

Abstract
Recent progress in cellular reprogramming technology and lineage-specific cell differentiation has provided great opportunities for translational research. Because virus-based gene delivery is not a practical reprogramming protocol, protein-based reprogramming has been receiving attention as a safe way to generate reprogrammed cells. However, the poor efficiency of the cellular uptake of reprogramming proteins is still a major obstacle. Here, we reported key factors which improve the cellular uptake of these proteins. Purified red fluorescent proteins fused with 9xLysine (dsRED-9K) as a cell penetrating peptide were efficiently delivered into the diverse primary cells. Protein delivery was improved by the addition of amodiaquine. Furthermore, purified dsRED-9K was able to penetrate all cell lineages derived from mouse embryonic stem cells efficiently. Our data may provide important insights into the design of protein-based reprogramming or differentiation protocols.
Keywords: Amodiaquine (AQ), Cell Penetrating Peptide (CPP), Differentiation, Polylysine (9K), Reprogramming
INTRODUCTION
Autologous cell replacement therapy has emerged as an alternative approach to treat a wide range of degenerative diseases. Recently, rapid progress in the field of reprogramming of somatic cells to induced pluripotent stem cells (iPSC) has allowed cell-based therapy to be plausible (1). In order to fully exploit the potential of iPSC, it is important to establish an efficient and safe way to facilitate its differentiation into specific cell lineages. In the last decade, retroviral or lentiviral introduction of key transcription factor-encoding genes and their overexpression have been extensively used in the specific cell lineage differentiation (2). However, this genetic engineering causes insertional mutagenesis and unpredictable genetic dysfunction (2, 3).
To overcome this safety issue, direct delivery of key proteins using cell penetrating peptides (CPPs) into the cells has been extensively studied. CPPs, also known as protein transduction domains (PTDs), are short peptides comprising 5–30 amino acids (4), which contain a high proportion of basic amino acids (e.g., arginine or lysine) (5). They have the ability to pass through tissue and cell membranes without interacting with specific receptors (6), and thus are attractive candidates to transport biologically active cargos, such as small molecules, DNAs, siRNAs, and polypeptides. Although the mechanisms of cellular internalization of CPPs have not been fully characterized, it is generally accepted that most CPP-cargo conjugates enter cells by either direct translocation (energy-independent) or endocytosis (energy-dependent) (7, 8).
Recently, CPP-based protein reprogramming was explored to generate iPSCs from human and mouse fibroblasts (9–12). Among these studies, Kim et al., (10) and Park et al., (12) successfully generated human and mouse iPSCs, respectively, by treating human fibroblasts and mouse hepatocytes with total cell extracts from HEK293 cells expressing each of the four reprogramming factors fused with 9R. Although human and mouse iPSCs could be successfully generated by protein-based reprogramming, the efficiency of this approach is extremely low (below 0.001%) and reprogramming takes a longer time as compared to virus-based methods. Therefore, improvements that increase the efficiency of protein-based reprogramming are necessary for the method to be practical.
Here, we expressed red fluorescent proteins (dsRED) fused with 9xArginine (9R) or 9xLysine (9K) in E. coli, purified using Ni-affinity chromatography, and checked its cell penetrating ability into fibroblast cells. We observed a better penetrating capacity of the purified dsRED-9K protein as compared to the purified ds-RED-9R protein and whole extracts from HEK293 cells transfected with a dsRED-9K mammalian expression vector. We determined the optimal concentration and treatment time of dsRED-9K protein for efficient delivery. Moreover, we found that amodiaquine (AQ) markedly increased internalization of dsRED-9K proteins into cells. Finally, we found that purified dsRED-9K proteins were able to penetrate efficiently into all the different cell lineages expected in the course of neuronal differentiation from mouse embryonic stem cells (mESCs).
