating the combined and private drivers of tumor pathology (B). Microsatellite instability (MSI) analysis was conducted using Mantis, with both metastases (blue) plotted against the recommended threshold for MSI-high tumors (red dashed line) (C). This was checked by immunohistochemistry for PD-L1 (D,F). Images are presented alongside H&E stains (E,G) and in magnified insets.
Table 3.
Filtered variants of interest
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP/dbVAR ID | LOH | Tumor tissue detection (VAF) |
|---|---|---|---|---|---|---|---|---|
| IGF2R | 6:160515930 GRCh37 |
c.3949dup | p.Asp1317GlyfsTer5 | Frameshift | LOF, dominant negative | Yes | Primary (NA) Left lung metastasis (0.00) Right lung metastasis (0.70) |
|
| PTPRD | 9:8341226 GRCh37 |
c.3772G>A | p.Ala1258Thr | Missense | Probably damaging | No | Primary (NA) Left lung metastasis (0.40) Right lung metastasis (0.38) |
|
| PTEN | 10:89717672 GRCh37 |
NM_000314.6 c.697C>T |
NP_000305.3 p.Arg233Ter |
Stop gain | LOF | rs121909219 | Yes | Primary (0.00) Left lung metastasis (0.48) Right lung metastasis (0.68) |
| MEN1 | 11:64572093 GRCh37 |
NM_130799.2 c.1561delC |
NP_000235.2 p.Arg521GlyfsTer43 |
Frameshift | LOF | rs794728642 | Yes | Primary (NA) Left lung metastasis (0.00) Right lung metastasis (0.26) |
| RB1 | 13:48941648 GRCh37 |
NM_000321.2 c.958C>T |
NP_000312.2 p.Arg320Ter |
Stop gain | LOF, LOH | rs121913300 | Yes | Primary (0.91) Left lung metastasis (0.63) Right lung metastasis (0.70) |
| TP53 | 17:7577548 GRCh37 |
NM_000546.5 c.733G>A |
NP_000537.3 p.Gly245Ser |
Missense | LOF, LOH | rs28934575 | Yes | Primary (0.90) Left lung metastasis (0.58) Right lung metastasis (0.74) |
| MSH2 | 2:47672676 GRCh37 |
c.1277-9_1292del | Splice critical | LOF, LOH | Yes | Primary (0.95) Left lung metastasis (0.46) Right lung metastasis (0.63) |
||
| MLH1 | 3:37058999 GRCh37 |
NM_000249.3 c.793C>T |
NP_000240.1 p.Arg265Cys |
Missense | LOF | rs63751194 | Yes | Primary (0.03) Left lung metastasis (0.00) Right lung metastasis (0.00) |
(LOF) Loss of function, (LOH) loss of heterozygosity, (NA) not applicable, (VAF) variant allele frequency.
Microsatellite Instability (MSI) Analysis and Immunological Assessment of Tumors
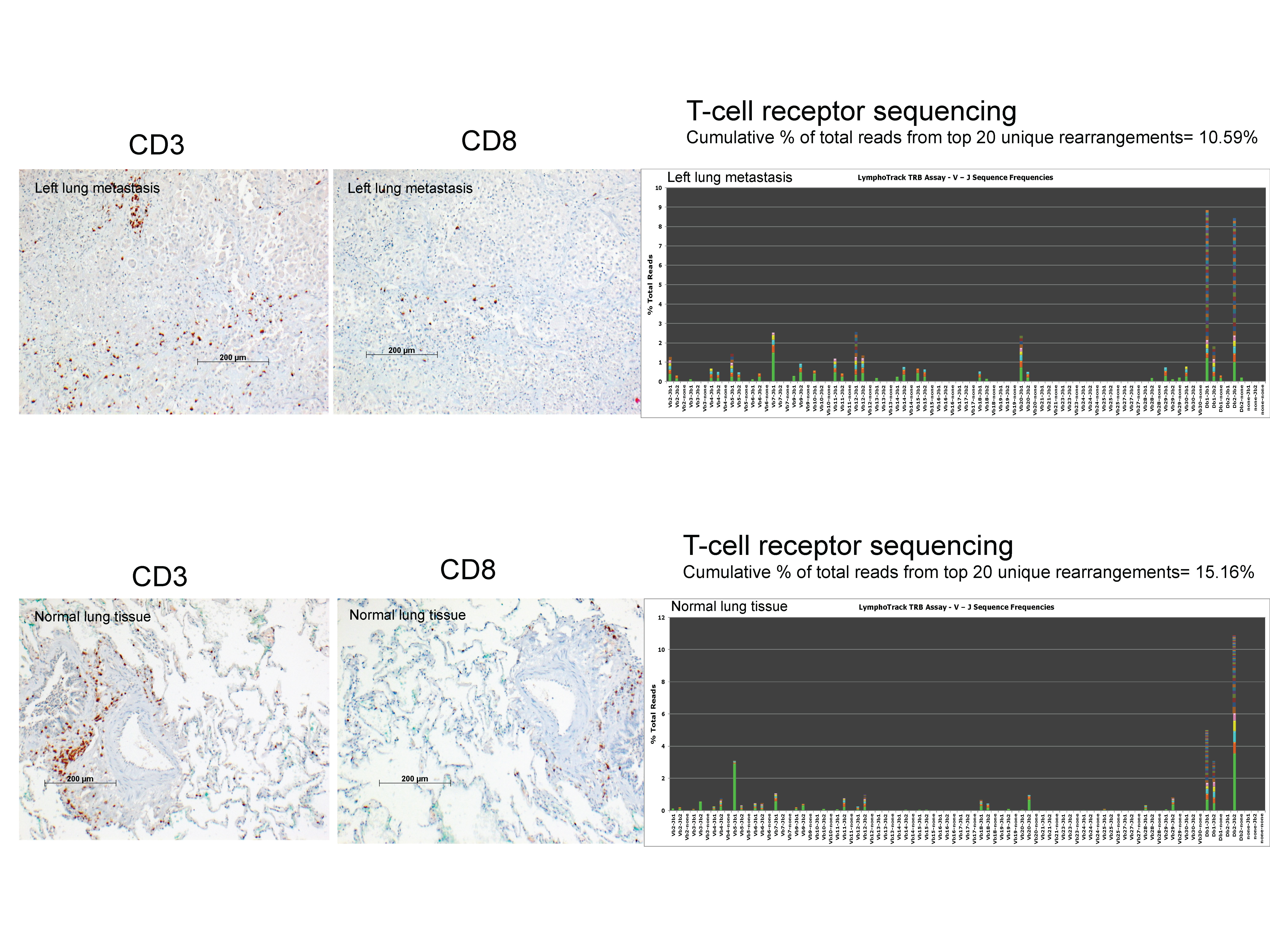
Given the deletion detected in MSH2, we assessed both metastatic tumors for MSI, and confirmed its presence in both cases (Fig. 3C). Immunohistochemical staining of metastatic tumors revealed the absence of MSH2 but the presence of MLH1, PMS2, and MSH6 (data not shown). Interestingly, subsequent immunohistochemical staining for PD-L1 revealed marked increases in staining positivity for the LLM (70%) and RLM (20%) compared to the primary (1%) (Fig. 3D–G; shown with corresponding hematoxylin and eosin [H&E] stains). As per the primary tumor, PD-L1-positive cells appeared larger than negative cells (Fig. 3D,F, insets). We next examined the LLM for the degree of T-cell infiltration by immunohistochemistry and T-cell receptor sequencing. These analyses revealed little to no immune infiltrate within the tumor (Supplemental Fig. S2).
