

Abstract
Peritoneal malignant mesothelioma is a rare disease with a generally poor prognosis and poor response to chemotherapy. To improve survival there is a need for increased molecular understanding of the disease, including chemotherapy sensitivity and resistance. We here present an unusual case concerning a young woman with extensive peritoneal mesothelioma who had a remarkable response to palliative chemotherapy (platinum/pemetrexed). Tumor samples collected at surgery before and after treatment were analyzed on the genomic and transcriptional levels (exome sequencing, RNA-seq, and smallRNA-seq). Integrative analysis of single nucleotide and copy-number variants, mutational signatures, and gene expression was performed to provide a comprehensive picture of the disease. LATS1/2 were identified as the main mutational drivers together with homozygous loss of BAP1 and PBRM1, which also may have contributed to the extraordinary chemotherapy response. The presence of the S3 mutational signature is consistent with homologous recombination DNA repair defects due to BAP1 loss. Up-regulation of the PI3K/AKT/mTOR pathway after treatment, supported by deactivated PTEN through miRNA regulation, is associated with cancer progression and could explain chemotherapy resistance. The molecular profile suggests potential benefit from experimental targeting of PARP, EZH2, the PI3K/AKT/mTOR pathway and possibly also from immune checkpoint inhibition. In addition to providing the molecular background for this unusual case of peritoneal mesothelioma, the results show the potential value of integrative genomic analysis in precision medicine.
Keywords: peritoneal mesothelioma
INTRODUCTION
Mesothelioma is a rare and aggressive malignancy associated with advanced age and asbestos exposure, that most often arises from the mesothelial lining of the pleura, and less commonly from the peritoneum (7%–30%) (Kim et al. 2017). It has a poor prognosis with survival ranging from 7 to 27 mo depending on the histological subtype (Yap et al. 2017). Platinum-based chemotherapy is the first-line treatment for unresectable disease. However, efficacy is often low and disease progression is common (Mott 2012; Kim et al. 2017). To improve treatment, increased molecular understanding of the disease is needed, including chemotherapy sensitivity and resistance. We here present a case of a young patient with peritoneal mesothelioma showing an extraordinary chemotherapy response. Using integrative genomic analysis, we aimed to identify genomic drivers and molecular determinants of chemotherapy response, and to suggest potential new therapeutic options.
RESULTS
Clinical Presentation
The case concerns a 19-yr-old female with no known asbestos exposure, presenting with abdominal pain and cachexia, who in 2013 was diagnosed with disseminated malignant peritoneal mesothelioma, epitheloid subtype (Figs. 1 and 2). Routine molecular analysis classified the tumor as KRAS, BRAF, and NRAS wild-type and microsatellite stable. At surgery, the disease was deemed unresectable with a peritoneal cancer index (PCI) of 39, signifying complete involvement of the peritoneal cavity. Palliative chemotherapy (cisplatin-pemetrexed, 10 cycles; carboplatin-pemetrexed, 6 cycles; carboplatin monotherapy 2 cycles) resulted in excellent clinical and radiological response, but treatment had to be discontinued because of allergic reactions. In 2015, laparotomy revealed PCI 16, and cytoreductive surgery and hyperthermic intraperitoneal chemotherapy (cisplatin and doxorubicin) was performed. In 2016, lymph node recurrences (aortic and pelvic) were resected, and in 2018, upon radiological progression of a new pelvic recurrence and appearance of a single liver metastasis, rechallenge with carboplatin-pemetrexed was administered, currently with progressive disease. Tumor biopsies and blood samples were obtained following written informed consent at the surgical procedures in 2013 (T1) and 2015 (T2).
Figure 1.
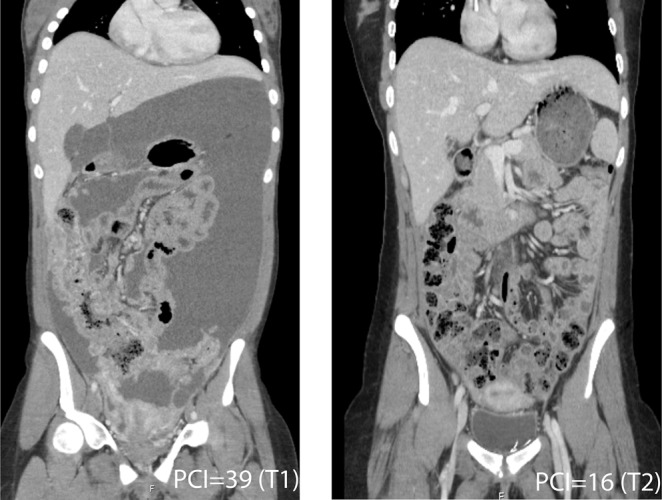
CT images before (T1, PCI = 39) and after (T2, PCI = 16) platinum/pemetrexed chemotherapy.
Figure 2.

Stained sections of tumor sample before treatment (T1). (Upper panel) Hematoxylin and eosin (HE1: 100×, HE2: 200×); (lower panel) (100×) the epithelial marker Ber-EP4 (negative) and the mesothelioma marker calretinin (positive).
Genomic Analyses
Whole-exome sequencing revealed a total of 121 somatic variants in the T1 and 429 in the T2 tumor sample (Fig. 3), representing >3 times increase in number of mutations. Four and eight known protein-coding cancer-relevant mutations were identified in T1 and T2, respectively (Table 1). No cancer-relevant germline mutations were detected.