RESULTS
Purified dsRED-9K protein can efficiently penetrate different cells
In order to compare the capability of CPP to cross the cell membrane, we made constructs expressing dsRED proteins fused with either 9R or 9K. The scheme for the three vectors is shown in Fig. 1A, where dsRED vector alone did not contain CPP. Plasmids pET15bdsRED, pET15bdsRED-9R, and pET15bdsRED-9K were each transformed into E. coli BL21(DE3)pLysS cells and the expression level was checked after induction with IPTG. Each protein was efficiently purified using Ni-affinity chromatography as described in the Materials and Methods section. Fig. 1B shows the penetrative capabilities of dsRED proteins with different CPP-tags in different mammalian cell lines, including HEK293 cells, human newborn fibroblasts, and mouse embryonic fibroblasts. While dsRED alone did not show any fluorescence in all tested cells, both dsRED-9R and dsRED-9K proteins were shown to be present in the cytoplasm, supporting the functional role of CPP in the penetration and delivery of proteins into mammalian cells. In addition, we found that dsRED-9K proteins could penetrate the cells more readily than dsRED-9R proteins, implying greater delivery efficiency of 9K as a CPP. Previously, total extracts from cells expressing each of the reprogramming proteins fused with CPP were used to generate human iPSCs with low reprogramming efficiency (10). However, the use of total extracts imposed limitations due to the exerted cytotoxicity (12). We compared cell penetrating efficiency of purified dsRED-9K to that of the whole extracts from HEK293 cells expressing dsRED-9K. As shown in Fig. 1C, purified dsRED-9K proteins could penetrate cells more efficiently and were scattered throughout the cytoplasm of human fibroblast cells compared to whole cell extracts. dsRED-9K proteins from whole cell extracts showed weaker signals in cell bodies with red clumps around the cell periphery and lots of puncta, possibly due to aggregation and
Fig. 1.
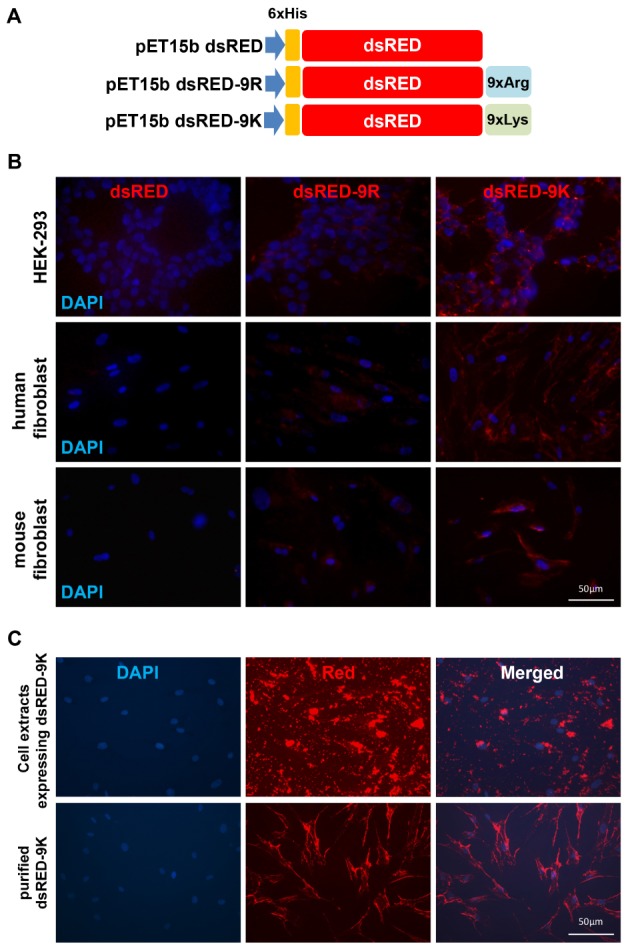
(A) Schematic diagram of E. coli expression vectors for dsRED, dsRED-9R, and dsRED-9K. (B) Cell penetrating capacity of dsRED, dsRED-9R, and dsRED-9K in both an immortalized cell line and primary cells which were cultured in 24-well plates and treated with 20 μg/ml of either dsRED-9R or dsRED-9K proteins. After 6 hrs of incubation, fluorescent images were captured with matching exposure to test the efficiency of penetration. Penetrations of both dsRED-9R and dsRED-9K, but not dsRED alone, were observed in all three target cell types. (C) Penetrating efficiency of purified dsRED-9K was compared to that of the extracts prepared from HEK293 cells expressing pCMV dsRED-9K. Human fibroblasts on 12-well plates were treated with purified dsRED-9K and whole cell extracts of HEK293 cells transfected with dsRED-9K construct. After 6 hrs of incubation, fluorescent images were captured. Purified dsRED-9K showed better distribution of signals in the cell body and surface.
