

Abstract
We report a case of early-onset pancreatic ductal adenocarcinoma in a patient harboring biallelic MUTYH germline mutations, whose tumor featured somatic mutational signatures consistent with defective MUTYH-mediated base excision repair and the associated driver KRAS transversion mutation p.Gly12Cys. Analysis of an additional 730 advanced cancer cases (N = 731) was undertaken to determine whether the mutational signatures were also present in tumors from germline MUTYH heterozygote carriers or if instead the signatures were only seen in those with biallelic loss of function. We identified two patients with breast cancer each carrying a pathogenic germline MUTYH variant with a somatic MUTYH copy loss leading to the germline variant being homozygous in the tumor and demonstrating the same somatic signatures. Our results suggest that monoallelic inactivation of MUTYH is not sufficient for C:G>A:T transversion signatures previously linked to MUTYH deficiency to arise (N = 9), but that biallelic complete loss of MUTYH function can cause such signatures to arise even in tumors not classically seen in MUTYH-associated polyposis (N = 3). Although defective MUTYH is not the only determinant of these signatures, MUTYH germline variants may be present in a subset of patients with tumors demonstrating elevated somatic signatures possibly suggestive of MUTYH deficiency (e.g., COSMIC Signature 18, SigProfiler SBS18/SBS36, SignatureAnalyzer SBS18/SBS36).
Keywords: neoplasm of the breast, neoplasm of the pancreas, pancreatic adenocarcinoma
INTRODUCTION
Oxidative DNA damage leads to formation of 8-oxoguanine, which during DNA replication, results in 8-OxoG:A mispairing and subsequent C:G>A:T transversion mutations (Lejbkowicz et al. 2012; Aretz et al. 2013). The MUTYH DNA glycosylase excises mismatched adenine from the 8-OxoG:A complex (Cheadle and Sampson 2007). Colorectal tumors from patients with germline biallelic MUTYH aberrations and MUTYH-associated polyposis (MAP) feature base excision repair (BER) deficiency mutational signatures enriched for C:G>A:T transversion mutations (Viel et al. 2017). Novel associations of this transversion signature have also been identified in a subset of germline MUTYH carriers with pancreatic neuroendocrine tumors and adrenocortical tumors displaying somatic inactivation of the second allele (Pilati et al. 2017; Scarpa et al. 2017).
We report a case of early-onset pancreatic ductal adenocarcinoma (PDAC) in a patient harboring biallelic MUTYH germline mutations, whose tumor featured somatic mutational signatures consistent with defective MUTYH-mediated BER and the associated driver KRAS transversion mutation p.Gly12Cys. Analysis of an additional 730 advanced cancer cases (N = 731) was undertaken to determine whether the mutational signatures were also present in tumors from germline MUTYH heterozygote carriers or if instead the signatures were seen only in those with biallelic loss of function. This review of our cohort revealed two female patients with breast cancer each carrying a single heterozygous pathogenic germline MUTYH variant associated with loss of heterozygosity (LOH) in the tumor and the same somatic signatures. Therefore, we describe three cases with germline or combined germline/somatic biallelic MUTYH aberrations and C:G>A:T mutational signatures previously linked to defective MUTYH in tumors not classically associated with MAP syndrome. These novel associations illustrate that both biallelic and a monoallelic MUTYH pathogenic germline variants can contribute to the mutational landscape of an individual's cancer and should therefore be considered when elevated somatic C:G>A:T transversion signatures possibly suggestive of MUTYH deficiency (e.g., COSMIC Signature 18, SigProfiler SBS18/SBS36, SignatureAnalyzer SBS18/SBS36) are identified.
RESULTS
Genomic profiling of a PDAC case with germline biallelic compound heterozygous MUTYH variants revealed markedly elevated Catalogue of Somatic Mutations in Cancer (COSMIC) Signature 18. These findings prompted review of our entire cohort of advanced cancers (N = 731) for MUTYH status and mutational signatures. Briefly, tumor with matched normal blood whole-genome sequencing (WGS) and tumor whole-transcriptome sequencing was performed in 731 patients with advanced cancers of diverse origins (Supplemental Table S1A). Mutational signatures were analyzed using the R package signIT (Zhao et al. 2017), and the reference matrices from COSMIC (Alexandrov et al. 2013; Forbes et al. 2017), SigProfiler (Alexandrov et al. 2018), and SignatureAnalyzer (Haradhvala et al. 2018) (please refer to Methods and Supplemental Methods for details).
Biallelic Germline MUTYH Variants and Pancreatic Ductal Adenocarcinoma
Clinical History
We report a female (Patient #1) of Chinese ancestry initially diagnosed with Stage IIB distal PDAC at the age of 45 yr.
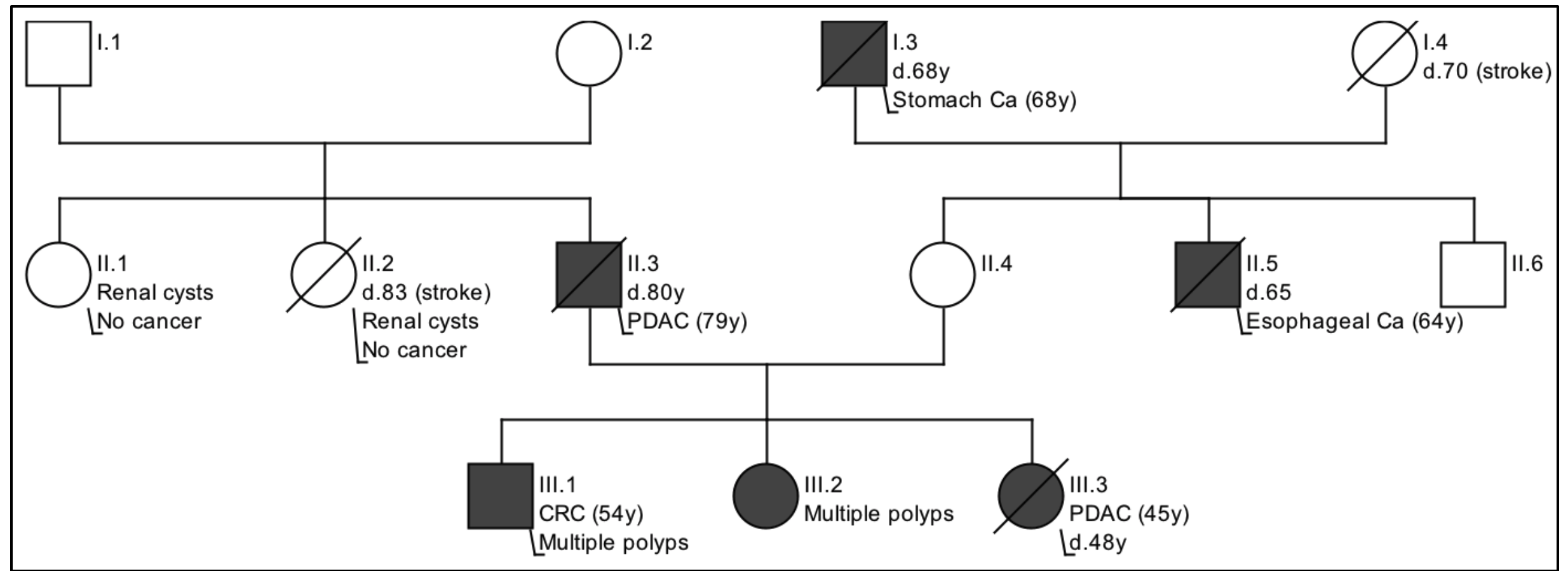
Of note, the patient had a prior colonoscopy and gastroscopy at age 43 for epigastric pain, which reported three colonic tubular adenomas, one rectal tubular adenoma, and one gastric fundic gland polyp. She was evaluated at the Hereditary Cancer Program for her early-onset PDAC and positive family history of gastrointestinal cancers. Her father was diagnosed with pancreatic cancer at the age of 79 yr. Her brother was diagnosed with colorectal cancer on a reported background of 30 tubular adenomas at the age of 54 yr (Supplemental Fig. S1 for pedigree).