Figure 3.
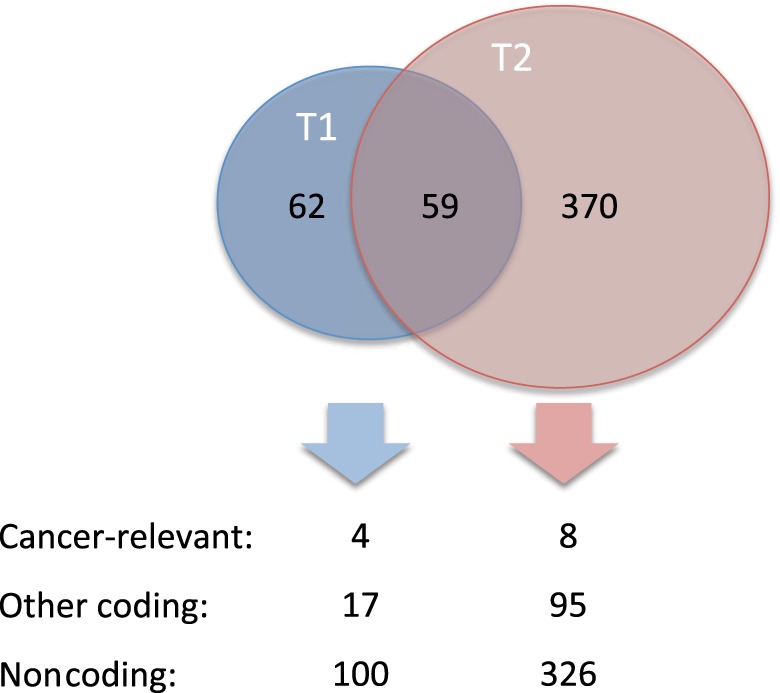
Venn diagram and overview of somatic mutations detected in the tumor tissue samples harvested before (T1) and after (T2) chemotherapy. Cancer-relevant mutations include coding variants in known tumor-suppressor genes and proto-oncogenes (coding implies those that alter the protein code or mutations at canonical splice sites). Noncoding mutations refer to silent and nonexonic mutations.
Table 1.
Known cancer-relevant mutations (in tumor-suppressor genes and proto-oncogenes) detected in the tumor samples harvested prechemotherapy (T1) and postchemotherapy (T2)
| Gene | HGVS protein | Genomic change | AF pre | AF post | Variant consequence | Predicted effect | dbSNP ID |
|---|---|---|---|---|---|---|---|
| LATS2 | p.Val901SerfsTer43 | 13:g.21553902T>TC | 0.55 | 0.53 | Frame shift | - | - |
| LATS1 | p.Trp879Ter | 6:g.149997831C>T | 0.66 | 0.55 | Stop gained | Damaging | - |
| MECOM | p.Pro88Ser | 3:g.169099088G>A | 0.34 | 0.19 | Missense | Damaging | - |
| OPCML | p.Gln49Glu | 11:g.132812843G>C | 0.04 | - | Missense | Tolerated | - |
| GATA3a | p.Pro189Leu | 10:g.8100592C>T | - | 0.13 | Missense | Mixed | - |
| EPHA3 | p.Gly114Ter | 3:g.89259196G>T | - | 0.15 | Stop gained | Damaging | - |
| GLI1 | p.Cys177Trp | 12:g.57859035C>G | - | 0.04 | Missense | Damaging | - |
| CABLES1 | p.Gln631His | 18:g.20837322G>C | - | 0.03 | Missense | Tolerated | - |
| CASC1 | p.Gly242Ter | 12:g.25297577C>A | - | 0.05 | Stop gained | Tolerated | - |
| ANAPC1 | p.Gln451His | 2:g.112615888C>G | - | 0.04 | Missense | Tolerated | 79100806 |
The predicted effect represents the majority prediction of several algorithms included in dbNSFP v3.5. AF, allelic fraction.
aAlthough not considered a proto-oncogene or tumor suppressor per se, we also include the transcription factor GATA3 because of its reported association to breast cancer.
LATS1/2 were mutated in both samples with an allelic fraction (AF) ∼0.5. These genes encode the large tumor suppressor 1/2 protein kinases and are commonly mutated in pleural and peritoneal mesothelioma (Murakami et al. 2011; Miyanaga et al. 2015; Sheffield et al. 2015; Woodard et al. 2017; Yap et al. 2017). The stop-gained (LATS1 p.Trp879Ter), and frame-shift (LATS2 p.Val901SerfsTer43) mutations are situated in the kinase domains, likely causing loss of protein functions. MECOM (MDS1 and EVI-1 complex locus) was also mutated in both samples (AF 0.34 and 0.19). The encoded protein, EVI-1, is a zinc finger transcription factor and oncoprotein involved in cell proliferation (Hirai 1999), functioning as a transcriptional activator, stimulating e.g., GATA2 and GATA3 promoters (Fuchs 2006). The missense mutation is located in the first zinc-finger domain of the protein, probably affecting DNA binding (Kurokawa et al. 1998). GATA3 (GATA binding protein 3) and EPHA3 (ephrin receptor A3) were mutated only in T2. The transcription factor GATA3 suppresses metastasis through reversal of epithelial-mesenchymal transition (EMT) (Yan et al. 2010). The missense mutation located in the second transactivating domain of the protein may alter binding to cofactors during transcription. The EPHA3 gene, encoding a receptor tyrosine kinase, carried a stop-gained mutation in the ligand-binding domain, causing loss of protein function. EPHA3 is involved in cell adhesion, cytoskeleton organization and apoptosis (Janes et al. 2014).