Optimization of incubation times for dsRED-9K protein delivery into the cells
Determining the optimal incubation time for protein delivery into the cells might be important. A time course of purified dsRED-9K delivery was compiled to determine the optimal condition for protein delivery. As shown in Fig. 2A, purified dsRED-9K proteins could penetrate within 30 min of treatment and showed increasing penetration rates with time. At 8.5 hrs of treatment, cell penetration of dsRED-9K proteins reached to its maximum (Fig. 2B), and thereafter the intensity of red fluorescence decreased. Therefore, we treated cells with dsRED proteins for 6–8 hrs for the rest of the study.
Fig. 2.

(A) Penetration time-course of purified dsRED-9K was checked. HEK293 cells were plated in a 12-well plate and treated with 20 μg of dsRED-9K in their culture medium for 24 hrs. Fluorescent images were acquired 8 times in 24 hrs with matching exposure. (B) Images were analyzed by CellProfiler (version 2.1.1; cellprofiler.org) for measuring the fluorescence intensity. Three independent fields from each time point were used with the matching exposure. Each cell was identified by object identification modules, followed by measurement of red fluorescent intensity.
Amodiaquine enhanced the 9K-mediated penetrating efficiency
Recently, it has been shown that the antimalarial drug chloroquine showed enhanced cellular uptake and inhibited the degradation of macromolecules, such as nucleic acids and peptides in cells (13, 14). Another antimalarial drug, AQ, is a well characterized drug and is known to inhibit cell intoxication by interacting with proteins involved in lysosomal function (15). In this respect, we tested the effect of AQ on the cellular uptake of dsRED-9K in mammalian cells. Both human fibroblasts (Fig. 3A) and Chinese hamster ovary (CHO) cells (Fig. 3B) were co-treated with dsRED-9K proteins and different concentrations of AQ (10 μM to 100 μM) for 6 hrs. As shown in Fig. 3A and 3B, treatment with AQ increased the penetration efficiency of dsRED-9K proteins into both CHO cells and fibroblasts in a dose-dependent manner.