Cellular Staining of Mitochondria
We hypothesized that the large swollen cells reported in Figures 1 and 3 were oncocytes; a common ACC cell type, characterized by an accumulation of mitochondria, and often exhibiting mass haploidization (Corver et al. 2012). Careful assessment of our patient's primary and metastatic tumor tissue revealed this to be the case, with oncocytes packed with SDHB-positive mitochondria (Fig. 4). Only one somatic variation, predicted pathogenic in silico by PolyPhen and SIFT, was observed in the mitochondrial genome—namely, MT-ND5:c.400G>A, a component of complex I.
Figure 4.

Tumor cells are oncocytic and are heavily populated with mitochondria. Cell type in the primary and metastatic tumor tissue was assessed for oncocytic potential, by size, and appearance by H&E (A,C,E), as well as for the presence of mitochondria by SDHB staining (B,D,F).
Germline Genomic Analysis
We assessed the germline for any inherited, deleterious mutations in MMR or mitochondrial genes. No variants of interest were detected in MMR genes, except apparent polymorphisms recorded in dbSNP and 1000 Genomes, in MSH3 (c.162-179del) and PMS2 (c.706-4del). Similarly, a variant of unknown significance (conflicting in silico data but reported in dbSNP), in complex III gene MT-CYB (c.20C>T), was detected. No germline mutations were detected in the commonly mutated TP53.
Liquid Biopsy
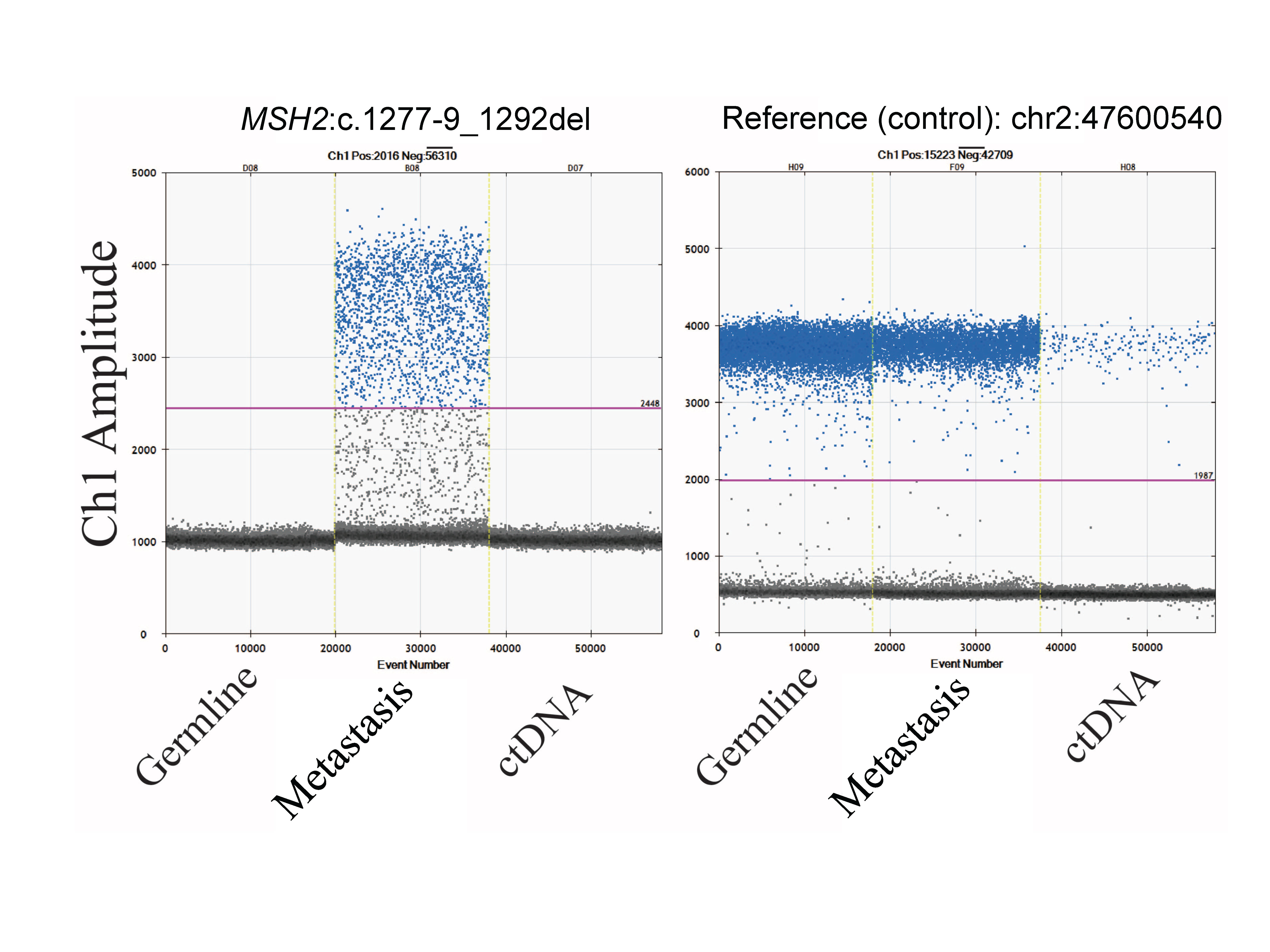
To enable early detection of cancer recurrence, we conducted a serial liquid biopsy of ctDNA, using ddPCR probes against the somatic MSH2 c.1277-9_1292del deletion, and a reference control, targeting Chr 2:47600540. The MSH2 deletion was detected only in the tumor tissue–derived DNA and not in any of the 10 subsequent liquid biopsies collected between June 2015 and November 2017 (Fig. 5; Supplemental Fig. S3). The control targeted allele was present in all tissues and liquid biopsy time points. The TP53, RB1, and MEN1 mutations were also analyzed in the germline, metastatic tumor DNA, and ctDNA and only detected in the metastases (data not shown). The data strongly support the clinical conclusion that the patient remains in remission, with no detectable signs of early disease progression.