Similar to the increase of mutations found in T2, copy-number analysis showed a threefold increase in copy-number segments compared to T1 (Fig. 4), indicating increased genomic instability. Both samples had heterozygous loss of segments in Chromosome 6 and 13, containing the mutated genes LATS1/2, respectively. Additionally, the samples had homozygous loss of a segment in Chromosome 3 containing two important tumor-suppressor genes BAP1 (BRCA1-associated-protein-1) and PBRM1 (Protein-polybromo-1), involved in homologous recombination (HR) DNA repair, and chromatin remodeling (Wilson and Roberts 2011; Yu et al. 2014). Furthermore, a mutational signature associated with failure of HR repair and response to platinum therapy (S3), was found in T1 and T2 (Fig. 5). Two additional signatures (S23 and S24) were present only in T2, exhibiting C>T and C>A mutations that most likely are induced by the treatments (Szikriszt et al. 2016). The tumor mutational burden (TMB) was low (0.87 and 3.82 mutations/Mb in T1 and T2, respectively), consistent with microsatellite stable tumor.
Figure 4.

Copy-number aberration plot from FACETS of samples harvested before (T1) and after (T2) treatment. The chromosomes are depicted with copy-number segments in blue, graded after cellular fraction (cf; dark blue: high cf, light blue: low cf). Heterozygous loss of segments in T1 and T2 were found in Chromosomes 1, 2, 3, 6, 13, and 22, and gains in Chromosome 6 (cf = 0.7). Additionally, T2 had losses in Chromosomes 9, 19, 21, and X, and gains in Chromosomes 1 and 5 (cf = 0.35). The arrows point to homozygous loss in Chromosome 3 (0.3 Mb, cf = 0.7) containing BAP1 and PBRM1, as well as heterozygous loss in Chromosomes 6 and 13 containing LATS1 and LATS2, respectively.
Figure 5.

Relative contribution of known mutational signatures in T1 and T2. Mutational signatures are different combinations of mutation types generated by different biological processes that improve our understanding of cancer etiology with potential implications for prevention and treatment. Signatures found in both tumor samples were associated with aging (S1), failure of HR DNA repair and responders to platinum treatment (S3). Signatures associated with C>T mutations (S23) and C>A mutations (S24) were found only in T2 and are probably related to treatment, whereas a signature of T>C mutations (S5) was found exclusively in T1. See also https://cancer.sanger.ac.uk/cosmic/signatures for a more thorough explanation of the proposed etiologies and associated cancer types underlying each signature.
Total RNA sequencing was performed on high-quality RNA extracted from T1 (n = 3) and T2 samples (n = 1). Differential expression analysis revealed 6228 differentially expressed genes, of which 51% were down-regulated after treatment. Among the top up-regulated genes were AKT3, a key regulator of the PI3K/AKT/mTOR pathway, HLA-A (major histocompatibility complex) that presents antigens to cytotoxic T-cells, the adhesion G-protein coupled receptor G6, and fibronectin (FN1), a glycoprotein of the extracellular matrix that binds to integrins in the cell membrane (Supplemental Table S1). The top down-regulated genes are involved in sodium channels (SCNN1A and SCN9A), plasma membrane integrity and cytoskeletal structure (SPTBN2), cell motility (SPINT2), HR DNA repair (SPIDR) and regulation of Rho GTPase-activity (ARHGAP44) (Supplemental Table S2). In addition, we found increased expression of mesenchymal markers (e.g., FN1) after treatment and reduced expression of epithelial markers (Supplemental Table S3).
Ingenuity pathway analysis (IPA, QIAGEN) was performed with differentially expressed genes as input. The most significantly altered canonical pathways after treatment were related to immune regulation and cancer progression (Supplemental Fig. S1). Interleukin signaling and signaling in lymphocytes and macrophages were activated, indicating an increased presence of immune cells in T2. In fact, CD8, a marker for cytotoxic T-cells, was threefold up-regulated (Padj = 0.03) in T2. The PI3K/AKT/mTOR pathway also showed increased activity through p70S6k and eIF4, supported by deactivation of PTEN signaling, and is consistent with AKT3 up-regulation. In addition, increased activity of pathways concerning cell adhesion and cytoskeleton organization may indicate altered cell plasticity and motility, which might be related to the mutated GATA3 and EPHA3.
By miRNA sequencing, 139 differentially expressed miRNAs (greater than or equal to twofold change) were identified in T2 compared to T1 (19 up-regulated and 120 down-regulated; Supplemental Fig. S2). Among the up-regulated miRNAs were miR-29, miR-19, and miR-21 known to target and repress PTEN (Zhang et al. 2010; Liang et al. 2011; Tumaneng et al. 2012) and promote EMT (Han et al. 2012; Jiang et al. 2014; Li et al. 2015). miR-29 is also regulated by GATA3 (Chou et al. 2013) that was mutated only in the post treatment tumor. Interestingly, a high number of immune-cell specific miRNAs (miR-142 [Sun et al. 2015], miR-155 [Dudda et al. 2013], miR-342 [Czimmerer et al. 2016], miR-150 [Zhou et al. 2007]) were up-regulated, pointing to an increase of immune cells in T2. Because mir-155 is required for CD8+ T-cell responses to cancer, the up-regulation is consistent with increased expression of CD8 and HLA-A in the tumor after treatment.