Distal pancreatectomy and partial splenectomy revealed a 6-cm moderately differentiated ductal adenocarcinoma within the pancreatic tail (Supplemental Fig. S2). Margins were clear, but there was lymphovascular and perineural invasion as well as one of four lymph nodes involved. There was no evidence of intratumoral lymphocytes. The final pathologic stage was pT2N1. The patient's primary tumor was assessed for DNA mismatch repair protein expression by immunohistochemistry and intact staining was noted in the MLH1, MSH2, MSH6, and PMS2 proteins (data not shown).
She received adjuvant chemotherapy with 5-fluorouracil, oxaliplatin, and irinotecan on a clinical trial and completed all 12 planned cycles. Posttreatment imaging and CA19-9 showed no evidence of disease recurrence. Three months after completing adjuvant chemotherapy, routine surveillance CT detected signs of local recurrence. Patient #1 received radiotherapy to the pancreatic remnant, 50 Gy in 25 fractions, with concurrent capecitabine. Restaging FDG-PET performed 1 mo after completion of therapy revealed multiple liver metastases. The patient consented to participate in the Personalized OncoGenomics study and underwent ultrasound-guided liver biopsy, which confirmed the diagnosis of metastatic PDAC.
She commenced first-line palliative chemotherapy with gemcitabine and nab-paclitaxel and had an excellent radiological partial response and biochemical response after six cycles of treatment. Six months later, she was switched to nivolumab for progression, but she did not respond to immunotherapy. The patient unfortunately passed away from complications of her metastatic disease at age 48.
Germline Analysis
Clinical germline genetic testing identified compound heterozygote pathogenic variants in MUTYH (c.996G>A, p.Ser332Ser and c.815G>A, p.Gly272Glu; NM_001048171). Blood DNA WGS with analysis of 98 cancer predisposition genes also identified the MUTYH variants (Table 1) and no other pathogenic variants (Table 1; Supplemental Table S1B,C). Somatic transcriptome data showed the synonymous variant to cause abnormal splicing and the pathogenic variants to be in trans (see Somatic Analysis below).
Table 1.
MUTYH pathogenic germline single-nucleotide variants (SNVs) and somatic MUTYH status
| General | Germline | Somatic | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Id | Tumor type | Chromosome and genomic position | HGVS DNA reference | HGVS protein reference | Variant type | dbSNP | Copy category | Zygosity in tumor | DNA ALT/total | RNA ALT/total | TCGA percentile | Sig18 exposure | Sig18 proportion |
| #1 | PAAD | Chr 1:45797481C>T | c.996G>A | p.Ser332Ser | Synonymous | rs372673338 | Neutral | Heterozygous | 26/53 | 2/15 | 16 | 5683 | 0.4702 |
| #1 | PAAD | Chr 1:45797914C>T | c.815G>A | p.Gly272Glu | Missense | rs730881833 | Neutral | Heterozygous | 18/45 | 20/29 | 16 | 5683 | 0.4702 |
| #2 | IDC | Chr 1:45797760T>C | c.892-2A>G | Splice acceptor | rs77542170 | Loss | Homozygous | 33/49 | 80/121 | 56 | 830 | 0.1668 | |
| #3 | IDC | Chr 1:45797228C>T | c.1145G>A | p.Gly382Asp | Missense | rs36053993 | Loss | Homozygous | 49/55 | 113/125 | 20 | 1735 | 0.0857 |
| #4 | ACC | Chr 1:45797228C>T | c.1145G>A | p.Gly382Asp | Missense | rs36053993 | Neutral | Heterozygous | 22/51 | NA | NA | 308 | 0.2081 |
| #5 | ATRT | Chr 1:45798475T>C | c.494A>G | p.Tyr165Cys | Missense | rs34612342 | Neutral | Heterozygous | biop2: 33/74 biop1: 27/68 |
biop2: 41/75 biop1: 52/101 |
23 | 76 | 0.0536 |
| #6 | LUAD | Chr 1:45797760T>C | c.892-2A>G | Splice acceptor | rs77542170 | Neutral | Heterozygous | 37/67 | 9/28 | 18 | 50 | 0.0059 | |
| #7 | RCC | Chr 1:45798475T>C | c.494A>G | p.Tyr165Cys | Missense | rs34612342 | Gain | Heterozygous | 37/76 | 59/126 | 22 | 20 | 0.0047 |
| #8 | IDC | Chr 1:45798475T>C | c.494A>G | p.Tyr165Cys | Missense | rs34612342 | Gain | Heterozygous | 62/90 | 91/120 | 56 | 21 | 0.0029 |
| #9 | ULMS | Chr 1:45797760T>C | c.892-2A>G | Splice acceptor | rs77542170 | Neutral | Heterozygous | 50/101 | 84/221 | 69 | 46 | 0.0056 | |
| #10 | PAAD | Chr 1:45797760T>C | c.892-2A>G | Splice acceptor | rs77542170 | Neutral | Heterozygous | 45/95 | 72/231 | 72 | 23 | 0.0031 | |
| #11 | PAAD | Chr 1:45797760T>C | c.892-2A>G | Splice acceptor | rs77542170 | Loss | Heterozygous | 19/44 | 13/50 | 37 | 18 | 0.0011 | |
| #12 | LUAD | Chr 1:45797760T>C | c.892-2A>G | Splice acceptor | rs77542170 | Neutral | Heterozygous | 19/44 | 36/97 | 58 | 48 | 0.0028 | |
Variant annotations with GRCh37 (hg19) genomic positions and MUTYH transcript NM_001048171 positions. All variants are heterozygous in the germline. The Cancer Genome Atlas (TCGA) MUTYH expression percentile is provided in comparison to the TCGA average for all cancers. COSMIC Signature 18 (Sig18) exposures are shown in this table, please refer to Supplemental Tables S2–S4 for complete mutational signatures profiles using reference matrices from COSMIC, SigProfiler, and Signature Analyzer.
(ACC) Adrenocortical carcinoma, (ATRT) atypical teratoid/rhabdoid tumor, (IDC) breast invasive ductal carcinoma, (LUAD) lung adenocarcinoma, (NA) not available, (PAAD) pancreatic adenocarcinoma, (RCC) renal cell carcinoma, (TCGA) The Cancer Genome Atlas, (ULMS) uterine leiomyosarcoma.