Fig. 3.
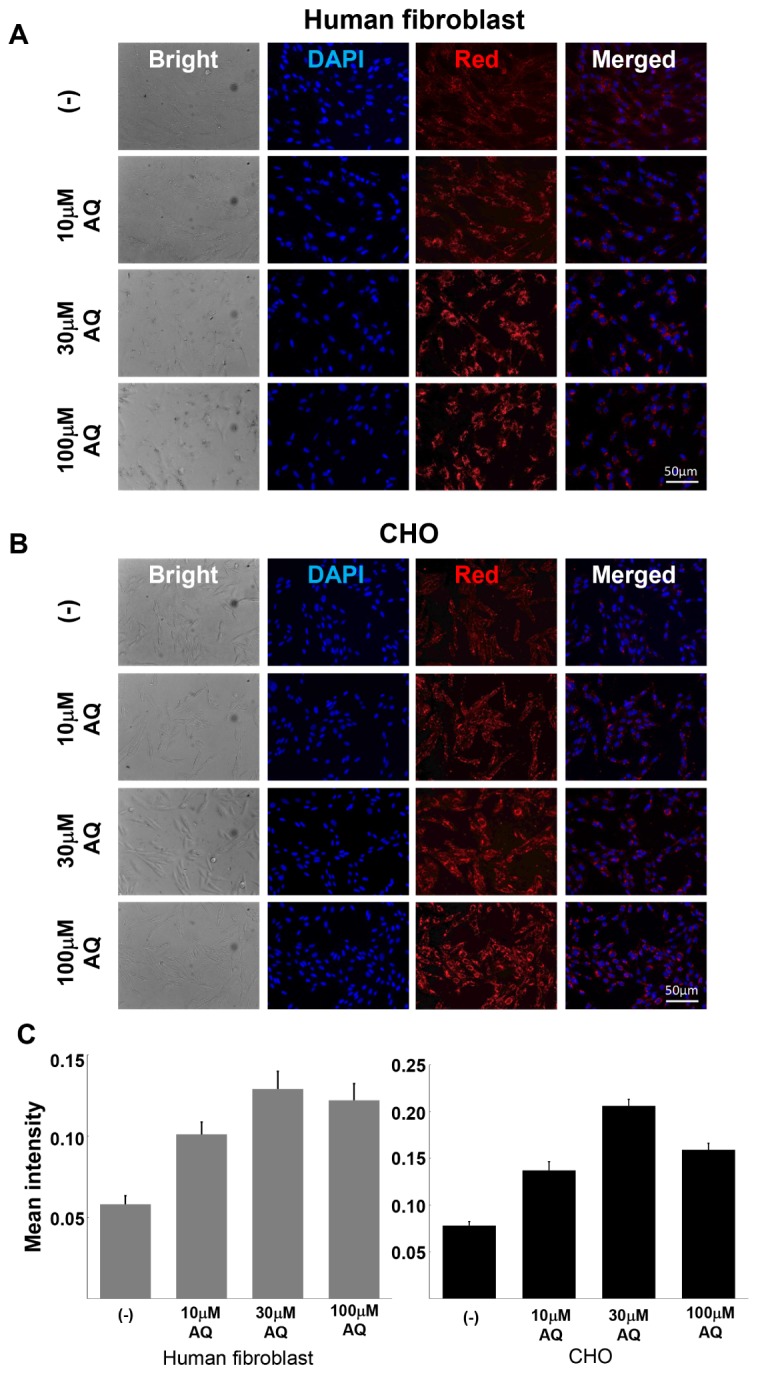
Transduction ability of dsRED-9K was enhanced by amodiaquine (AQ). Human fibroblasts (A) and CHO cells (B) were pre-treated with different concentrations of AQ and then treated with 20 μg/ml of dsRED-9K. After 6 hrs of incubation, fluorescent images were captured. AQ further increased the penetrating efficiency of the purified dsRED-9K proteins in a dose-dependent manner. (C) Images were analyzed by CellProfiler.
dsRED-9K protein was able to penetrate all cell lineages derived by in vitro differentiation of mESCs
It is also necessary to determine the optimal condition for protein delivery into mESC-derived cells during in vitro differentiation. For this, we checked whether dsRED-9K can penetrate mESC-derived cells (e.g. ESC, embryonic bodies (EB), neural precursor (NPC), and fully differentiated cells). The pluripotent mESCs were differentiated using our neuronal differentiation protocol described in the Materials and Methods section (Fig. 4A). Then, mESC-derived cells were treated with purified dsRED-9K proteins for 6 hrs in the presence of 10 μM AQ. As shown in Fig. 4B, we found that the purified dsRED-9K protein was efficiently delivered into mESC-derived cells at all stages, including ESCs (Fig. 4B), EBs (Fig. 4C), NPCs (Fig. 4D), and neurons (Fig. 4E). The different cell types were stained with their corresponding markers after differentiation. Nestin and Tuj1 were used for neural progenitor cells and fully differentiated neurons, respectively. Together, these findings strongly support the fact that proteins fused with 9K can be efficiently delivered into ESC-derived cells at any stage, implying that candidate proteins (e.g. transcription factors) fused with 9K might be used in order to enhance differentiation efficiency.
Fig. 4.