DISCUSSION
Loss-of-function mutations in tumor suppressors LATS1/2 together with LOH of both genes would leave the tumor cells with only one copy of LATS1/2, the majority harboring the mutations. LATS1/2 proteins are involved in the Hippo signaling pathway, which plays an essential role in tissue growth control and is commonly deregulated in cancer (Gomez et al. 2014). Loss of functional LATS1/2 will activate transcriptional coactivators (YAP/TAZ) to promote cell migration, proliferation, and survival (Fig. 6; Meng et al. 2016). The homozygous loss of tumor suppressors BAP1 and PBRM1, found in both tumor samples may also contribute to cancer progression, because defective chromatin remodeling and HR repair over time will give rise to genomic instability (Thompson and Schild 2001; Hopson and Thompson 2017) and increased mutation rate, consistent with the findings in our study. Taken together, LATS1/2 mutations and loss of BAP1 and PBRM1 are likely genomic drivers of the disease.
Figure 6.

Simplified overview of the main pathways (Hippo, PI3K/AKT/mTOR, DNA repair, and chromatin remodeling) affected by the molecular alterations found by integrative genomic analysis in this case of peritoneal mesothelioma. Depicted mutations and copy-number loss (stars) were present in tumor samples before (T1) and after (T2) chemotherapy, whereas components marked up- or down-regulated (triangles) refer to the state in T2 compared to T1.
In addition to being mutational drivers, the combined loss of BAP1 and PBRM1 may explain the observed extraordinary chemotherapy response. HR repair-deficient cancer cells (here due to BAP1 loss) are not capable of repairing the massive DNA damage caused by chemotherapy and will in most cases undergo cell death. In addition, PBRM1 loss could further increase sensitivity, because BAF180 (encoded by PBRM1) as part of the SWI/SNF complex might play a critical role in protecting DNA from damage (Freeman et al. 2014). The majority of mesothelioma patients harbor BAP1 alterations with heterozygous loss being detected in about 30%–60% of cases (Bott et al. 2011; Borczuk et al. 2016; Joseph et al. 2017; Leblay et al. 2017; Hmeljak et al. 2018), whereas homozygous deletion seem to be a rare event in peritoneal mesothelioma and infrequent also in pleural mesothelioma (which is far more extensively studied). However, biallelic inactivation of BAP1, which also includes heterozygous deletion combined with an inactivating mutation, seems to be a more common occurrence, and often correlates with loss of BAP1 expression assessed by immunohistochemistry (Joseph et al. 2017; Leblay et al. 2017). It is therefore unlikely that loss of BAP1 alone could explain the exceptional chemotherapy response in this case. Mutations and homozygous deletions of PBRM1 are rarely detected in peritoneal mesothelioma, whereas heterozygous deletion is reported at a frequency of 35% (Borczuk et al. 2016). Simultaneous complete loss of BAP1 and PBRM1 has not previously been described in peritoneal mesothelioma, and seems to be a rare event also in clear-cell renal cell carcinomas in which loss of gene expression of BAP1 and PBRM1 rarely coexist (Peña-Llopis et al. 2012). Complete deletion of PBRM1 alone or in combination with BAP1 in this case could therefore represent a distinct genotype that could be hypothesized to confer increased sensitivity to DNA damaging agents. Interestingly, in a 2015 case report otherwise very similar to our study, BAP1 and PBRM1 aberrations were not detected, and the response to similar treatment was poor (Sheffield et al. 2015). The suggested immune response in T2 could also contribute to the therapeutic effect, as it could keep the tumor in a state of functional dormancy (Mittal et al. 2014). Infiltrating CD8-expressing T lymphocytes are associated with improved clinical outcomes in a broad range of tumor types (Barnes and Amir 2017).
No standard-of-care is defined in platinum/pemetrexed-resistant peritoneal mesothelioma, and targeted treatments must be considered experimental. Tumors with defective HR repair (i.e., BAP1 loss) may be sensitive to PARP inhibitors (McCabe et al. 2006), and a trial recently opened for use of niraparib in BAP1 deficient cancers, including mesothelioma (NCT03207347), suggesting PARP inhibition as a potential experimental treatment option. Another alternative based on BAP1 and PBRM1 deficiency is inhibition of EZH2 (enhancer of zeste homolog 2), the enzymatic subunit of polycomb repressive complex 2 (PRC2) (Kim and Roberts 2016; Morel et al. 2017). The inhibitor exploits synthetic lethality by selectively killing chromatin remodeling-deficient tumor cells that rely on PRC2 for survival (Morel et al. 2017). A functional dependency of EZH2 activity has been described for PBRM1 and BAP1 aberrations in tumors and cell lines, respectively (Kim et al. 2015; LaFave et al. 2015). The EZH2 inhibitor tazemetostat is being explored clinically, including in BAP1-deficient mesothelioma (NCT02860286), indicating that this could be a novel therapeutic possibility. Increased activation of the PI3K/AKT/mTOR pathway in T2 may suggest targeting this commonly deregulated pathway in mesothelioma (Suzuki et al. 2009). Clinical benefit was observed for PI3K-mTOR inhibitor apitolisib in two cases of peritoneal mesothelioma (Dolly et al. 2017) and AKT-inhibitors are effective in mesothelioma cell lines (Yamaji et al. 2017). Importantly, increased activity in this pathway could explain platinum resistance at rechallenge (Ohmichi et al. 2005). Immune check-point inhibition is being explored in pleural mesothelioma (NCT03048474, NCT02716272), but in our case the evidence to support such treatment is not conclusive. Low TMB, consistent with the tumor being microsatellite stable, would suggest restricted likelihood of immunotherapy response (Goodman et al. 2017), as would a more mesenchymal phenotype in T2 (Shields et al. 2017). On the other hand, increased CD8 expression in T2 could indicate increased cytotoxic T-cell infiltration, which is associated with response to immune checkpoint inhibitors (Tumeh et al. 2014; Shields et al. 2017). A recent study in patients with clear cell renal cell carcinoma showed that loss of functional PBRM1 was associated with increased overall survival after anti-programmed cell death protein-1 treatment (Miao et al. 2018). Total loss of PBRM1 in our case might therefore suggest response to such treatment.