Somatic Analysis
A tetraploid model (four copies) with an estimated tumor content of 51% was used for the PDAC. Tumor genomic features such as single-nucleotide variants (SNVs), indels, structural variants (SVs), copy-number variants (CNVs), and mutational signatures were analyzed in conjunction with gene expression from transcriptome data. In comparison to The Cancer Genome Atlas (TCGA) data set, the overall coding mutational burden was moderate with 88 coding SNVs (73rd percentile for all TCGA cancers and 76th percentile for TCGA pancreatic cancers) and six coding indels (73rd percentile for all TCGA cancers and first percentile for TCGA pancreatic cancers). The total number of somatic SNVs (coding and noncoding) for Patient #1 was 12087, or 3.78 SNVs per Mb. There were 86 SVs (60th percentile among our local database of 626 cancers of diverse origins). Please refer to Supplemental Table S2A–D for additional genomic data.
As mentioned previously, tumor transcriptome data showed the germline variants to be in trans and expressed (Fig. 1A). The targeted assembly pipeline (TAP) (Chiu et al. 2018) on the transcriptome showed the synonymous germline variant (c.996G>A, p.Ser332Ser) to create a novel canonical AG acceptor splice site removing 42 bp and 14 amino acids (Fig. 1B; Supplemental Table S2E). Because of the creation of this novel acceptor splice site and subsequent splicing out of the c.996G>A variant, the variant was only present in two out of 15 RNA-seq reads spanning the Chr 1:45797481 genomic site. The second pathogenic variant (c.815G>A, p.Gly272Glu) was present in 20 out of 29 RNA-seq reads covering the Chr 1:45797914 genomic site. The novel exon junction at Chr 1:45797479 was supported by 12 reads, whereas the canonical junction at Chr 1:45797521 was supported by 19 reads (Supplemental Table S2E).
Figure 1.

Patient #1 compound heterozygous MUTYH germline variants. (A) Integrated Genomics Viewer (IGV) (Thorvaldsdóttir et al. 2013) capture of whole-genome sequencing of tumor and matched blood DNA with whole-transcriptome sequencing of tumor RNA data at the genomic region encompassing the two MUTYH variants (c.996G>A, p.Ser332Ser and c.815G>A, p.Gly272Glu; NM_001048171). Paired-end transcriptome reads showing the germline variants to be in trans with read pairs containing the p.Gly272Glu variant (exon 10) but not the p.Ser332Ser variant (exon 12) (red box). The splicing aberration caused by p.Ser332Ser variant was not visually apparent in IGV and required transcriptome TAP for detection and characterization. TAP was used to assemble reads into contigs. Contig 1 shows aberrant splicing and contig 2 shows normal splicing. (B) Schematic of TAP analysis results showing the synonymous germline variant (c.996G>A, p.Ser332Ser) creating a novel canonical AG acceptor splice site at Chr 1:45797480–45797481 and removing 42 bp (14 amino acids).
The PDAC tumor featured the highest Signature 18 and SBS36 signatures of our cohort using three different published signatures references matrices (COSMIC, SigProfiler, and SignatureAnalyzer) with a proportion of mutations contributed of 41%–48% (4962–5769 SNVs) (Figs. 2 and 3; Supplemental Table S2F–H). KRAS increased expression (94th percentile compared to the TCGA average) was associated with copy gains (+3 copies) and the driver KRAS p.Gly12Cys (c.34G>T, NM_033360) transversion mutation (3/6 copies). Given the predicted mutational signatures probability from each reference matrix, the Bayesian probability (Supplemental Table S2I) for the KRAS transversion mutation to be caused by the transversion signatures previously associated with MUTYH deficiency is 84% for Signature 18 (COSMIC), 75% for combined SBS18/SBS36 signatures (SigProfiler), and 62% for combined SBS18/SBS36 signatures (SignatureAnalyzer).
Figure 2.

Comparison of mutation signatures exposures of MUTYH germline or combined germline/somatic biallelic aberrations against 731 cancer genomes of mixed origins. The cohort distribution of mutation signature exposures for each signature is shown using signature composition reference matrices from COSMIC, SigProfiler, and SignatureAnalyzer. The mutation fraction exposures (proportion of all mutations contributed by each signature) of Patient #1, Patient #2, and Patient #3 are superimposed. Signatures previously associated with MUTYH-mediated BER deficiency are highlighted by a rectangle. Patient #1 (PDAC, pink diamond), Patient #2 (breast cancer, green diamond), and Patient #3 (breast cancer, blue diamond) demonstrated germline or combined germline/somatic biallelic MUTYH aberrations. Functional biallelic MUTYH loss of function was present in tumors of Patient #1, Patient #2, and Patient #3 and all three patients displayed elevated outlier (>1.5 interquartile range above third quartile) signatures previously associated with defective MUTYH (COSMIC Signature 18, SigProfiler SBS18 and SBS36, and SignatureAnalyzer SBS18 and SBS36).
Figure 3.

Mutation catalogs and COSMIC mutational signatures of MUTYH germline or combined germline/somatic biallelic aberrations. Complete catalogs of SNVs from WGS of tumors were classified based on variant and 3′/5′ contexts into 96 categories (left side) and mutation catalogs were used to calculate the contribution of each signature to mutational burden (right side). The proportion of mutations or mutation count in each of the 96 categories is shown here as a barplot (left side), whereas the signIT Markov Chain Monte Carlo simulation result for COSMIC mutational signatures is shown as a violin plot distribution (right side). COSMIC Signature 18 composition is shown at the top for reference. Patient #1 (germline MUTYH c.815G>A, p.Gly272Glu; c.996G>A, p.Ser332Ser; NM_001048171) and Patient #2 (germline MUTYH c.892-2A>G) tumors showed increased Signature 18 somatic mutation burden, as exemplified by the resemblance of their mutation catalogs to COSMIC reference Signature 18. Other mutational processes contributed most to the mutational burden of Patient #3's tumor.
The tumor harbored several somatic events that have previously been associated with PDAC (Bailey et al. 2016). A 10-Mb deletion at 9p led to homozygous copy loss of tumor-suppressor genes (TSGs) CDKN2A, CDKN2B, and MTAP, and a 51-Mb deletion at 18q led to heterozygous copy loss of TSGs SMAD2 and SMAD4. In addition to the gain-of-function (GOF) KRAS mutation (p.Gly12Cys), somatic small mutations of interest included the TP53 frameshift indel with a deletion copy loss leading to biallelic loss of function, as well as heterozygous frameshift indels in two epigenetic TSGs: ARID1A and SETD2. Including subclonal variants, 53/95 (56%) of moderate to high impact variants in protein coding genes were due to tranversion events (Supplemental Table S2C). Fusions of potential biological relevance included a Chr 12 duplication leading to an inversion-fusion of GPRC5A and CCDC91 associated with a copy gain (+2 copies) and elevated GPRC5A expression (96th percentile compared to the TCGA average). BER pathway genes OGG1 and NUDT1 had low percentile expression (third percentile for both) and OGG1 had a copy loss (−1 copy). Please refer to Supplemental Table S2 for additional genomic data.
Germline MUTYH Carriers with Somatic Second Hits in Breast Cancer
Analysis of an additional 730 advanced cancer cases (N = 731) was undertaken to determine whether the mutational signatures seen in Patient #1's PDAC were also present in tumors from germline MUTYH heterozygote carriers or, if instead, the signatures were only seen in the case of biallelic loss of function.