Figure 5.

Liquid biopsy confirms patient is in remission. Liquid biopsy for ctDNA was acquired between June 2015 and November 2017. Droplet digital PCR probes were obtained to interrogate MSH2:c.1277-9_1292del (blue), in the germline, primary tumor, metastases, and across all ctDNA samples. We used a control probe to interrogate a wild-type Chr 2:47600540 (red). Presented here are the droplet digital PCR counts on a log10 scale.
DISCUSSION
We have conducted an extensive molecular characterization of tumor and serial liquid biopsies in an aggressive ACC case with two lung metastases. The primary and metastatic tumors were characterized by a mass hyperhaploidization event in the primary, and mutational signatures were associated with defects in MMR.
We hypothesize that this was driven by a driver-somatic, loss-of-function (LOF) deletion, detected in MSH2. In the primary ACC, it was found in association with a somatic missense mutation in MLH1. Digenic alterations in both proteins have been previously reported in hereditary nonpolyposis colorectal tumors (Wei et al. 2011); however, the MLH1 mutation leads to a functional protein, albeit at reduced capacity compared to wild type (Perera and Bapat 2008). Both proteins are critical for normal MMR, although MSH2, as part of the MutS complex, is essential for single base pair repairs and DNA loop repairs encompassing indels. It is the latter function that can be associated with DNA slippage and consequently, MSI, as seen in the colorectal tumors above and others, including the metastatic ACC case presented herein (Cortes-Ciriano et al. 2017). Furthermore, LOF MSH2 deletions, induce a “mutator phenotype,” which leads to (i) disrupted methylation of CpG islands, possibly accounting for the deamination of 5-methylcytosine signature, usually observed in aging individuals (Tubbs and Nussenzweig 2017), and (ii) the elevated accumulation of mutations in oncogenes and tumor suppressor genes, including often in TP53 (Xiniarianos et al. 2002).
p53 is a transcription factor considered the guardian of the genome, being implicated in regulating genes that control cell cycle and death, maintaining genome stability, and mediating cellular responses to genotoxic insults and hypoxia, among other functions (Menendez et al. 2006). It is essential for arresting the cell cycle during G1, to allow for necessary DNA repair, prior to DNA replication (Menendez et al. 2006). Our patient's somatic, mutational-hotspot TP53 (p.Gly245Ser) variant, is known to disrupt DNA binding and, consequently, transcription (Lepre et al. 2017). Dysfunctional p53 allows the cycle cell to continue unabated, even in the presence of erroneously mutated DNA (Menendez et al. 2006; Lepre et al. 2017). pRB is also essential for controlling the cell cycle and is the third most mutated protein in ACC (Ragazzon et al. 2014). Our patient's LOF mutations in RB1 and TP53 may allow for the uncontrolled proliferation of cancerous lesions.
Intriguingly, p53 is also essential for mitochondrial genome stability, as well as mitochondrial respiration and energy production. Normal cells under aerobic conditions rely heavily on oxidative phosphorylation (OXPHOS), which for every molecule of pyruvate generates 36 molecules of adenosine triphosphate (ATP) (Zheng 2012). The alternative pathway is glycolysis, which produces just two ATPs under anaerobic conditions. Over the last 10 years, there has been renewed interest in energy metabolism in cancer, and emerging data is challenging the decades-old dogma that there is a preference to activate the glycolysis pathway while shutting down OXPHOS (for reviews, see Pavlova and Thompson 2016; Corbet and Feron 2017; Anderson et al. 2018). Indeed, the majority of cancers do rely upon glycolysis to promote proliferation over differentiation (Fernandez-de-Cossio-Diaz and Vazquez 2017; Yu et al. 2017), as compared to OXPHOS, it is quicker and generates more metabolites that are used as building blocks downstream for nucleic acids, proteins, and lipids (Zheng 2012; Yu et al. 2017). Further, glycolysis appears to be essential for cancer survival, as too much ATP generated by OXPHOS inhibits glycolysis and may result in more reactive oxygen species (ROS), which are dangerous to cancer cells (Zheng 2012; Anderson et al. 2018; Cameron et al. 2018). However, increasing data show that some cancers rely upon functioning OXPHOS and that this pathway is not entirely dependent upon glucose-derived pyruvate to function, and can, in fact, decouple itself from glycolysis. The citric acid cycle can feed off lactate, fatty acids, and amino acids such as glutamine to maintain OXPHOS in cancer (Pavlova and Thompson 2016; Corbet and Feron 2017). Metabolic processes in cancer, including that of the patient presented herein, require deeper investigation. However, based on our available data, we hypothesize that a metabolic switch has occurred from OXPHOS to glycolysis in our patient. The reasons are twofold. First, mitochondrial DNA is susceptible to OXPHOS-produced ROS, yet our patient exhibits only one somatic mutation. Second, our patient exhibits dysfunctional p53 and the somatic p.Arg233Ter mutation in PTEN. Both are known to induce a “hypoxia”-like microenvironment that favors glycolysis while suppressing OXPHOS (Lu et al. 2009; Zheng 2012; He et al. 2013; Anderson et al. 2018). The evidence for the latter point in the literature is consequent activation of HIF1α, which induces transcription factors to facilitate adaptation to a hypoxic environment, as well as up-regulating nine of 10 glycolysis enzymes and blocking OXPHOS by inhibiting the conversion of pyruvate to acetyl CoA (Zheng 2012). Normally, p53 inhibits HIF1α expression, inhibits glycolysis, and increases SCO2 expression in the inner mitochondrial membrane, leading to increased OXPHOS (Zheng 2012; Anderson et al. 2018). Our patient's mutations in TP53 and PTEN have also been shown to induce activation of oncogene AKT, another factor which stimulates expression of HIF1α (Zheng 2012).