In conclusion, integrative molecular analysis revealed LATS1/2 as the main mutational drivers of the disease together with total loss of both BAP1 and PBRM1, which also may contribute to the extraordinary chemotherapy response. The molecular profile suggests potential benefit from experimental targeting of PARP, EZH2, the PI3K/AKT/mTOR pathway, and possibly also from immune checkpoint inhibition.
METHODS
Patient Samples
Tumor tissue was harvested at two timepoints, before (T1, n = 3) and after (T2, n = 1) chemotherapy. The tissue samples were frozen in liquid nitrogen immediately after resection and stored at −80°C. Two HE-stained sections per sample were assessed for tumor content (>50%) by a pathologist. The tissue samples were homogenized and disrupted using TissueLyzer LT from QIAGEN. DNA/RNA was then extracted from the lysate using the AllPrep DNA/RNA/miRNA Universal Kit automated on the QIAcube (QIAGEN). DNA/RNA concentrations were evaluated using ThermoFisher NanoDrop spectrophotometer and RNA integrity was evaluated by Agilent Technologies Bioanalyzer RNA 6000 Nano kit. All tumor samples had high nucleic acid purity (Abs260/280 >1.8) and RIN values around 9. DNA from EDTA-blood of the patient was used as normal reference for WES-sequencing.
Whole-Exome Sequencing (WES)
WES of DNA from two fresh frozen biopsies and one matching control sample (blood) was performed at the Genomics Core Facility Oslo (Oslo University Hospital, Norway). Library preparation was performed using SureSelectXT Human All Exon V6 + Cosmic (Agilent), following manufacturer's instructions. Exome libraries were sequenced paired-end 2 × 100 bp using sequencing by synthesis (SBS) chemistry v3 on an Illumina HiSeq2500. Raw sequencing data was converted to FASTQ files and demultiplexed using the Illumina bcl2fastq v2 software. Sequencing coverage data is presented in Table 2.
Table 2.
Mean coverage data of exome sequencing
| Sample | Mean coverage | % bases ≥100 |
|---|---|---|
| Normal | 170.93 | 78.4 |
| T1 | 245.97 | 88.8 |
| T2 | 233.71 | 85.4 |
We applied a bioinformatics pipeline to detect acquired single nucleotide variants and short insertions and deletions in the two tumor samples. Initially, sequence reads of the control sample and the tumor samples were aligned to the human reference genome (build b37 with an added decoy contig) using BWA-mem v0.7.15 (Li and Durbin 2009). Next, marking of duplicates was performed with Picard tools (v.2.5.0); GATK tools (v3.7) were used for two-step local realignment around INDELS (each tumor-normal sample pair was processed jointly), followed by base quality recalibration and calculation of coverage statistics (McKenna et al. 2010). Somatic SNV detection on the two tumor-normal pairs was performed with MuTect and Strelka (Saunders et al. 2012; Cibulskis et al. 2013). Strelka alone was used for somatic INDEL detection. The total set of somatic variants identified by MuTect and Strelka were further limited to those with a minimum sequencing depth of 20 (both normal and tumor samples). For copy-number detection, we used FACETS, which performs allele-specific copy-number analysis corrected for tumor purity, ploidy, and clonal heterogeneity (Shen and Seshan 2016). Finally, we applied the Personal Cancer Genome Reporter (Nakken et al. 2018) an annotation pipeline intended for clinical interpretation of somatic SNVs/InDels and copy-number aberrations, which is built upon a combination of Ensembl's Variant Effect Predictor (VEP) and vcfanno (McLaren et al. 2016; Pedersen et al. 2016). Germline variants (SNVs/InDels) in the control sample were identified using the outlined “Best Practices for Germline SNP & Indel Discovery in Whole Genome and Exome Sequence” developed through GATK/Broad Institute. Specifically, we called germline variants with GATK's HaplotypeCaller tool (v3.7) on the existing alignment for the control sample. The variants were subsequently annotated using an in-house developed cancer predisposition report tool (https://github.com/sigven/cpsr).
Mutational Signatures
We assessed the relative contribution of 30 established mutational signatures (COSMIC) within the somatic base substitution sets of the tumor samples. Specifically, we applied the deconstructSigs framework (PMID:26899170), in which signatures with a weight less than 0.06 were discarded, and the search space per tumor sample were limited to a maximum of six mutational signatures (default settings in v 1.8.0).