(A) mESCs were differentiated using the neuronal differentiation protocol described in the Materials and Methods section. The formation of typical cell types expected in the course of neuronal differentiation were observed. (B) The penetration of dsRED-9K was checked in mouse ES cells cultured on 24-well plates and confirmed by detection of the emitted red fluorescence. (C) ES cells differentiated to embryoid bodies (EB). 20 μg/ml of dsRED-9K was added to the culture media and the delivery of dsRED-9K was confirmed by detection of the emitted red fluorescence. (D, E) EBs were differentiated to neurons. Neural precursors (D) and neurons (E) were collected at day 16 and day 25, respectively. 20 μg/ml of dsRED-9K was added to the culture media and the delivery of dsRED-9K was confirmed by detection of the emitted red fluorescence. Nestin and Tuj1 correspond to dsRED-9K proteins in neural precursors and neurons, respectively. The dsRED-9K protein can penetrate all the cell types formed during the course of in vitro mESC differentiation.
DISCUSSION
Stem cell research and its application have the great potential for the treatment of intractable human diseases. Reprogramming somatic cells to iPSCs or differentiation of stem cells to specific cell lineages require genetic manipulation such as DNA- or virus-mediated delivery of key transcription factors. However, since genetic manipulation causes unpredictable chromosomal integrations (3), these genetically altered stem cells and differentiated cells are not suitable for clinical or biomedical uses. Thus, it is important to establish novel protocols for generating reprogrammed cells without genetic alterations. Recently, the use of CPP capable of crossing the cell membrane have been paid attentions as an alternative way for gene delivery (5, 16).
Previously, treatment of whole cell extracts from HEK293 cells expressing each of 4 reprogramming factors fused with 9R into fully differentiated fibroblast cells could successfully generated human iPSCs (10), which could subsequently differentiate to functional dopaminergic neurons and rescue motor deficits in Parkinson’s disease (PD) rat model (17). In addition, mouse iPSCs generated by protein-based reprogramming protocol showed better genomic and functional integrity than those produced by lentiviral or retroviral delivery (12). However, protein-based reprogramming efficiency was too low to be practical. Therefore, improvements that increase the efficiency are necessary for the method to become feasible.
It has been proposed that different CPPs have been developed for the delivery of macromolecules to cells (18). However, there are currently no optimized CPPs for defined transcription factors used in reprogramming and differentiation of cell lineages, because the efficiency varies substantially depending on cell type and cargo molecules. The experiments presented here have defined some critical aspects affecting protein delivery to mammalian cells, including: (i) dsRED-9K proteins could penetrate more efficiently into cells than dsRED-9R proteins, (ii) purified dsRED-9K proteins could penetrate cells more efficiently and were scattered throughout the cytoplasm compared to whole cell extracts containing dsRED-9K proteins, (iii) the red signal began to appear within 30 min of treatment with dsRED-9K protein and reached its maximum at 8.5 hrs, (iv) Addition of AQ further enhanced the delivery efficiency of dsRED-9K, and finally (v) dsRED-9K protein could be efficiently delivered to all ESC-derived cells generated by in vitro differentiation.
Although total extracts from four HEK293 cells expressing each of the four reprogramming proteins fused with 9R had been successfully used for reprogramming (10), the study had its limitations due to low efficiency and toxicity. Our results in this experiment indicated that purified dsRED-9K proteins could penetrate more efficiently and were distributed more evenly in human fibroblast cells compared to total extracts. Therefore, using purified proteins will allow us to generate iPSCs and specific cell lineages through delivery of defined transcription factors efficiently.
Previously, CQ has been shown to enhance cellular uptake and inhibit the degradation of macromolecules such as nucleic acids and peptides in cells (13, 14). AQ is a well characterized anti-malarial drug which shares a similar backbone structure with CQ, and is known to inhibit cell intoxication by interacting with proteins involved in lysosomal function (15). In our previous report, a lower concentration of AQ was shown to be a potent agonist of its target nuclear receptor, Nurr1 (19). We tested the effect of AQ on the cellular uptake of dsRED-9K. As expected, AQ increased the penetrating efficiency of dsRED-9K proteins into both Chinese hamster ovary cell (CHO) and human fibroblasts (Fig. 3). However, high concentration of AQ induced cytotoxicity in a cell-type dependent manner (20). Therefore, the concentration of AQ should be optimized for protein-based reprogramming or differentiation of cells.