Active glycolysis may account for the qualitatively large presence of mitochondria that we observed in our swollen oncocytes, which may have occurred to compensate for the mass reduction in energy production due to the postulated suppression of OXPHOS, as described elsewhere (Corver et al. 2012). The cell cycle is an energy-intensive process, particularly for DNA replication and cell division. The lack of energy production may disturb mitotic spindle formation and function, resulting in unbalanced mitosis, with loss of chromosomes, as postulated by Corver et al. (2012). As multiple cell divisions occur, more chromosomes are lost, until the genome becomes near-haploid, which may be less energy-intensive to maintain than a diploid genome (Corver et al. 2012). Alternatively, mass haploidization/LOH may occur earlier than mitosis, with LOH having been observed to occur early in DNA replication (Pedersen and De 2013). It has been hypothesized that entering a largely haploid state is a cancer-survival mechanism (Corver et al. 2012). It is noteworthy that in our patient, several chromosomes remained diploid, similar to those reported previously (Corver et al. 2012). This may provide a tumor survival advantage, such as for Chromosome 7, which contains oncogenes cMET, EGFR, and BRAF (Corver et al. 2012). Similar oncocytic phenotypes are observed in disparate cancer types; however, they are rare overall, which is consistent with the various mutations and metabolic preferences that occur across cancer.
With MSH2 as the postulated driver behind these complex series of events in our patient, we applied serial liquid biopsies between June 2015 (first metastatic resection) and November 2017 to monitor for relapse. Two other studies have applied liquid biopsy to ACC with very limited success. Creemers et al. (2017), detected somatic mutations in just one of the three patients with ACC, and observed decreases in mutation load following surgery in that patient. Garinet et al. (2018) similarly was able to track mutations in the ctDNA in just two of the eight patients with ACC. This may have been due to the larger tumor load, leading to higher levels of detectable ctDNA; however, tumor load was also high in cases where mutations were not detected (Garinet et al. 2018). Garinet et al. (2018) postulated that this could be due to (i) slow progression of the tumor, (ii) low quantities of input ctDNA, or (iii) that ACC may be a poor secretor of ctDNA, such as seen in glioma or renal cell carcinoma (Bettegowda et al. 2014). In the latter cancer type, a significant relationship was observed between ctDNA detectability and radiographic tumor burden in 34 patients, but not between ctDNA detectability and treatment response (Maia et al. 2017). Larger cohort studies are needed to confirm data for renal cell carcinoma, and we suggest the same is required for ACC before the utility of liquid biopsy in this cancer type is confirmed. Given the issues of ctDNA sensitivity outlined above, we chose ddPCR to sensitively monitor for the MSH2 mutation, rather than NGS. ddPCR confirmed that the patient remained in remission. Our techniques were sound through the ability to detect the reference control allele in every germline, tumor, and ctDNA sample. It would have been preferable to obtain liquid biopsy at the time of primary ACC resection when the tumor burden would have been high. Unfortunately, blood was not preserved for the study herein at that time.
It is quite remarkable that our patient remains in remission. As stated previously, survival rates for ACCs are very low. Further, our patient had a high-grade Ki67 proliferation score of 60%–75% and a Weiss score of 5. In one study, 38% of patients with a Weiss score of >3 had passed away within 5 yr (Lucon et al. 2002). However, long-term survival is not unheard of in the literature, with cases of nonmetastatic disease, through to recurrent disease, surviving from 12 to 28 yr (Hermsen et al. 2008). Immunologically, our results suggest a tumor intrinsic induction of immune invasion as demonstrated by the constitutive activation of PD-L1 and lack of tumor-infiltrating lymphocytes. Such tumors have been defined as type III by Teng et al. (2015). Although speculative, the presence of high TMB, MSI, strong PD-L1 staining, but low T-cell numbers suggests there may be a rationale for this patient to receive a combination treatment in the event of relapse that would recruit T lymphocytes into the tumor microenvironment (e.g., anti-CTLA4 [ipilimumab]), but also inhibit PD-L1 if progression had occurred (e.g., anti-PD-1 [pembrolizumab]) (Pai et al. 2017; Eggermont et al. 2018).
Our data, however, uncover further interesting pathways that could be explored for targeted drug therapy particularly in the ACC setting, including mitochondrial metabolism and eventually p53. As targeted drug development and liquid biopsy continue to expand, with the shift from treating cancer based on its tissue of origin to its molecular profile (personalized medicine), therapeutic options against these targets are becoming, or may become, available (Porporato et al. 2018). This is desperately needed for ACC, as mitotane is the only drug approved for its treatment, and response rates are very low.
In conclusion, we postulate that the somatic deletion in MMR gene MSH2, in a patient who presented with primary ACC and dual lung metastases, has induced a mutator phenotype and MSI. These events were associated with deactivation of p53 and PTEN, which has led to the change of mitochondrial energy production to favor glycolysis over OXPHOS. In turn, there has been an accumulation of mitochondria within the cancerous cells to compensate for the energy reduction, leading to mass haploidization/LOH. Follow-up experimentation is needed to confirm this hypothesis. Liquid biopsy has confirmed the clinical data that our patient remains in remission; however, combination immunotherapy appears to be the preferred treatment option in the event of relapse. Genomic stratification of rare/poorly studied malignancies is an effective means of identifying new therapeutic options, which, when applied with liquid biopsy, could help improve treatment plans and prognoses.
METHODS
Next-Generation Sequencing
Primary ACC tissue had previously been archived in formalin-fixed, paraffin-embedded (FFPE) blocks. Genomic DNA was extracted at Garvan Molecular Genetics, using QIAGEN's DNeasy kit (69504). Library preparation and targeted capture of 170 genes were conducted using the TruSight Tumor 170 Kit (Illumina OP-101-1003), with sequencing then performed using Illumina's NextSeq 500 System to 512× depth (Supplemental Table S2) at the Kinghorn Centre for Clinical Genomics (KCCG).