RNA Sequencing
Total RNA sequencing of high-quality RNA from four tumor samples, before (n = 3) and after (n = 1) chemotherapy, was performed at the Genomics Core Facility Oslo (Oslo University Hospital). RNA sequencing libraries were generated using TruSeq Stranded Total RNA Gold Sample Preparation Kit v2 (Illumina Inc.), and 1 µg total RNA starting material according to the manufacturer's instructions. The libraries were quality controlled using Agilent Tape-station for size distribution, and quantified using Agilent qPCR kit for Illumina sequencing libraries. Paired-end sequencing (2 × 75 bp) has been performed on an Illumina NextSeq500 sequencer using v2 chemistry. Raw sequencing data was converted to FASTQ files and demultiplexed using the Illumina bcl2fastq v2 software.
Salmon was used for mapping reads to the reference genome, using default parameters (Patro et al. 2017). As reference genome, we used the Ensemble annotation based on the genome build GRCh38, found at ftp://ftp.ensembl.org/pub/release-90/fasta/homo_ sapiens//cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz. To normalize between samples the TMM (trimmed mean of M values) function of the edgeR R/bioconductor package was used on the number of reads per transcript (Robinson et al. 2010; Robinson and Oshlack 2010). To identify transcripts differentially expressed after treatment, we used the voom function of the limma R/bioconductor using default parameters (Law et al. 2014; Ritchie et al. 2015). Transcripts with Benjamini Hochberg false discovery rate values (q-values) less than 0.1 were taken to functional analysis using IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). R code can be found at GitHub repository https://github.com/staaln/mesothelioma.
smallRNA Sequencing
RNA from two tumor samples (before and after treatment) with high quality were used to prepare small RNA NGS libraries (RIN > 6), using TruSeq Small RNA Library preparation protocol. Successfully prepared libraries were sequenced using Illumina HiSeq 2500 High-Throughput Sequencer using single end sequencing (50 bp).
3′ adapter sequences were automatically identified and trimmed; reads were quality-filtered (Q 33) and reads within length of 18 and 27 nt were retained for mapping using sRNAbench (Rueda et al. 2015), fastx-toolkit and custom perl scripts. Reads were mapped to MirGeneDB (Fromm et al. 2015) using bowtie1.2 (Langmead et al. 2009), requiring an 18 nt seed sequence of zero mismatches to avoid cross-mapping. Mapped reads were counted using “summarizeOverlaps()” function from the “GenomicAlignments” Bioconductor package (Lawrence et al. 2013).
ADDITIONAL INFORMATION
Data Deposition and Access
All data that support the findings of this study and that do not compromise research participant privacy are available as Supplemental Data. All data that may compromise research participant privacy, subject to GDPR regulations (e.g., raw sequence data), are available for inspection upon request to the corresponding author on nondisclosure terms. The variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV000886625–SCV000886627.
Ethics Statement
The patient was included following informed consent in an ongoing observational study on peritoneal surface malignancies at Oslo University Hospital (NCT02073500). Additionally, she has consented to publication of this case report (specifically including radiological and histological images).
Author Contributions
C.L.-A. and K.F. conceived of and planned the study and drafted the manuscript. This highly multidisciplinary project required essential intellectual input regarding data generation and interpretation from all coauthors: surgical and oncological management (S.G.L. and Br.D.), radiology (L.J.), pathology (Be.D.), multilevel molecular analysis (Si.N., St.N., B.F., L.B.A., T.W.A., A.T.K., E.H.). All authors contributed to the critical revision of the manuscript.
Funding
This research is funded by the Norwegian Cancer Society (grant number 4499184) and B.F. of the South-Eastern Norway Regional Health Authority (grant number 2014041).
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Barnes TA, Amir E. 2017. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer 117: 451–460. 10.1038/bjc.2017.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borczuk AC, Pei J, Taub RN, Levy B, Nahum O, Chen J, Chen K, Testa JR. 2016. Genome-wide analysis of abdominal and pleural malignant mesothelioma with DNA arrays reveals both common and distinct regions of copy number alteration. Cancer Biol Ther 17: 328–335. 10.1080/15384047.2016.1145850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, Creaney J, Lake RA, Zakowski MF, Reva B, et al. 2011. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet 43: 668–672. 10.1038/ng.855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. 2013. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol 15: 201–213. 10.1038/ncb2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219. 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czimmerer Z, Varga T, Kiss M, Vázquez CO, Doan-Xuan QM, Rückerl D, Tattikota SG, Yan X, Nagy ZS, Daniel B, et al. 2016. The IL-4/STAT6 signaling axis establishes a conserved microRNA signature in human and mouse macrophages regulating cell survival via miR-342-3p. Genome Med 8: 63 10.1186/s13073-016-0315-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolly SO, Migali C, Tunariu N, Della-Pepa C, Khakoo S, Hazell S, de Bono JS, Kaye SB, Banerjee S. 2017. Indolent peritoneal mesothelioma: PI3K-mTOR inhibitors as a novel therapeutic strategy. ESMO Open 2: e000101 10.1136/esmoopen-2016-000101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudda JC, Salaun B, Ji Y, Palmer DC, Monnot GC, Merck E, Boudousquie C, Utzschneider DT, Escobar TM, Perret R, et al. 2013. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 38: 742–753. 10.1016/j.immuni.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MD, Mazu T, Miles JS, Darling-Reed S, Flores-Rozas H. 2014. Inactivation of chromatin remodeling factors sensitizes cells to selective cytotoxic stress. Biologics 8: 269–280. 10.2147/btt.S67046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm B, Billipp T, Peck LE, Johansen M, Tarver JE, King BL, Newcomb JM, Sempere LF, Flatmark K, Hovig E, et al. 2015. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu Rev Genet 49: 213–242. 10.1146/annurev-genet-120213-092023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs O. 2006. [EVI1 and its role in myelodysplastic syndrome, myeloid leukemia and other malignant diseases]. Cas Lek Cesk 145: 619–624. [PubMed] [Google Scholar]
- Gomez M, Gomez V, Hergovich A. 2014. The Hippo pathway in disease and therapy: cancer and beyond. Clin Transl Med 3: 22 10.1186/2001-1326-3-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. 2017. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 16: 2598–2608. 10.1158/1535-7163.Mct-17-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Wang Y, Liu M, Bi X, Bao J, Zeng N, Zhu Z, Mo Z, Wu C, Chen X. 2012. MiR-21 regulates epithelial-mesenchymal transition phenotype and hypoxia-inducible factor-1α expression in third-sphere forming breast cancer stem cell-like cells. Cancer Sci 103: 1058–1064. 10.1111/j.1349-7006.2012.02281.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H. 1999. The transcription factor Evi-1. Int J Biochem Cell Biol 31: 1367–1371. 10.1016/S1357-2725(99)00064-3 [DOI] [PubMed] [Google Scholar]
- Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, Tarpey P, Danilova L, Drill E, Gibb EA, et al. 2018. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov 8: 1548–1565. 10.1158/2159-8290.Cd-18-0804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopson S, Thompson MJ. 2017. BAF180: its roles in DNA repair and consequences in cancer. ACS Chem Biol 12: 2482–2490. 10.1021/acschembio.7b00541. [DOI] [PubMed] [Google Scholar]
- Janes PW, Slape CI, Farnsworth RH, Atapattu L, Scott AM, Vail ME. 2014. EphA3 biology and cancer. Growth Factors 32: 176–189. 10.3109/08977194.2014.982276 [DOI] [PubMed] [Google Scholar]
- Jiang H, Zhang G, Wu JH, Jiang CP. 2014. Diverse roles of miR-29 in cancer (review). Oncol Rep 31: 1509–1516. 10.3892/or.2014.3036 [DOI] [PubMed] [Google Scholar]
- Joseph NM, Chen YY, Nasr A, Yeh I, Talevich E, Onodera C, Bastian BC, Rabban JT, Garg K, Zaloudek C, et al. 2017. Genomic profiling of malignant peritoneal mesothelioma reveals recurrent alterations in epigenetic regulatory genes BAP1, SETD2, and DDX3X. Mod Pathol 30: 246–254. 10.1038/modpathol.2016.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Roberts CW. 2016. Targeting EZH2 in cancer. Nat Med 22: 128–134. 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et al. 2015. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med 21: 1491–1496. 10.1038/nm.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Bhagwandin S, Labow DM. 2017. Malignant peritoneal mesothelioma: a review. Ann Transl Med 5: 236 10.21037/atm.2017.03.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H. 1998. The oncoprotein Evi-1 represses TGF-β signalling by inhibiting Smad3. Nature 394: 92–96. 10.1038/27945 [DOI] [PubMed] [Google Scholar]
- LaFave LM, Béguelin W, Koche R, Teater M, Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T, Konstantinoff K, et al. 2015. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med 21: 1344–1349. 10.1038/nm.3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblay N, Leprêtre F, Le Stang N, Gautier-Stein A, Villeneuve L, Isaac S, Maillet D, Galateau-Sallé F, Villenet C, Sebda S, et al. 2017. BAP1 is altered by copy number loss, mutation, and/or loss of protein expression in more than 70% of malignant peritoneal mesotheliomas. J Thorac Oncol 12: 724–733. 10.1016/j.jtho.2016.12.019 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang S, Yan W, Yang J, Qin YJ, Lin XL, Xie RY, Wang SC, Jin W, Gao F, et al. 2015. MicroRNA-19 triggers epithelial-mesenchymal transition of lung cancer cells accompanied by growth inhibition. Lab Invest 95: 1056–1070. 10.1038/labinvest.2015.76 [DOI] [PubMed] [Google Scholar]
- Liang Z, Li Y, Huang K, Wagar N, Shim H. 2011. Regulation of miR-19 to breast cancer chemoresistance through targeting PTEN. Pharm Res 28: 3091–3100. [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, et al. 2006. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66: 8109–8115. 10.1158/0008-5472.Can-06-0140 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. 2016. The Ensembl Variant Effect Predictor. Genome Biol 17: 122 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Guan KL. 2016. Mechanisms of Hippo pathway regulation. Genes Dev 30: 1–17. 10.1101/gad.274027.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bossé D, Wankowicz SM, Cullen D, et al. 2018. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359: 801–806. 10.1126/science.aan5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal D, Gubin MM, Schreiber RD, Smyth MJ. 2014. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol 27: 16–25. 10.1016/j.coi.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanaga A, Masuda M, Tsuta K, Kawasaki K, Nakamura Y, Sakuma T, Asamura H, Gemma A, Yamada T. 2015. Hippo pathway gene mutations in malignant mesothelioma: revealed by RNA and targeted exon sequencing. J Thorac Oncol 10: 844–851. 10.1097/jto.0000000000000493 [DOI] [PubMed] [Google Scholar]
- Morel D, Almouzni G, Soria JC, Postel-Vinay S. 2017. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann Oncol 28: 254–269. 10.1093/annonc/mdw552 [DOI] [PubMed] [Google Scholar]