Pluripotent stem cells like ESC and iPSC are able to differentiate into diverse cell types. In general, differentiation towards specific cell lineages requires genetic manipulation, mainly by overexpression of critical transcription factors involved in the lineage-specific development. For instance, many transcription factors have been known to play important roles in the dopamine (DA) neuron development (21) and their overexpression in ESCs or iPSCs by genetic manipulation was shown to facilitate DA neuron differentiation (22, 23). This genetic engineering has been mostly mediated by DNA- or virus-based delivery of key transcription factors, which results in clinically or medically unfavorable cell sources. Thus, it is important to establish the differentiation protocol without causing any genetic change. In this context, our findings about efficient delivery of dsRED-9K proteins into ESCs, EBs, and NP cells (Fig. 4) may provide an insight on how to introduce necessary transcription factors more effectively into cells in the future.
Taken together, a deeper understanding of the intracellular mechanisms related to the protein penetration and further optimization of protein delivery to specific cell lineages may provide medically safe cell sources for studying and treatment of human diseases in the future.
MATERIALS AND METHODS
Plasmid DNA constructions
dsRED, dsRED-9R and dsRED-9K DNAs were amplified by PCR and cut with Xho I and then cloned into the pET15b plasmid (Novagen), yielding three plasmids, pET15b dsRED, pET15b dsRED-9R, and pET15b dsRED-9K. The forward and reverse primer sequences for PCR amplification of dsRED-9K for mammalian expression were 5′-AAT CAG GAA TTC ATG GCC TCC TCC GAG AAC GTC ATC AC-3′ and 5′-GAA AAC CTC GAG TTA CTT CTT TTT CTT CTT CTT CTT CTT CTT CAG GAA CAG GTG GTG GCG GCC CTC-3′, respectively. Amplified PCR products were digested with EcoRI and XhoI and then cloned into the pcDNA3.1/myc-His vector (Invitrogen, USA), resulting in pCMV dsRED-9K plasmid. The EndoFree Plasmid Midi Kit (Qiagen) was used to prepare plasmids for transfection experiments. The integrity of all sequences was verified by DNA sequence analyses.
Expression and purification of recombinant proteins
pET15b dsRED, pET15b dsRED-9R and pET15b dsRED-9K plasmids were transformed into E. coli BL21(DE3)pLysS cells. Protein expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) at a final concentration of 1 mM. To maximize the soluble form of recombinant proteins, cells were further cultured overnight at 22°C. Cell pellets were washed with phosphate buffered saline (PBS), resuspended in a buffer containing 50 mM sodium phosphate (pH 8.0), 300 mM NaCl and 10 mM imidazole, and sonicated on ice. After centrifugation, the supernatant was loaded onto the column containing Ni-NTA resin (Qiagen), washed with a buffer consisting 50 mM sodium phosphate (pH 8.0), 300 mM NaCl and 20 mM imidazole, and eluted with a gradient of imidazole concentrations, from 50 mM to 500 mM. Fractions containing each protein were pooled and dialyzed against PBS. The protein concentration was determined by the bicinchoninic acid (BCA) protein assay (Pierce).
Cell Cultures
Human newborn fibroblasts (HNF) were purchased from ATCC (CCL-117) and cultured at 37°C and 5% CO2 in Dulbecco’s modified Minimal Essential Medium (DMEM; Life Technologies), supplemented with 10% fetal bovine serum (FBS; Hyclone), 2 mM L-glutamine (Life Technologies), 1xNon-essential amino acids (NEAA; Life Technologies), 1 mM β-mercaptoethanol, 100 U/ml penicillin and 100μg/ml streptomycin (Life Technologies) in a humidified atmosphere with 5% CO2 at 37°C.