Using reference matrices from COSMIC, SigProfiler, and SignatureAnalyzer, somatic mutational signatures were analyzed in terms of mutation counts (number of SNVs) and proportions (fraction between 0 and 1) contributed to the overall mutational burden. Mutation counts (mean exposures) and mutation fractions (proportion contributed by each signature) higher than 1.5 interquartile range above the third quartile were considered outliers. Among germline MUTYH carriers (N = 12; Table 1) all three cases of biallelic MUTYH aberrations (Patient #1, Patient #2, and Patient #3) were outliers for the proportion of mutations contributed by transversion signatures previously associated with defective MUTYH (Figs. 2, 3; Supplemental Tables S2F–H, S3F–H, S4E–G).
Patient #2 Overview
Patient #2 is a female patient of North-East Asian ancestry diagnosed with metastatic invasive ductal breast carcinoma (4 × 5-cm mass on ultrasound, Nottingham grade 2/3, estrogen receptor 8/8, progesterone receptor 7/8, and HER2–) at the age of 32 yr. Two bone lesions at L5 and T10 were seen on MRI and PET imaging. She was treated with doxorubicin and cyclophosphamide for six cycles, followed by mastectomy and axillary node resection. Family history was negative for MAP-associated cancers. Patient #2 was found to carry a single germline MUTYH splice site variant (c.892-2A>G; NM_001048171). No other pathogenic variants were identified on clinical multigene panel testing or analysis of 97 additional cancer susceptibility genes by WGS. Their tumor featured a somatic copy loss of MUTYH (triploid model, tumor content 38%), resulting in the splice site variant being homozygous in the tumor.
The tumor had an unremarkable coding mutational burden with 24 nonsynonymous coding SNVs (29th percentile among all TCGA cancers and 54th percentile among TCGA breast cancer data set), one coding indel (24th percentile among all TCGA cancers and 28th percentile among TCGA breast cancer data set), and 116 SVs (73rd percentile among our local database of 583 cancer cases) (Supplemental Table S3A–D). The total number of somatic SNVs (coding and noncoding) for Patient #2 was 4979, or 1.52 SNVs per Mb.
The targeted assembly pipeline (TAP) (Chiu et al. 2018) on the transcriptome showed two mechanisms of abnormal splicing associated with this germline variant. The first mechanism leads to a 9-bp deletion due to the canonical splice site acceptor disruption and selection of a downstream canonical AG acceptor splice site at Chr 1:45797750–45797751 (new exon start at Chr 1:45797749). The second mechanism leads to marked intron 10 and intron 11 retention (Fig. 4; Supplemental Table S3E).
Figure 4.

Germline MUTYH founder Asian splice site variant impact on splicing. The Asian founder mutation MUTYH disrupting the canonical acceptor splice site at exon 11 (c.892-2A>G) was found in six patients (Patient #2, Patient #6, Patient #9, Patient #10, Patient #11, and Patient #12). (A) Integrated Genomic Viewer (IGV) (Thorvaldsdóttir et al. 2013) capture of whole-genome sequencing of tumor and matched blood DNA with whole-transcriptome sequencing of tumor RNA data at the genomic region encompassing the MUTYH variant (c.892-2A>G; NM_001048171).Transcriptome data showing abnormal splicing removing 9 bp from exon 11 (19 reads with a black line) and marked intron 10 and intron 11 retention. (B) Schematic of abnormal splicing removing 9 bp from exon 11. (C) Schematic of abnormal splicing resulting in intron retention.
Despite the overall low mutation counts (4979 SNVs), the contribution of COSMIC Signature 18, SigProfiler SBS36, and SignatureAnalyzer SBS36 signatures was elevated (Table 1; Supplemental Table S3F–H), suggesting that some degree of MUTYH-mediated BER deficiency could be present in the tumor because of the combined germline and somatic biallelic MUTYH aberrations. COSMIC Signature 1 (1203 SNVs or 24.2% of all SNVs), associated with age, contributed most to overall mutational burden, followed by Signature 18 (830 SNVs or 16.7% of all SNVs) and nonspecific Signature 5 (757 SNVs or 15.2% of all SNVs) (Figs. 2 and 3; Supplemental Table S4F–H).
Somatic genomic features of interest included PIK3CA p.His1047Arg (heterozygous, 2/3 copies) GOF mutation, ADGRA2 amplification (+9 copies), AKT1 copy gain (+1 copy), and AKT3 amplification (+5 copies). Additional CNVs of potential clinical relevance included amplification of MDM4 (+5 copies) and single-copy gains of MDM2, CDK4, CDK6, and AURKA. BER gene OGG1 had low expression (second percentile), but both OGG1 and NUDT1 were copy neutral. Please refer to Supplemental Table S3 for additional data.
Patient #3 Overview
Patient #3 is a female patient of European ancestry diagnosed at the age of 69 yr with invasive left ductal breast carcinoma pT2N1aMX grade 3 (estrogen receptor/progesterone receptor positive, HER2–). She was initially treated with partial left mastectomy and left axillary lymph node dissection followed by standard care adjuvant therapy doxorubicin, cyclophosphamide, and paclitaxel, but paclitaxel was replaced by docetaxel following a severe allergic reaction to paclitaxel. This regimen was followed by adjuvant letrozole and radiation therapy (16 cycles). In addition to the pathogenic germline MUTYH variant (c.1145G>A, p.Gly382Asp), this patient also carries a heterozygous pathogenic germline CHEK2 mutation (NM_007194:c.1100delC, p.Thr367Metfs, rs555607708). Family history revealed a maternal aunt with premenopausal breast cancer at the age of 30 yr who underwent bilateral mastectomy.
The tumor had an elevated coding mutational burden with 92 nonsynonymous coding SNVs (74th percentile among all TCGA cancers and 93rd percentile among TCGA breast cancer data set), four coding indels (59th percentile among all TCGA cancers and 69th percentile among TCGA breast cancer data set), and 248 SVs (93rd percentile among our local database of 626 cancer cases) (Supplemental Table S4A–D). The total number of somatic SNVs (coding and noncoding) for Patient #3 was 20237, or 6.32 SNVs per Mb.
The tumor had a somatic MUTYH copy loss (triploid model, tumor content 70%) resulting in the germline variant being homozygous in the tumor. Although COSMIC Signature 18 had a significant contribution (1735 or 8.6% of all SNVs), Signature 2 (6621 or 32.7% of all SNVs) and Signature 13 (3512 or 17.4% of all SNVs), associated with the APOBEC family of cytidine deaminases, were the main contributors to overall mutational burden (Table 1; Figs. 2, 3; Supplemental Tables S4A,E–G). Deletion copy loss (−1 copy) of BRCA1 and BRCA2 was observed in the tumor. Furthermore, the germline CHEK2 c.1100delC heterozygous variant was homozygous in the tumor by reason of a somatic deletion LOH event. However, Signature 3, associated with homologous recombination deficiency, only very modestly contributed to overall mutational burden (304 or 1.5% of all SNVs).