For normal or metastatic tissues, DNA was extracted from fresh-frozen tissues using the AllPrep DNA/RNA/Protein Mini Kit (QIAGEN 8004) according to the manufacturer's instructions. Whole-genome sequencing (WGS) to intended coverage depths of 30× (normal tissue) and 90× (metastatic tumor tissue) (Supplemental Table S2) was performed using Illumina's HiSeq X platform at KCCG.
Bioinformatics
Targeted Sequencing of Primary Tumor
Raw fastq files were uploaded to BaseSpace before being transferred to the cloud-based genomic analysis platform, DNAnexus (www.dnanexus.com). Paired-end short reads were aligned to the hs37d5 reference genome, using BWA-MEM (v0l7.10-r789) (Li and Durbin 2009) and duplicates marked using Novosort (default settings), and then processed according to a series of quality scores (Supplemental Table S1). Following the Genome Analysis Toolkit (GATK) best practices guidelines (Van der Auwera et al. 2013), read alignment and quality were improved using GATK IndelRealigner and GATK BaseRecalibrator (v3.3) (Van der Auwera et al. 2013). SNVs and insertions/deletions (indels) were called using VarDictJava (v1.4.6) (Lai et al. 2016), with copy number variants (CNVs) determined using CNVkit (v0.9.1), against a pooled reference panel of 60 FFPE germline controls (Talevich et al. 2016). Tumor purity was assessed using PureCN (v1.10.0) with R (v3.6) (Riester et al. 2016) prior to annotation with Ensembl Variant Effect Predictor (VEP, v87).
WGS of Germline and Metastatic Tumors
Raw fastq files were processed through BWA-MEM and GATK as above. Germline SNVs and short (<50-bp) indels were identified using GATK HaplotypeCaller (v3.3) in GVCF mode (Van der Auwera et al. 2013), with somatic variants identified using Strelka (v1.0.14) (Saunders et al. 2012). Variants were then annotated with VEP, converted into a database using GEMINI (Paila et al. 2013), and imported into Seave (Gayevskiy et al. 2018) for filtration and prioritization. This was performed according to <1% frequency in 1000 Genomes and Exome Aggregation Consortium (ExAC) databases, conservation (assessed by GERP), functional impact (PolyPhen-2 and SIFT), biological likelihood through candidate gene and pathway analysis, and association with known Catalogue of Somatic Mutations in Cancer (COSMIC) variants.
Tumor purity, CNVs, and structural variants were identified using PURPLE (https://github.com/hartwigmedical/hmftools/tree/master/purity-ploidy-estimator) (v1.1.0) and Manta (v0.27.1), respectively (Chen et al. 2016), and then annotated using Ensembl genes. SomaticSignatures (v.2.16.0) (Gehring et al. 2015) was used to link observed SNVs to mutation generating processes, and assessment of MSI was conducted using Mantis (v1.0.3) (Kautto et al. 2017).
Bcftools intersect (https://samtools.github.io/bcftools/bcftools.html) was used to compare the two metastases for conserved and novel variants.
T-Cell Receptor Sequencing
Normal- and tumor-derived DNA was analyzed for T-cell clonality for TCR-β rearrangements using the Lymphotrack TRB assay panel (Invivoscribe; cat. no. 72250019) according to manufacturer's instructions, with libraries sequenced by using Illumina's MiSeq v2 (500 cycle). A clonality estimate was calculated from the sum of the top 20 unique TCR-β rearrangements.
Fluorescence In Situ Hybridization
Ploidy assessment was conducted according to manufacturer's instructions, using the ZytoLight SPEC BCR/ABL1 Dual Color Dual Fusion Probe, targeting t(9;22)(9q34.1;q11.2) (Zytovision Z-2111-200).
Immunohistochemistry
All immunohistochemistry was conducted on 5-µm sections of tumor and normal tissue. Morphological analysis was assessed using H&E, before analysis of any tumor immunosuppression activity, using the 22C3 pharmDx Kit (Agilent) against programmed cell death ligand-1 (PD-L1), according to manufacturer's instructions. T-cell infiltration was assessed with anti-CD3 (Roche 790-43141) and CD8 (Agilent M7103) antibodies according to the manufacturer's instructions, as was qualitative assessment of mitochondrial staining using a mouse monoclonal antibody against SDHB (Abcam ab14714).
Liquid Biopsy
Blood samples were obtained from the patient quarterly between June 2015 and November 2017, into Streck BCT tubes (Streck), with immediate separation of plasma. ctDNA was immediately extracted from 4 mL plasma aliquots using the Circulating Nuclei Acid Kit (QIAGEN 55114) according to manufacturer's instructions, and frozen at −80°C. Somatic variants of interest were probed by droplet digital PCR (ddPCR), using the Bio-Rad QX200 Droplet Digital PCR System. Probes were obtained from Thermo Fisher Scientific.
ADDITIONAL INFORMATION
Data Deposition and Access
Genomic data have been deposited at the European Genome-phenome Archive (EGA; http://www.ebi.ac.uk/ega/) under accession number EGA00001003497.
Ethics Statement
Blood and tumor samples were obtained from the patient with informed consent and with prior ethics approval, granted through St. Vincent's Hospital Sydney Human Research Ethics Committee, NSW, Australia (protocol X13-0109).
Acknowledgments
We would like to thank Mr. Peter Priestley and the Kinghorn Centre for Clinical Genomics for technical assistance. The research was undertaken with assistance and services from the National Computational Infrastructure, which is supported by the Australian Government.
Author Contributions
This work was conducted in the Kinghorn Centre of Clinical Genomics’ Tumour Genomics laboratory and Garvan's Hormones and Cancer Group, led by M.E.D., A.I.M., and M.J.C. M.J.M., M.P., and M.J.C. led the study design of this project, carrying out most of the genomics analyses and interpretation. M.J.M. produced this manuscript and led the processing of germline and metastatic tumor samples with C.-L.C., and the design, development, and interpretation of liquid biopsy assay, under the expert direction of Y.Y.C. and R.C.Y.L. S.Q.W. performed the TCR sequencing. E.H. processed the primary tumor sample. M.P. conducted initial genomics analyses and characterization and identified the therapeutic option. N.S. collated all clinical reports and aided in the write-up of clinical data. T.J.T. extracted DNA from patient blood or tumor tissue, and J.G., M.W., M.-E.A.G., C.P., and V.G. expertly aided in further interpretation of genomic data, with V.G. also aiding with genomic filtering and prioritizing using his recently published platform, Seave. S.Q.W., W.B., P.E., and R.L. conducted pathology staining and/or provided expert reports and interpretations for H&E, PD-L1, CD3, CD8, SDHB, Ki67, and FISH. D.M.T., D.N.W., and A.I.M. provided expert clinical guidance and interpretations of the data.