- Mott FE. 2012. Mesothelioma: a review. Ochsner J 12: 70–79. [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Mizuno T, Taniguchi T, Fujii M, Ishiguro F, Fukui T, Akatsuka S, Horio Y, Hida T, Kondo Y, et al. 2011. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res 71: 873–883. 10.1158/0008-5472.Can-10-2164 [DOI] [PubMed] [Google Scholar]
- Nakken S, Fournous G, Vodák D, Aasheim LB, Myklebost O, Hovig E. 2018. Personal Cancer Genome Reporter: variant interpretation report for precision oncology. Bioinformatics 34: 1778–1780. 10.1093/bioinformatics/btx817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. 2005. Mechanisms of platinum drug resistance. Trends Pharmacol Sci 26: 113–116. 10.1016/j.tips.2005.01.002 [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Layer RM, Quinlan AR. 2016. Vcfanno: fast, flexible annotation of genetic variants. Genome Biol 17: 118 10.1186/s13059-016-0973-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, Leng N, Pavía-Jiménez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, et al. 2012. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 44: 751–759. 10.1038/ng.2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda A, Barturen G, Lebrón R, Gómez-Martin C, Alganza Á, Oliver JL, Hackenberg M. 2015. sRNAtoolbox: an integrated collection of small RNA research tools. Nucleic Acids Res 43: W467–W473. 10.1093/nar/gkv555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28: 1811–1817. 10.1093/bioinformatics/bts271 [DOI] [PubMed] [Google Scholar]
- Sheffield BS, Tinker AV, Shen Y, Hwang H, Li-Chang HH, Pleasance E, Ch'ng C, Lum A, Lorette J, McConnell YJ, et al. 2015. Personalized oncogenomics: clinical experience with malignant peritoneal mesothelioma using whole genome sequencing. PLoS One 10: e0119689 10.1371/journal.pone.0119689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Seshan VE. 2016. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44: e131 10.1093/nar/gkw520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields BD, Mahmoud F, Taylor EM, Byrum SD, Sengupta D, Koss B, Baldini G, Ransom S, Cline K, Mackintosh SG, et al. 2017. Indicators of responsiveness to immune checkpoint inhibitors. Sci Rep 7: 807 10.1038/s41598-017-01000-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Oravecz-Wilson K, Mathewson N, Wang Y, McEachin R, Liu C, Toubai T, Wu J, Rossi C, Braun T, et al. 2015. Mature T cell responses are controlled by microRNA-142. J Clin Invest 125: 2825–2840. 10.1172/jci78753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Murakami H, Kawaguchi K, Tanigushi T, Fujii M, Shinjo K, Kondo Y, Osada H, Shimokata K, Horio Y, et al. 2009. Activation of the PI3K-AKT pathway in human malignant mesothelioma cells. Mol Med Rep 2: 181–188. 10.3892/mmr_00000081 [DOI] [PubMed] [Google Scholar]
- Szikriszt B, Póti Á, Pipek O, Krzystanek M, Kanu N, Molnár J, Ribli D, Szeltner Z, Tusnády GE, Csabai I, et al. 2016. A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol 17: 99 10.1186/s13059-016-0963-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LH, Schild D. 2001. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Res 477: 131–153. 10.1016/S0027-5107(01)00115-4 [DOI] [PubMed] [Google Scholar]
- Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, Fitamant J, Bardeesy N, Camargo FD, Guan KL. 2012. YAP mediates crosstalk between the Hippo and PI3K-TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol 14: 1322–1329. 10.1038/ncb2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. 2014. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515: 568–571. 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Roberts CW. 2011. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11: 481–492. 10.1038/nrc3068 [DOI] [PubMed] [Google Scholar]
- Woodard GA, Yang YL, You L, Jablons DM. 2017. Drug development against the hippo pathway in mesothelioma. Transl Lung Cancer Res 6: 335–342. 10.21037/tlcr.2017.06.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji M, Ota A, Wahiduzzaman M, Karnan S, Hyodo T, Konishi H, Tsuzuki S, Hosokawa Y, Haniuda M. 2017. Novel ATP-competitive Akt inhibitor afuresertib suppresses the proliferation of malignant pleural mesothelioma cells. Cancer Med 6: 2646–2659. 10.1002/cam4.1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. 2010. GATA3 inhibits breast cancer metastasis through the reversal of epithelial–mesenchymal transition. J Biol Chem 285: 14042–14051. 10.1074/jbc.M110.105262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Aerts JG, Popat S, Fennell DA. 2017. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer 17: 475–488. 10.1038/nrc.2017.42 [DOI] [PubMed] [Google Scholar]
- Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, Barbour H, Corbeil L, Hébert J, Drobetsky E, et al. 2014. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci 111: 285–290. 10.1073/pnas.1309085110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. 2010. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non–small cell lung cancer (NSCLC). Clin Chim Acta 411: 846–852. 10.1016/j.cca.2010.02.074 [DOI] [PubMed] [Google Scholar]
- Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. 2007. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci 104: 7080–7085. 10.1073/pnas.0702409104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study and that do not compromise research participant privacy are available as Supplemental Data. All data that may compromise research participant privacy, subject to GDPR regulations (e.g., raw sequence data), are available for inspection upon request to the corresponding author on nondisclosure terms. The variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV000886625–SCV000886627.