Mouse embryonic stem cells (MESCs) were cultured in MESC medium, which contained DMEM supplemented with 15% FBS, 2 mM L-glutamine, 1x NEAA, 1mM β-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin and 2,000 U/ml leukemia inhibitory factor (LIF; Chemicon). mESCs were maintained on mitomycine C (10 μg/ml, Sigma-Aldrich, St. Louis, MO, USA)-treated MEF feeder layers.
Preparation of whole extracts from HEK293 cells
Using LipofectamineTM (Life Technologies), 2 μg of pCMV dsRED-9K plasmid DNA was transfected to 4 × 105 HEK293 cells. HEK293 cells were washed in PBS and resuspended in cold cell lysis buffer containing 100 mM HEPES (pH 8.2), 50 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol and protease inhibitors, followed by incubation for 30–45 min on ice. Cells were sonicated by a Labsonic-M pulse sonicator fitted with a 3-mm-diameter probe on ice until all cells were lysed. Lysates were centrifuged at 15,000 g for 15 min at 4°C. The supernatant was collected and filtered through a 0.2 μm pore-size filter membrane.
In vitro differentiation of mouse embryonic stem cells
mESCs were harvested using 0.05% trypsin-EDTA solution (Life Technologies). Subsequently, cells were transferred to low attachment bacterial culture dishes and cultured in MESC medium without LIF for 4 days for embryoid body (EB) formation. On day 4, EBs were transferred to cell culture plates and cultured in neural inducing ITSFn medium, which contained DMEM/F12 supplemented with 50 μg/ml apotransferrin (Sigma Aldrich), 5 μg/ml insulin (Sigma Aldrich), 30 nM sodium selenite (Sigma Aldrich), 250 ng/ml fibronectin (Sigma Aldrich), 100 U/ml penicillin and 100 μg/ml streptomycin. After 10–12 days of selection, neural precursors were harvested by trypsinization and plated on plates coated with 15 μg/ml poly-L-ornithine (Sigma Aldrich) and 1μg/ml fibronectin (Sigma Aldrich). Neural precursors were expanded for 4 days in N3 medium (DMEM/F-12 with 100 μg/ml apotransferrin, 5 μg/ml insulin, 30 nM sodium selenite, 20 nM progesterone, 100 nM putrescine, 1 μg/ml laminin, 100 U/ml penicillin and 100 μg/ml streptomycin) supplemented with 10 ng/ml basic fibroblast growth factor (bFGF) (Life Technologies). By removing bFGF and adding 200 μM ascorbic acid (Sigma Aldrich), cells were differentiated into neuronal phenotypes. Neurons were cultured up to 28 days.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 15 min at room temperature. After rinsing with PBS, cells were incubated with blocking/permeabilizing solution, which consists of PBS with 10% donkey serum and 0.1% Triton X-100 for 1 hr at room temperature. Subsequently, cells were incubated with primary antibodies overnight at 4°C. Primary antibodies used in this study were anti-nestin (1:1000; BD Sciences, USA) and anti-beta-III tubulin (1:500; Covance). In order to visualize the primary antibodies, cells were incubated with fluorescent-labeled secondary antibodies (Alexa 488-Alexa 594-, or Alexa 647-labeled IgG; Life Technologies) for 1 hr at room temperature. Images of stained cells were captured using an Olympus IX-70 inverted microscope, equipped with an Orca-ER CCD digital camera (Hamamatsu) and OpenLab software (Improvision). ImageJ (Wayne Rasband National Institute of Health, USA) was used for image analysis. In addition, images were analyzed by CellProfiler (version 2.1.1; cellprofiler.org) for measuring fluorescent intensity. Three independent fields from each time point were used with matching exposure.