Funding
This work was supported by a Cancer Institute NSW Early Career Fellowship (grant number 13/ECF/1-46), Cancer Institute NSW Career Development Fellowship (grant number 2019/CDF004), NSW Health Early-Mid Career Fellowship, Cancer Council NSW (grant number RG 15-19), and philanthropic donations from the Kinghorn Foundation.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Enzo Lalli
Ralph DeBerardinis
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Agosta C, Laugier J, Guyon L, Denis J, Bertherat J, Libé R, Boisson B, Sturm N, Feige JJ, Chabre O, et al. 2018. MiR-483-5p and miR-139-5p promote aggressiveness by targeting N-myc downstream-regulated gene family members in adrenocortical cancer. Int J Cancer 143: 944–957. 10.1002/ijc.31363 [DOI] [PubMed] [Google Scholar]
- Anderson NM, Mucka P, Kern FH. 2018. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9: 216–237. 10.1007/s13238-017-0451-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armignacco R, Cantini G, Canu L, Poli G, Ercolino T, Mannelli M, Luconi M. 2018. Adrenocortical carcinoma: the dawn of a new era of genomic and molecular biology analysis. J Endocrinol Invest 41: 499–507. 10.1007/s40618-017-0775-y [DOI] [PubMed] [Google Scholar]
- Aronova A, Min IM, Crowley MJP, Panjwani SJ, Finnerty BM, Scognamiglio T, Liu YF, Whitsett TG, Garg S, Demeure MJ, et al. 2018. STMN1 is overexpressed in adrenocortical carcinoma and promotes a more aggressive phenotype in vitro. Ann Surg Oncol 25: 792–800. 10.1245/s10434-017-6296-2 [DOI] [PubMed] [Google Scholar]
- Atallah S, Al-Assaf H, Xu Y, El-Sayed S. 2017. Adrenocortical carcinoma: patterns of care and role of adjuvant radiation therapy—a population-based study and review of the literature. Curr Oncol 24: e316–e322. 10.3747/co.24.3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babińska A, Pęksa R, Wiśniewski P, Świątkowska-Stodulska R, Sworczak K. 2017. Diagnostic and prognostic role of SF1, IGF2, Ki67, p53, adiponectin, and leptin receptors in adrenal cortical tumors. J Surg Oncol 116: 427–433. 10.1002/jso.24665 [DOI] [PubMed] [Google Scholar]
- Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. 2010. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376: 687–697. 10.1016/S0140-6736(10)61121-X [DOI] [PubMed] [Google Scholar]
- Berruti A, Baudin E, Gelderblom H, Haak HR, Propiglia F, Fassnacht M, Pentheroudakis G, EMSO Guidelines Working Group. 2012. Adrenal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 23: vii131–vii138. 10.1093/annonc/mds231 [DOI] [PubMed] [Google Scholar]
- Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. 2014. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6: 224ra24 10.1126/scitranslmed.3007094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TC, Murtha TD, Rubinstein JC, Korah R, Carling T. 2018a. SLC12A7 alters adrenocortical carcinoma cell adhesion properties to promote an aggressive invasive behavior. Cell Commun Signal 16: 27–39. 10.1186/s12964-018-0243-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TC, Nicolson NG, Korah R, Carling T. 2018b. BCL9 upregulation in adrenocortical carcinoma: a novel Wnt/β-catenin signaling activating even driving adrenocortical malignancy. J Am Coll Surg 226: 988–995. 10.1016/j.jamcollsurg.2018.01.051 [DOI] [PubMed] [Google Scholar]
- Cameron ME, Yakovenko A, Trevino JG. 2018. Glucose and lactate transport in pancreatic cancer: glycolytic metabolism revisited. J Oncol 2018: 1–6. 10.1155/2018/6214838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. 10.1093/bioinformatics/btv710 [DOI] [PubMed] [Google Scholar]
- Corbet C, Feron O. 2017. Cancer cell metabolism and mitochondria: nutrient plasticity for TCA cycle fueling. Biochim Biophys Acta Rev Cancer 1868: 7–15. 10.1016/j.bbcan.2017.01.002 [DOI] [PubMed] [Google Scholar]
- Cortes-Ciriano I, Lee S, Park WT, Kim TM, Park PJ. 2017. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 8: 15180–15191. 10.1038/ncomms15180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corver WE, Ruano D, Weijers K, den Hartog WC, van Nieuwenhuizen MP, de Miranda N, van Eijk R, Middeldorp A, Jordanova ES, Oosting J, et al. 2012. Genome haploidisation with Chromosome 7 retention in oncocytic follicular thyroid carcinoma. PLoS One 7: e38287–e38298. 10.1371/journal.pone.0038287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creemers SG, Korpershoek E, Atmodimedjo PN, Dinjens WNM, van Koetsveld PM, Feelders RA, Hofland LJ. 2017. Identification of mutations in cell-free circulating tumor DNA in adrenocortical carcinoma: a case series. J Clin Endocrinol Metab 102: 3611–3615. 10.1210/jc.2017-00174 [DOI] [PubMed] [Google Scholar]
- Dawson SJ, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, et al. 2013. Analysis of circulating tumor DNA to monitor breast cancer. N Engl J Med 368: 1199–1209. 10.1056/NEJMoa1213261 [DOI] [PubMed] [Google Scholar]
- Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A, Carlino MS, et al. 2018. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med 378: 1789–1901. 10.1056/NEJMoa1802357 [DOI] [PubMed] [Google Scholar]
- El Ghorayeb E, Grunenwald S, Nolet S, Primeau V, Côté S, Maugard CM, Lacroix A, Gaboury L, Bourdeau I. 2016. First case report of an adrenocortical carcinoma caused by a BRCA2 mutation. Medicine 95: e4756–e4759. 10.1097/MD.0000000000004756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Else T. 2012. Association of adrenocortical carcinoma with familial cancer susceptibility syndromes. Mol Cell Endocrinol 35: 66–70. 10.1016/j.mce.2011.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Else T, Lerario AM, Everett J, Haymon L, Wham D, Mullane M, Wilson TL, Rainville I, Rana H, Worth AJ, et al. 2017. Adrenocortical carcinoma and succinate dehydrogenase gene mutations: an observational case series. Eur J Endocrinol 177: 439–444. 10.1530/EJE-17-0358 [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A, Jarzab B, et al. 2012. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 366: 2189–2197. 10.1056/NEJMoa1200966 [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Kroiss M, Allolio B. 2013. Update in adrenocortical carcinoma. J Clin Endocrinol Metab 98: 4551–4564. 10.1210/jc.2013-3020 [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, Kroiss M, Quinn DI, Hesseltine E, Ronchi CL, et al. 2015. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol 16: 426–435. 10.1016/S1470-2045(15)70081-1 [DOI] [PubMed] [Google Scholar]