Other somatic events of interest included copy gain (+2 copies) with PIK3CA p.His1047Arg (heterozygous, 4/5 copies) GOF mutation, FGFR2 amplification (+58 copies), CCND1 amplification (+19 copies), and AKT3 amplification (+4 copies). Additional CNVs of potential clinical relevance included amplifications of MDM4 (+8 copies), MDM2 (+4 copies), CDK2/CDK4 (+3 copies), and AURKA (+3 copies). BER pathway genes NUDT1 and OGG1 had low percentile expression (2nd and 15th percentile, respectively) and NUDT1 had copy neutral LOH, whereas OGG1 had a copy loss (−1 copy). Please refer to Supplemental Table S4 for additional data.
MUTYH Asian Founder Splice Site Variant
The Asian founder MUTYH splice site mutation c.892-2A>G (rs77542170) (Taki et al. 2015) was identified in six out of 731 patients (minor allele frequency 0.0041, or 0.41%). According to the gnomAD database, this splice site variant has a minor allele frequency of 0.11% (0.0011) in the general population and 1.5% (0.015) in the East Asian population (Lek et al. 2016). Given the frequency of this variant in our cohort, we conducted single-nucleotide polymorphism principal component analysis using the EthSeq R package (Romanel et al. 2017) to show that all patients carrying this pathogenic germline variant were of East Asian ancestry (Supplemental Table S5). The ethnic composition of our cohort explains the large number of carriers identified with the MUTYH c.892-2A>G variant. Transcriptome TAP of the tumors from patients carrying the c.892-2A>G splice site mutation consistently identified the same two abnormal splicing mechanisms described in Figure 4 (other MUTYH carriers transcriptome assembly data not shown).
DISCUSSION
We observed mutational signatures featuring a strong C:G>A:T transversion phenotype that have previously been associated with defective MUTYH in tumors from patients with biallelic germline or combined germline and somatic MUTYH loss of function (N = 3). Such signatures, collectively referred to as SBS18/SBS36 signatures, include COSMIC Signature 18, SigProfiler SBS18/SBS36, and SignatureAnalyzer SBS18/SBS36 (Pilati et al. 2017; Scarpa et al. 2017; Viel et al. 2017; Alexandrov et al. 2018). The study of Patient #1 (germline compound heterozygote) provides evidence for the contribution of aberrant MUTYH function to the PDAC genomic landscape. Zhou et al. reported a high frequency of KRAS p.Gly12Cys transversion mutations in pancreatic cancer (80/126, 63%) (Kairupan et al. 2005; Fokkema et al. 2011; Win et al. 2011; Zhou et al. 2016). The KRAS p.Gly12Cys variant was seen in 2% of colorectal cancers, and, of those, 25% had germline biallelic pathogenic MUTYH variants (Jones et al. 2004; Aimé et al. 2015). As activated KRAS is a frequent and early driver of PDAC, germline mutations in MUTYH have been hypothesized to be associated with development of pancreatic adenocarcinoma cancer on this basis, although this had not been demonstrated prior to our study (Smith et al. 2009). Given the predominant contribution of SBS18/SBS36 signatures to the PDAC mutational landscape in the setting of germline biallelic MUTYH mutations (Patient #1), the oncogenic driver KRAS transversion was likely caused by MUTYH deficiency. A Bayesian probability approach consistently ranked the transversion signatures previously associated with germline or combined germline/somatic MUTYH impairment (COSMIC Signature 18 [Pilati et al. 2017; Scarpa et al. 2017], SigProfiler SBS36 [Alexandrov et al. 2018], and SignatureAnalyzer SBS36 [Alexandrov et al. 2018]) at the top (Supplemental Table S2I).
In all the MUTYH mutation carriers (n = 9) in which a wild-type copy was retained in the tumor, SBS18/SBS36 signatures were not predominant or were inconsistent between the methods (COSMIC, SigProfiler, and SignatureAnalyzer). These results suggest that MUTYH is haplosufficient with respect to MUTYH function in a monoallelic state, but that biallelic complete loss of MUTYH function can cause SBS18/SBS36 signatures to arise even in tumors not classically associated with MAP syndrome (Table 1).
We identified strong evidence for MUTYH-mediated BER deficiency contributing to oncogenesis in our PDAC case with germline biallelic MUTYH aberrations. Patient #2 and Patient #3 both carry a heterozygous germline MUTYH pathogenic variant (c.892-2A>G and c.1145G>A (p.Gly382Asp), respectively). and their breast cancer tumor displayed a somatic MUTYH copy loss causing the germline variant to become functionally homozygous (2/2 remaining copies, triploid model). While APOBEC signatures were the main contributors to Patient # 3's mutational landscape, signatures associated with age contributed most to Patient #2's somatic landscape. Although the SBS18/SBS36 signatures contribution remained modest in the two breast cancer cases accounting for 16.7%–19.8% and 8.9%–10.3% of all mutations in Patient #2 and Patient #3, respectively, the rarity of these characteristic signatures in the setting of combined germline and somatic biallelic MUTYH inactivation suggests that MUTYH deficiency is likely the main determinant of C:G>A:T transversion mutations in these tumors. There is debate in the literature as to whether or not inactivation of MUTYH predisposes to breast cancer (Wasielewski et al. 2010; Boesaard et al. 2014). Although evidence of somatic SBS18/SBS36 signatures in two breast cancer cases does not resolve this question, our findings suggest that germline MUTYH heterozygous mutations can occasionally contribute to somatic mutational evolution in the presence of secondary somatic MUTYH loss of function. Even though other known germline cancer predisposition variants such as the CHEK2 c.1100delC in Patient #3 are stronger determinants of lifetime breast cancer risk, a minor influence of germline MUTYH pathogenic variant carrier status cannot be excluded.
MUTYH is one of two BER DNA glycosylase genes that have been linked to recessive familial adenomatous polyposis syndromes and specific somatic mutational signatures. NTHL1 is associated with a C:G>T:A mutational phenotype (Weren et al. 2015). MUTYH is associated with the 8-OxoG BER pathway and a C:G>A:T transversion phenotype (Viel et al. 2017). The 8-OxoG BER pathway is redundant and, therefore, it is possible that multiple genomics hits in this pathway, including NUDT1 and OGG1, may be required for the SBS18/SBS36 C:G>A:T transversion signatures to arise in presence of oxidative DNA damage. NUDT1-null mice showed increased tumorigenesis, but the number of spontaneous mutations was not increased compared to wild-type mice (Tsuzuki et al. 2001). When the NUDT1 knockout was superimposed on a mismatch repair–deficient background, tumors displayed enrichment for C:G>A:T transversion mutations (Egashira et al. 2002). OGG1-null mice showed increased spontaneous tumorigenesis and transversion mutations (Klungland et al. 1999; Minowa et al. 2000; Sakumi et al. 2003). However, double knockout of OGG1 and NUDT1 did not increase tumorigenesis, which suggests the oncogenic effects of OGG1 deficiency may be counteracted by NUDT1 deficiency (Sakumi et al. 2003). Finally, MUTYH-null mice showed increased intestinal tumorigenesis and C:G>A:T transversion mutations (Sakamoto et al. 2007). To date, germline variation in OGG1 and NUDT1 has not been associated with a Mendelian hereditary cancer predisposition, and MUTYH remains the only validated cancer predisposition gene linked to the SBS18/SBS36 signatures with a strong phenotype of C:G>A:T transversions (Mur et al. 2018).