ACKNOWLEDGEMENTS
This work was supported by St. Jude Institutional Funds (to M-J Han), the Global Excellent Technology Innovation Program (Project No. 10052590 to K. Han), the Bio & Medical Technology Development Program (NRF-2016M3A9B4918833 to C-H Park), and Basic Science Research Program (NRF-2016R1A2B4007640 to C-H Kim).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Yamanaka S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell. 2007;1:39–49. doi: 10.1016/j.stem.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 4.Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385–393. doi: 10.1016/j.molmed.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 5.El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 7.Herbig ME, Weller K, Krauss U, Beck-Sickinger AG, Merkle HP, Zerbe O. Membrane surface-associated helices promote lipid interactions and cellular uptake of human calcitonin-derived cell penetrating peptides. Biophys J. 2005;89:4056–4066. doi: 10.1529/biophysj.105.068692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 9.Nemes C, Varga E, Polgar Z, Klincumhom N, Pirity MK, Dinnyes A. Generation of mouse induced pluripotent stem cells by protein transduction. Tissue Eng Part C Methods. 2014;20:383–392. doi: 10.1089/ten.tec.2013.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim D, Kim CH, Moon JI, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, Wei W, Zhu S, et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell. 2009;4:16–19. doi: 10.1016/j.stem.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Park H, Kim D, Kim CH, et al. Increased genomic integrity of an improved protein-based mouse induced pluripotent stem cell method compared with current viral-induced strategies. Stem Cells Transl Med. 2014;3:599–609. doi: 10.5966/sctm.2013-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang S, Coles DJ, Esposito A, Mitchell DJ, Toth I, Minchin RF. Cellular uptake of self-assembled cationic peptide-DNA complexes: multifunctional role of the enhancer chloroquine. J Control Release. 2009;135:159–165. doi: 10.1016/j.jconrel.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 14.Shiraishi T, Nielsen PE. Enhanced delivery of cell-penetrating peptide-peptide nucleic acid conjugates by endosomal disruption. Nat Protoc. 2006;1:633–636. doi: 10.1038/nprot.2006.92. [DOI] [PubMed] [Google Scholar]
- 15.Qiao S, Tao S, Rojo de la Vega M, et al. The antimalarial amodiaquine causes autophagic-lysosomal and proliferative blockade sensitizing human melanoma cells to starvation- and chemotherapy-induced cell death. Autophagy. 2013;9:2087–2102. doi: 10.4161/auto.26506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frankel AD, Bredt DS, Pabo CO. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science. 1988;240:70–73. doi: 10.1126/science.2832944. [DOI] [PubMed] [Google Scholar]
- 17.Rhee YH, Ko JY, Chang MY, et al. Protein-based human iPS cells efficiently generate functional dopamine neurons and can treat a rat model of Parkinson disease. J Clin Invest. 2011;121:2326–2335. doi: 10.1172/JCI45794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guidotti G, Brambilla L, Rossi D. Cell-Penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38:406–424. doi: 10.1016/j.tips.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Kim CH, Han BS, Moon J, et al. Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2015;112:8756–8761. doi: 10.1073/pnas.1509742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heidari R, Babaei H, Eghbal MA. Amodiaquine-induced toxicity in isolated rat hepatocytes and the cytoprotective effects of taurine and/or N-acetyl cysteine. Res Pharm Sci. 2014;9:97–105. [PMC free article] [PubMed] [Google Scholar]
- 21.Smidt MP, Burbach JP. How to make a mesodiencephalic dopaminergic neuron. Nat Rev Neurosci. 2007;8:21–32. doi: 10.1038/nrn2039. [DOI] [PubMed] [Google Scholar]
- 22.Chung S, Hedlund E, Hwang M, et al. The homeodomain transcription factor Pitx3 facilitates differentiation of mouse embryonic stem cells into AHD2-expressing dopaminergic neurons. Mol Cell Neurosci. 2005;28:241–252. doi: 10.1016/j.mcn.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Andersson E, Tryggvason U, Deng Q, et al. Identification of intrinsic determinants of midbrain dopamine neurons. Cell. 2006;124:393–405. doi: 10.1016/j.cell.2005.10.037. [DOI] [PubMed] [Google Scholar]