- Fernandez-de-Cossio-Diaz J, Vazquez A. 2017. Limits of aerobic metabolism in cancer cells. Sci Rep 7: 13488–13495. 10.1038/s41598-017-14071-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. 2013. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31: 1023–1031. 10.1038/nbt.2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garinet S, Nectoux J, Neou M, Pasmant E, Joinot A, Sibony M, Orhant L, Pipoli da Fonseca J, Perlemoine K, Bricaire L, et al. 2018. Detection and monitoring of circulating tumor DNA in adrenocortical carcinoma. Endocr Relat Cancer 25: L13–L17. 10.1530/ERC-17-0467 [DOI] [PubMed] [Google Scholar]
- Gayevskiy V, Roscioli T, Dinger ME, Cowley MJ. 2018. Seave: a comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics 35: 122–125. 10.1093/bioinformatics/bty540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring JS, Fischer B, Lawrence M, Huber W. 2015. SomaticSignatures: inferring mutations signatures from single-nucleotide variants. Bioinformatics 31: 3673–3675. 10.1101/010686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano A, Rapa I, Duregon E, Votta A, Giorcelli J, Buttigliero C, Scagliotti GV, Volante M, Terzolo M, Papotti M. 2017. Tissue expression and pharmacological in vitro analyses of mTOR and SSTR pathways in adrenocortical carcinoma. Endocr Pathol 28: 95–102. 10.1007/s12022-017-9473-8 [DOI] [PubMed] [Google Scholar]
- Hantel C, Shapiro I, Poli G, Chiapponi C, Bidlingmaier M, Reincke M, Luconi M, Jung S, Beuschlein F. 2016. Targeting heterogeneity of adrenocortical carcinoma: evaluation and extension of preclinical tumor models to improve clinical translation. Oncotarget 7: 79292–79304. 10.18632/oncotarget.12685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasanovic A, Ruggiero C, Jung S, Rapa I, Signetti L, Ben Hadj M, Terzolo M, Beuschlein F, Volante M, Hantel C, et al. 2018. Targeting the multidrug transporter Patched potentiates chemotherapy efficiency on adrenocortical carcinoma in vitro and in vivo. Int J Cancer 143: 199–211. 10.1002/ijc.31296 [DOI] [PubMed] [Google Scholar]
- He X, Arrotta N, Radhakrishnan D, Wang Y, Romigh T, Eng C. 2013. Cowden syndrome–related mutations in PTEN associate with enhanced proteasome activity. Cancer Res 73: 3029–3040. 10.1158/0008-5472.CAN-12-3811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermsen IGC, Gelderblom H, Kievit J, Romijn JA, Haak HR. 2008. Extremely long survival in six patients despite recurrent and metastatic adrenal carcinoma. Eur J Endocrinol 158: 911–919. 10.1530/EJE-07-0723 [DOI] [PubMed] [Google Scholar]
- Hui W, Liu S, Zheng J, Fang Z, Ding Q, Feng C. 2018. Nutlin-3a as a novel anticancer agent for adrenocortical carcinoma with CTNNB1 mutation. Cancer Med 7: 1440–1449. 10.1002/cam4.1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautto EA, Bonneville R, Miya J, Yu L, Krook MA, Reeser JW, Roychowdhury S. 2017. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 8: 7452–7463. 10.18632/oncotarget.13918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroiss M, Quinkler M, Johanssen S, van Erp NP, Lankheet N, Pöllinger A, Laubner K, Strasburger CJ, Hahner S, Müller HH, et al. 2013. Sunitinib in refractory adrenocortical carcinoma: a phase II, single-arm, open-label trial. J Clin Endocrinol Metab 97: 3495–3503. 10.1210/jc.2012-1419 [DOI] [PubMed] [Google Scholar]
- Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, Dry JR. 2016. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 44: e108–e118. 10.1093/nar/gkw227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepre MG, Omar SI, Grasso G, Morbiducci U, Deriu MA, Tuszynski JA. 2017. Insights into the effect of the G245S single point mutation on the structure of p53 and the binding of the protein to DNA. Molecules 22: E1358–E1374. 10.3390/molecules22081358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libé R. 2015. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol 3: 45–52. 10.3389/fcell.2015.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SY, Lee JH, Diefench RJ, Kefford RF, Rizos H. 2018. Liquid biomarkers in melanoma: detection and discovery. Mol Cancer 17: 8–21. 10.1186/s12943-018-0757-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncar Z, Djukic V, Zivaljevic V, Pekmezovic T, Diklic A, Tatic S, Dundjerovic D, Olujic B, Slijepcevic N, Paunovic I. 2015. Survival and prognostic factors for adrenocortical carcinoma: a single institution experience. BMC Urol 15: 43–49. 10.1186/s12894-015-0038-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sharma LK, Bai Y. 2009. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res 19: 802–815. 10.1038/cr.2009.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucon AM, Pereira MA, Mendonça BB, Zerbini MC, Saldanha AS. 2002. Adrenocortical tumors: results of treatment and study of Weiss's score as a prognostic factor. Rev Hosp Clín Fac Med Sao Paulo 57: 251–256. 10.1590/S0041-87812002000600002 [DOI] [PubMed] [Google Scholar]