Beyond consideration for defective BER as a potential underlying etiology, SBS18/SBS36 transversion signatures ultimately arise from an excess of reactive oxygen species (ROS) and subsequent formation of 8-oxoguanine (8-OxoG) (Cheng et al. 1992). Increased oxidative damage lead to C:G>A:T transversion mutations even in the presence of intact MUTYH, but the frequency of such transversions is markedly increased by MUTYH deficiency (Sakai et al. 2006). Suzuki et al. (2010) recently suggested that MUTYH may actually increase C:G>A:T transversions in presence of 8-Oxo-dGTP. In light of this, SBS18/SBS36 signatures are most likely multifactorial in origin and depend on the state of oxidative stress and metabolism, as well as on the functionality of MUTYH and BER mechanisms.
We identified several known pathogenic MUTYH founder mutations within our cohort as well as previously described pathogenic variants (Table 1; NM_001048171). Founder variants included the European p.Gly382Asp and p.Tyr165Cys variants (Lejbkowicz et al. 2012; Aretz et al. 2013) and the Asian c.892-2A>G variant (Taki et al. 2015). Previously reported pathogenic variants included c.815G>A (p.Gly272Glu) and c.996G>A (p.Ser332Ser) (Kairupan et al. 2005; Fokkema et al. 2011; Win et al. 2011). Although classifications of c.996G>A and c.892-2A>G are less than uniform among reporting centers per ClinVar (Landrum et al. 2016), ranging from being a variant of uncertain significance to likely pathogenic or pathogenic, they are locally considered to be pathogenic alleles. The combined germline and somatic data presented herein support these classifications. The presence of markedly elevated SBS18/SBS36 transversion signatures in Patient #1 associated with abnormal splicing of c.996G>A and phasing of the biallelic compound heterozygous germline variants (c.996G>A, p.Ser332Ser and c.815G>A, p.Gly272Glu) support pathogenicity of the synonymous variant (p.Ser332Ser). Patient #2 carries a heterozygous germline MUTYH variant (c.892-2A>G) shown to cause aberrant splicing and their tumor featured somatic inactivation of the remaining allele resulting in biallelic MUTYH aberrations. The presence of the same transversion signatures in Patient #2 also supports pathogenicity of the Asian founder variant c.892-2A>G.
CONCLUSION
Our study demonstrates that tumors with biallelic MUTYH aberrations arising in the germline or combined germline and somatic contexts display a characteristic elevation of somatic C:G>A:T transversion signatures previously associated with defective MUTYH-mediated BER. Defective MUTYH is not the sole determinant of these signatures, but MUTYH germline variants may be present in a subset of patients with tumors demonstrating mutational signatures possibly suggestive of MUTYH deficiency (e.g., COSMIC Signature 18, SigProfiler SBS18/SBS36, or SignatureAnalyzer SBS18/SBS36). Further research in large cohorts will be important to elucidate the role of germline and somatic MUTYH aberrations, in relation to BER deficiency in diverse cancer types, in particular within populations where founder mutations have been identified.
METHODS
Patient written informed consent was obtained for the Personalized OncoGenomics trial at BC Cancer (NCT02155621), which was approved by the University of British Columbia/BC Cancer Research Ethics Board. Tumor with matched normal blood WGS and tumor whole-transcriptome sequencing was performed in 731 patients with advanced cancers of diverse origins (Supplemental Table S1A). The average depth of coverage was 80× and 40× for the tumor and normal genome, respectively. Library preparation, sequencing, and bioinformatics analyses were performed according to previously published protocols (Thibodeau et al. 2018). For each patient, tumor and matched normal samples were analyzed together to identify somatic LOH and copy-number alteration (CNA) regions using CNAseq (v0.0.8) (Jones et al. 2010) and APOLLOH (v0.1.2) (Ha et al. 2012) tools, which both utilize the hidden Markov model. The ensemble of LOH and CNA regions were compared to a set of theoretical ploidy models ranging from diploid to pentaploid and a set of theoretical tumor content varying by 10% intervals from the initial tumor content assessed on pathological examination. For example, if the tumor content was 44% on initial assessment, ploidy models (diploid, triploid, tetraploid, pentaploid) were assessed with theoretical tumor contents of 14%, 24%, 34%, 44%, 54%, 64%, 74%, 84%, and 94%. This results in a total collection of 40 models. The collected copy states are compared to the values for theoretical copy states in each model. The model which results in the lowest error and model complexity is selected upon manual review. Mutational burden was reported in terms of coding nonsynonymous mutation count as well as total SNVs count (coding and noncoding) and SNVs per Mb rate. The SNVs per Mb rate was calculated using an approximate hg19 genome assembly size of 3200 Mb. SV count (or SV mutational load) of a given case was compared to the cohort local database of SVs at the time of analysis. A case enrolled earlier in the study is compared to a smaller cohort (smaller local database) than a case enrolled later in the study (larger local database). Somatic mutations called by Strelka (Saunders et al. 2012) were classified into 96 classes based on the variant base and 3′ and 5′ context (trinucleotide). Using publicly available signatures reference matrices from COSMIC (Alexandrov et al. 2013), SigProfiler (Alexandrov et al. 2018), and SignatureAnalyzer (Haradhvala et al. 2018), the contribution of each signature was determined with the Bayesian R package signIT as previously described (Zhao et al. 2017).
To calculate the probability for driver KRAS transversion mutation (C[C>A]A transversion leading to p.Gly12Cys) to be caused by signatures previously associated with MUTYH deficiency (COSMIC Signature 18, SigProfiler SBS18/SBS36, and SignatureAnalyzer SBS18/SBS36) in Patient #1, Bayesian inference was used. For each signature, the posterior probability P(S|M) for the mutation to be caused by that given signature was calculated as follows:
in which P(M|S) is the prior probability of C[C>A]A in the reference signature matrix, P(S) is the proportion of mutation contributed by this signature, and P(M) is the total number of C[C>A]A events.
ADDITIONAL INFORMATION
Data Deposition and Access
Whole-genome sequencing and RNA-seq data (.bam files) for 11 out of 12 patients discussed in this study have been submitted to the European Genome-phenome Archive (EGA) (www.ebi.ac.uk/ega/home) under data accession numbers EGAD00001002597 (Patient #1), EGAD00001004710 (Patient #2), EGAD00001004711 (Patient #3), EGAD00001002037 (Patient #4), EGAD00001002985 (Patient #6), EGAD00001003023 (Patient #7), EGAD00001003635 (Patient #8), EGAD00001003733 (Patient #9), EGAD00001004666 (Patient #10), EGAD00001004713 (Patient #11), and EGAD00001004714 (Patient #12). Consent for data sharing does not allow for data deposition for Patient #5. This work, including data deposition, was approved by the Research Ethics Board at the BC Cancer, protocols H12-00137 and H14-00681. Written consent was obtained from the patients after discussion with their oncologists.