- Maia MC, Bergerot PG, Dizman N, Hsu J, Jones J, Lanman RB, Banks KC, Pal SK. 2017. Association of circulating tumor DNA (ctDNA) detection in metastatic renal cell carcinoma (mRCC) with tumor burden. Kidney Cancer 1: 65–70. 10.3233/KCA-170007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez D, Inga A, Resnick MA. 2006. The biological impact of the human master regulator p53 can be altered by mutations that change the spectrum and expression of its target genes. Mol Cell Biol 26: 2297–2308. 10.1128/MCB.26.6.2297-2308.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary B, Hrebien S, Morden JP, Beaney M, Fribbens C, Huang X, Liu Y, Bartlett CH, Koehler M, Cristofanilli M, et al. 2018. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun 9: 896–905. 10.1038/s41467-018-03215-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai SH, Carneiro BA, Chae YK, Costa RL, Kalyan A, Shah HA, Helenowski I, Rademaker AW, Mahalingam D, Giles FJ. 2017. Correlation of tumor mutational burden and treatment outcomes in patients with colorectal cancer. J Gastrointest Oncol 8: 858–866. 10.21037/jgo.2017.06.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paila U, Chapman BA, Kirchner R, Quinlan AR. 2013. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol 9: e1003153–e1003160. 10.1371/journal.pcbi.1003153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. 2016. The emerging hallmarks of cancer metabolism. Cell Metab 23: 27–47. 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, De S. 2013. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic Acids Res 41: 7615–7624. 10.1093/nar/gkt552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera S, Bapat B. 2008. The MLH1 variants p.Arg265Cys and p.Lys618Ala affect protein stability while p.Leu749Gln affects heterodimer formation. Hum Mutat 29: 332–343. 10.1002/humu.9523 [DOI] [PubMed] [Google Scholar]
- Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L. 2018. Mitochondrial metabolism and cancer. Cell Res 28: 265–280. 10.1038/cr.2017.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragazzon B, Libé R, Assié G, Tissier F, Barreau O, Houdayer C, Perlemoine K, Audebourg A, Clauser E, René-Corail F, et al. 2014. Mass-array screening of frequent mutations in cancers reveals RB1 alterations in aggressive adrenocortical carcinomas. Eur J Endocrinol 170: 385–391. 10.1530/EJE-13-0778 [DOI] [PubMed] [Google Scholar]
- Raymond VM, Else T, Everett JN, Long JM, Gruber SB, Hammer GB. 2013. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab 98: E119–E225. 10.1210/jc.2012-2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy-Lagunes DL, Lung B, Untch BR, Raj N, Hrabovsky A, Kelly C, Gerst S, Katz S, Kampel L, Chou J, et al. 2017. Complete responses to mitotane in metastatic adrenocortical carcinoma—a new look at an old drug. Oncologist 22: 1102–1106. 10.1634/theoncologist.2016-0459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riester M, Singh AP, Brannon AR, Yu K, Campbell CD, Chiang DY, Morrissey MP. 2016. PureCN: copy number calling and SNV classification using targeted short read sequencing. Source Code Biol Med 11: 13–25. 10.1186/s13029-016-0060-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero C, Lalli E. 2017. VAV2: a novel prognostic marker and a druggable target for adrenocortical carcinoma. Oncotarget 8: 88257–88258. 10.18632/oncotarget.21448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero C, Doghman-Bouguerra M, Ronco C, Benhida R, Rocchi S, Lalli E. 2018. The GRP78/BiP inhibitor HA15 synergizes with mitotane action against adrenocortical carcinoma cells through convergent activation of ER stress pathways. Mol Cell Endocrinol 474: 57–64. 10.1016/j.mce.2018.02.010 [DOI] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28: 1811–1817. 10.1093/bioinformatics/bts271 [DOI] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, Bastian BC. 2016. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 12: e1004873–e1004890. 10.1371/journal.pcbi.1004873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MW, Ngiow SF, Ribas A, Smyth MJ. 2015. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res 75: 2139–2145. 10.1158/0008-5472.CAN-15-0255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A. 2017. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168: 644–656. 10.1016/j.cell.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From FastQ data to high-confidence variants calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11.10.1–11.10.33. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Sant HP, Bouvy ND, Kazemier G, Bonier HJ, Hop WC, Feelders RA, de Herder WW, de Krijger RR. 2007. The prognostic value of two different histopathological scoring systems for adrenal cortical carcinomas. Histopathology 51: 239–245. 10.1111/j.1365-2559.2007.02747.x [DOI] [PubMed] [Google Scholar]
- Wang J, Bettegowda C. 2017. Applications of DNA-based liquid biopsy for central nervous system neoplasms. J Mol Diagn 19: 24–34. 10.1016/j.jmoldx.2016.08.007 [DOI] [PubMed] [Google Scholar]
- Wei W, Liu F, Liu L, Li Z, Zhang X, Jiang F, Shi Q, Zhou X, Sheng W, Cai S, et al. 2011. Distinct mutations in MLH1 and MSH2 genes in hereditary non-polyposis colorectal cancer (HNPCC) families from China. BMB Rep 44: 317–322. 10.5483/BMBRep.2011.44.5.317 [DOI] [PubMed] [Google Scholar]
- Wortmann S, Quinker M, Ritter C, Kroiss M, Johanssen S, Hahner S, Allolio B, Fassnacht M. 2010. Bevacizumab plus capecitabine as a salvage therapy in advanced adrenocortical carcinoma. Eur J Endocrinol 16: 349–356. 10.1530/EJE-09-0804 [DOI] [PubMed] [Google Scholar]
- Xiniarianos G, Liloglou T, Prime W, Sourvinos G, Karachistos A, Gosney JR, Spandidos DA, Field JK. 2002. P53 status correlates with the differential expression of the DNA mismatch repair protein MSH2 in non-small cell lung carcinoma. Int J Cancer 101: 248–252. 10.1002/ijc.10598 [DOI] [PubMed] [Google Scholar]
- Yu L, Chen X, Sun X, Wang L, Chen S. 2017. The glycolytic switch in tumors: how many players are involved? J Cancer 8: 3430–3440. 10.7150/jca.21125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. 2012. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (Review). Oncol Lett 4: 1151–1157. 10.3892/ol.2012.928 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
{kind=link}
{kind=link}
{kind=link}
Data Availability Statement
Genomic data have been deposited at the European Genome-phenome Archive (EGA; http://www.ebi.ac.uk/ega/) under accession number EGA00001003497.