Ethics Statement
The Personalized OncoGenomics project was approved by the University of BC Cancer (BCCA) Research Ethics Board (REB) (protocols H12-00137 and H14-00681). Informed written consent was obtained from the patients for comprehensive tumor profiling with transcriptome sequencing (tumor) and whole-genome sequencing (tumor and blood). The use of data sets is allowed for research reports and scientific publications. The protocol allows for data to be not only used for research reports and scientific publication but also to be made available to named investigators of institutions who agree by a data transfer agreement stating they will honor the same ethical and privacy principles required by the BCCA REB. Following informed consent, the patients underwent tumor biopsies and peripheral blood sampling as part of the Personalized OncoGenomics trial at the BC Cancer (Clinicaltrials.gov ID:NCT02155621).
Acknowledgments
We gratefully acknowledge the participation of our patients and families and the Personalized OncoGenomics (POG) team and the generous support of the BC Cancer Foundation and Genome British Columbia (project B20POG). We are also grateful for the funding support provided by the BC Cancer Foundation. We thank Jessica Nelson and Robyn Roscoe for project management, as well as Patrick Plettner and Young Chen for genomic data deposition. We acknowledge that the results published here are in part based upon data generated by the following projects and obtained from dbGaP (http://www.ncbi.nlm.nih.gov/gap): The Cancer Genome Atlas managed by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) (http://cancergenome.nih.gov); Genotype-Tissue Expression (GTEx) Project, supported by the Common Fund of the Office of the Director of the National Institutes of Health (https://commonfund.nih.gov/GTEx).
Author Contributions
S.J.M.J., K.A.S., M.A.M., and J.L. contributed to conception and design of this study. H.W., C.Cr., H.J.L., K.G., C.L., D.F.S., A.F., D.J.R., and K.A.S. provided and reviewed patients clinical data. Review of germline variants was contributed by S.Yo. and A.K. Pathological study was contributed by S.Yi., R.M., and D.F.S. Data production, analysis, and interpretation were contributed by M.L.T., E.Y.Z., C.R., C.Ch., Y.S., M.R.J., E.P., W.Z., R.H., P.E., J.K., S.E.K., G.T., E.M., M.B., Z.Z., D.B., K.L.M., Y.P.M., K.A.S., and S.J.M.J. Targeted transcriptome assembly pipeline and splicing analyses were contributed by I.B., R.C., and M.L.T. Mutational signatures methods, results, and figures were contributed by M.L.T. and E.Y.Z. Sequencing and review of methods were contributed by A.J.M. and M.L.T. Additional targeted sequencing and analyses (data not shown) were provided by D.H. and D.F.S. Manuscript writing and critical editing was contributed by M.L.T., D.J.R., K.A.S., and S.J.M.J. All the authors have read and approved this manuscript.
Funding
Funding, under the form of unrestricted philanthropic donations, was generously provided by the BC Cancer Foundation (NRF10229) and Genome British Columbia (project B20POG). We also acknowledge contributions toward equipment and infrastructure from Genome Canada and Genome British Columbia (projects 202SEQ, 212SEQ, 12002), Canada Foundation for Innovation (projects 20070, 30198, 30981, 33408), CIHR Foundation (FDN-143288) programs, and the BC Knowledge Development Fund. Additionally, the Michael Smith Foundation for Health Research and the Canadian Institutes of Health Research funding held by K.A.S. supported this work, and M.L.T. is supported by the University of British Columbia Clinician Investigator Program.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Aldo Scarpa
Serena Nik-Zainal
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Aimé A, Coulet F, Lefevre JH, Colas C, Cervera P, Flejou J-F, Lascols O, Soubrier F, Parc Y. 2015. Somatic c.34G>T KRAS mutation: a new prescreening test for MUTYH-associated polyposis? Cancer Genet 208: 390–395. 10.1016/j.cancergen.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L, Kim J, Haradhvala NJ, Huang MN, Ng AW, Boot A, Covington KR, Gordenin DA, Bergstrom E, Lopez-Bigas N, et al. 2018. The repertoire of mutational signatures in human cancer. bioRxiv 1: 1–27. 10.1101/322859 [DOI] [Google Scholar]
- Aretz S, Tricarico R, Papi L, Spier I, Pin E, Horpaopan S, Cordisco EL, Pedroni M, Stienen D, Gentile A, et al. 2013. MUTYH-associated polyposis (MAP): evidence for the origin of the common European mutations p.Tyr179Cys and p.Gly396Asp by founder events. Eur J Hum Genet 22: 923–929. 10.1038/ejhg.2012.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, Miller DK, Christ AN, Bruxner TJC, Quinn MC, et al. 2016. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531: 47–52. 10.1038/nature16965 [DOI] [PubMed] [Google Scholar]
- Boesaard EP, Vogelaar IP, Bult P, Wauters CA, van Krieken JHJ, Ligtenberg MJ, van der Post RS, Hoogerbrugge N. 2014. Germline MUTYH gene mutations are not frequently found in unselected patients with papillary breast carcinoma. Hered Cancer Clin Pract 12: 21 10.1186/1897-4287-12-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle JP, Sampson JR. 2007. MUTYH-associated polyposis—from defect in base excision repair to clinical genetic testing. DNA Repair 6: 274–279. 10.1016/j.dnarep.2006.11.001 [DOI] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 1992. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G->T and A->C substitutions. J Biol Chem 267: 166–172. [PubMed] [Google Scholar]
- Chiu R, Nip KM, Chu J, Birol I. 2018. TAP: a targeted clinical genomics pipeline for detecting transcript variants using RNA-seq data. BMC Med Genomics 11: 7–9. 10.1186/s12920-018-0402-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egashira A, Yamauchi K, Yoshiyama K, Kawate H, Katsuki M, Sekiguchi M, Sugimachi K, Maki H, Tsuzuki T. 2002. Mutational specificity of mice defective in the MTH1 and/or the MSH2 genes. DNA Repair 1: 881–893. 10.1016/S1568-7864(02)00113-1 [DOI] [PubMed] [Google Scholar]
- Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. 2011. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 32: 557–563. 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. 2017. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45: D777–D783. 10.1093/nar/gkw1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. 2012. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res 22: 1995–2007. 10.1101/gr.137570.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Kim J, Maruvka YE, Polak P, Rosebrock D, Livitz D, Hess JM, Leshchiner I, Kamburov A, Mouw KW, et al. 2018. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun 9: 1–9. 10.1038/s41467-018-04002-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Lambert S, Williams GT, Best JM, Sampson JR, Cheadle JP. 2004. Increased frequency of the k-ras G12C mutation in MYH polyposis colorectal adenomas. Br J Cancer 90: 1591–1593. 10.1038/sj.bjc.6601747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SJ, Laskin J, Li YY, Griffith OL, An J, Bilenky M, Butterfield YS, Cezard T, Chuah E, Corbett R, et al. 2010. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol 11: R82 10.1186/gb-2010-11-8-r82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kairupan CF, Meldrum CJ, Crooks R, Milward EA, Spigelman AD, Burgess B, Groombridge C, Kirk J, Tucker K, Ward R, et al. 2005. Mutation analysis of the MYH gene in an Australian series of colorectal polyposis patients with or without germline APC mutations. Int J Cancer 116: 73–77. 10.1002/ijc.20983 [DOI] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. 1999. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci 96: 13300–13305. 10.1073/pnas.96.23.13300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. 2016. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44: D862–D868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejbkowicz F, Cohen I, Barnett-Griness O, Pinchev M, Poynter J, Gruber SB, Rennert G. 2012. Common MUTYH mutations and colorectal cancer risk in multiethnic populations. Fam Cancer 11: 329–335. 10.1007/s10689-012-9516-8 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa O, Arai T, Hirano M, Monden Y, Nakai S, Fukuda M, Itoh M, Takano H, Hippou Y, Aburatani H, et al. 2000. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc Natl Acad Sci 97: 4156–4161. 10.1073/pnas.050404497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mur P, Jemth A-S, Bevc L, Amaral N, Navarro M, Valdés-Mas R, Pons T, Aiza G, Urioste M, Valencia A, et al. 2018. Germline variation in the oxidative DNA repair genes NUDT1 and OGG1 is not associated with hereditary colorectal cancer or polyposis. Hum Mutat 39: 1214–1225. 10.1002/humu.23564 [DOI] [PubMed] [Google Scholar]
- Pilati C, Shinde J, Alexandrov LB, Assié G, André T, Hélias-Rodzewicz Z, Ducoudray R, Le Corre D, Zucman-Rossi J, Emile J-F, et al. 2017. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J Pathol 242: 10–15. 10.1002/path.4880 [DOI] [PubMed] [Google Scholar]
- Romanel A, Zhang T, Elemento O, Demichelis F. 2017. EthSEQ: ethnicity annotation from whole exome sequencing data. Bioinformatics 33: 2402–2404. 10.1093/bioinformatics/btx165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai A, Nakanishi M, Yoshiyama K, Maki H. 2006. Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli. Genes Cells 11: 767–778. 10.1111/j.1365-2443.2006.00982.x [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Tominaga Y, Yamauchi K, Nakatsu Y, Sakumi K, Yoshiyama K, Egashira A, Kura S, Yao T, Tsuneyoshi M, et al. 2007. MUTYH-null mice are susceptible to spontaneous and oxidative stress–induced intestinal tumorigenesis. Cancer Res 67: 6599–6604. 10.1158/0008-5472.CAN-06-4802 [DOI] [PubMed] [Google Scholar]
- Sakumi K, Tominaga Y, Furuichi M, Xu P, Tsuzuki T, Sekiguchi M, Nakabeppu Y. 2003. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res 63: 902–905. [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28: 1811–1817. 10.1093/bioinformatics/bts271 [DOI] [PubMed] [Google Scholar]
- Scarpa A, Chang DK, Nones K, Corbo V, Patch A-M, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A, et al. 2017. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 543: 65–71. 10.1038/nature21063 [DOI] [PubMed] [Google Scholar]
- Smith LM, Sharif S, Brand R, Fink E, Lamb J, Whitcomb DC. 2009. MUTYH exon 7 and 13 mutations associated with colorectal cancer (MAP syndrome) are not commonly associated with sporadic pancreatic cancer. Pancreatology 9: 793–796. 10.1159/000199440 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Harashima H, Kamiya H. 2010. Effects of base excision repair proteins on mutagenesis by 8-oxo-7,8-dihydroguanine (8-hydroxyguanine) paired with cytosine and adenine. DNA Repair 9: 542–550. 10.1016/j.dnarep.2010.02.004 [DOI] [PubMed] [Google Scholar]
- Taki K, Sato Y, Nomura S, Ashihara Y, Kita M, Tajima I, Sugano K, Arai M. 2015. Mutation analysis of MUTYH in Japanese colorectal adenomatous polyposis patients. Fam Cancer 15: 261–265. 10.1007/s10689-015-9857-1 [DOI] [PubMed] [Google Scholar]
- Thibodeau ML, Zhao E, Reisle C, Zhang W, Thiessen N, Bleile D, Mungall AJ, Ma YP, Renouf DJ, Ng T, et al. 2018. Whole genome and whole transcriptome genomic profiling of a metastatic eccrine porocarcinoma. NPJ Precis Oncol 2: 8 10.1038/s41698-018-0050-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki T, Egashira A, Igarashi H, Iwakuma T, Nakatsuru Y, Tominaga Y, Kawate H, Nakao K, Nakamura K, Ide F, et al. 2001. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc Natl Acad Sci 98: 11456–11461. 10.1073/pnas.191086798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viel A, Bruselles A, Meccia E, Fornasarig M, Quaia M, Canzonieri V, Policicchio E, Urso ED, Agostini M, Genuardi M, et al. 2017. A specific mutational signature associated with DNA 8-oxoguanine persistence in MUTYH-defective colorectal cancer. EBioMedicine 20: 39–49. 10.1016/j.ebiom.2017.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasielewski M, Out AA, Vermeulen J, Nielsen M, van den Ouweland A, Tops CMJ, Wijnen JT, Vasen HFA, Weiss MM, Klijn JGM, et al. 2010. Increased MUTYH mutation frequency among Dutch families with breast cancer and colorectal cancer. Breast Cancer Res Treat 124: 635–641. 10.1007/s10549-010-0801-7 [DOI] [PubMed] [Google Scholar]
- Weren RDA, Ligtenberg MJL, Kets CM, de Voer RM, Verwiel ETP, Spruijt L, van Zelst-Stams WAG, Jongmans MC, Gilissen C, Hehir Kwa JY, et al. 2015. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 47: 668–671. 10.1038/ng.3287 [DOI] [PubMed] [Google Scholar]
- Win AK, Cleary SP, Dowty JG, Baron JA, Young JP, Buchanan DD, Southey MC, Burnett T, Parfrey PS, Green RC, et al. 2011. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer 129: 2256–2262. 10.1002/ijc.25870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao EY, Shen Y, Pleasance E, Kasaian K, Leelakumari S, Jones M, Bose P, Ch'ng C, Reisle C, Eirew P, et al. 2017. Homologous recombination deficiency and platinum-based therapy outcomes in advanced breast cancer. Clin Cancer Res 23: 7521–7530. 10.1158/1078-0432.CCR-17-1941 [DOI] [PubMed] [Google Scholar]
- Zhou L, Baba Y, Kitano Y, Miyake K, Zhang X, Yamamura K, Kosumi K, Kaida T, Arima K, Taki K, et al. 2016. KRAS, BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic cancers: pyrosequencing technology and literature review. Med Oncol 33: 32 10.1007/s12032-016-0745-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
{kind=link}
{kind=link}
Data Availability Statement
Whole-genome sequencing and RNA-seq data (.bam files) for 11 out of 12 patients discussed in this study have been submitted to the European Genome-phenome Archive (EGA) (www.ebi.ac.uk/ega/home) under data accession numbers EGAD00001002597 (Patient #1), EGAD00001004710 (Patient #2), EGAD00001004711 (Patient #3), EGAD00001002037 (Patient #4), EGAD00001002985 (Patient #6), EGAD00001003023 (Patient #7), EGAD00001003635 (Patient #8), EGAD00001003733 (Patient #9), EGAD00001004666 (Patient #10), EGAD00001004713 (Patient #11), and EGAD00001004714 (Patient #12). Consent for data sharing does not allow for data deposition for Patient #5. This work, including data deposition, was approved by the Research Ethics Board at the BC Cancer, protocols H12-00137 and H14-00681. Written consent was obtained from the patients after discussion with their oncologists.