

Abstract
Activation of transforming growth factor β (TGF‐β) combined with persistent hypoxia often affects the tumor microenvironment. Disruption of cadherin/catenin complexes induced by these stimulations yields aberrant extracellular matrix (ECM) production, characteristics of epithelial‐mesenchymal transition (EMT). Hypoxia‐inducible factors (HIF), the hallmark of the response to hypoxia, play differential roles during development of diseases. Recent studies show that localization of cadherin/catenin complexes at the cell membrane might be tightly regulated by protein phosphatase activity. We aimed to investigate the role of stabilized HIF‐1α expression by protein phosphatase activity on dissociation of the E‐cadherin/β‐catenin complex and aberrant ECM expression in lung cancer cells under stimulation by TGF‐β. By using lung cancer cells treated with HIF‐1α stabilizers or carrying doxycycline‐dependent HIF‐1α deletion or point mutants, we investigated the role of stabilized HIF‐1α expression on TGF‐β‐induced EMT in lung cancer cells. Furthermore, the underlying mechanisms were determined by inhibition of protein phosphatase activity. Persistent stimulation by TGF‐β and hypoxia induced EMT phenotypes in H358 cells in which stabilized HIF‐1α expression was inhibited. Stabilized HIF‐1α protein expression inhibited the TGF‐β‐stimulated appearance of EMT phenotypes across cell types and species, independent of de novo vascular endothelial growth factor A (VEGFA) expression. Inhibition of protein phosphatase 2A activity abrogated the HIF‐1α‐induced repression of the TGF‐β‐stimulated appearance of EMT phenotypes. This is the first study to show a direct role of stabilized HIF‐1α expression on inhibition of TGF‐β‐induced EMT phenotypes in lung cancer cells, in part, through protein phosphatase activity.
Keywords: epithelial‐mesenchymal transition, HIF‐1α, hypoxia, lung cancer, TGF‐β
1. INTRODUCTION
Mounting evidence suggests that the tumor microenvironment plays important roles in development and malignant progression of cancers and resistance of cancer cells to therapies. In normal tissue, crosstalk between epithelial cells and fibroblasts retains tight epithelial integrity through formation of an E‐cadherin/β‐catenin complex at the cell membrane1, 2 and maintains extracellular matrix (ECM) homeostasis.3, 4 Disruption of epithelial integrity causes aberrant phenotype alterations in proliferation, differentiation, adhesion, motility, and aberrant ECM production—all hallmarks of cancers.1, 2 Furthermore, dynamic imbalance in remodeling and homeostasis of ECM directly deregulates the behavior of cancer cells and cancer‐associated fibroblasts (CAF); as a consequence, cancers progress.3, 4
The tissue microenvironment often includes not only activated transforming growth factor β (TGF‐β) but is also affected by persistent hypoxia stimulation during development of cancer.5, 6 Recent studies show that TGF‐β‐induced β‐catenin translocation from E‐cadherin complexes at the cell membrane into the cytoplasm induces expression of mesenchymal genes such as fibronectin and collagen type I.7, 8, 9, 10, 11 Thus, disruption of epithelial integrity and aberrant ECM production are characteristics of epithelial‐mesenchymal transition (EMT) in cancers.12
Stabilized expression of hypoxia‐inducible factor 1α (HIF‐1α) protein has been postulated to be a hallmark of the response to hypoxia.13, 14 As a prognostic significance of HIF‐1α expression was reported in several types of cancer,15, 16 many clinical trials targeting inhibition of HIF‐1α expression have been carried out.17 Meanwhile, a recent study suggested that the stabilization of hypoxia‐inducible factor 2α (HIF‐2α) expression could prevent dissociation of the cadherin complex at the endothelial cell membrane through protein phosphatase activity, resulting in attenuation of acute lung injury.18 Thus, the exact role of stabilized HIF‐1α expression in aberrant ECM production in cancers remains elusive.
The present study aimed to determine the role of stabilized HIF‐1α expression in cadherin/catenin complex formation and inhibition of aberrant ECM production in lung cancer cells and fibroblasts under stimulation by TGF‐β.
2. MATERIALS AND METHODS
2.1. Materials
Monoclonal mouse anti‐HIF‐1α antibody (clone 54/HIF‐1α), purified anti‐β‐catenin antibody (clone 14/Beta‐Catenin), and FITC‐conjugated mouse anti‐E‐cadherin antibody (clone 36/E‐Cadherin) were purchased from BD Biosciences (Franklin Lakes, NJ, USA). Monoclonal mouse anti‐HIF‐2α antibody (clone #318404) was from R&D Systems (Minneapolis, MN, USA). Streptavidin (SAv) Alexa 594 (SAv‐594)‐conjugated anti‐mouse antibody, mouse monoclonal anti‐α‐smooth muscle actin (α‐SMA) antibody (clone 1A4), and RNAiMAX, or stealth RNAi negative control with Medium GC Duplex #2 were from Invitrogen Life Technologies (Carlsbad, CA, USA). Purified anti‐fibronectin antibody (clone TV‐1) was from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) Okadaic acid (OA), an inhibitor of protein phosphatase 2A subunit Cα (PP2A), mouse anti‐Smad2 antibody, and rabbit anti‐phospho‐Smad2 (Ser 465/467) antibody were from Cell Signaling Technology (Danvers, MA, USA). Biotinylated anti‐mouse IgG was from Vector Laboratories (Burlingame, CA, USA). Rabbit anti‐type I collagen (Col I) antibody was from Rockland (Limerick, PA, USA). Affinity‐isolated rabbit anti‐actin antibody was from Sigma‐Aldrich (St Louis, MO, USA). Hoechst33342 was from Dojindo (Kumamoto, Japan). PhosSTOP was from Roche Applied Science (Mannheim, Germany). Can Get Signal was from Toyobo Co. (Osaka, Japan). Peroxidase‐linked anti‐mouse or rabbit IgG were from Amersham Biosciences Inc. (Buckingham, UK). Doxycycline (Dox), pTRE‐Tight vector, and pTet‐On Advanced were from Clontech (Mountain View, CA, USA). Tyramide Signal Amplification Biotin System was from Perkin Elmer (Danvers, MA, USA). FG4592 (Roxadustat) was from Cayman Chemical (Ann Arbor, MI, USA).
2.2. Cells
H358 cells (with epithelial characteristics) and immortalized rat alveolar type II cell lines RLE‐6TN were purchased from ATCC. CC2512 cells (human adult normal lung fibroblasts) were from Lonza (Basel, Switzerland). The Dox‐dependent gene expression system was applied to H358 cells carrying pTet‐On Advanced (H358ON).10, 11 Cells were treated with TGF‐β (2 ng/mL) for the indicated period. FG4592 (FG) at 30 μg/mL, CoCl2 at 150 nmol/L, OA at 20 nmol/L, and/or Dox 1 μg/mL were also added to the cells for the indicated period. The cells were cultured under hypoxia (1% O2) for the indicated periods using a hypoxic chamber (Wakenyaku Co. Ltd, Kyoto, Japan).19, 20 The in vitro scratch assay was also carried out.21
2.3. Vectors and gene transfection
Hypoxia‐inducible factor 1α (NM_001530) was subcloned into the pTRE‐Tight vector (Clontech).19 For alanine (Ala) substitution of two proline (Pro) residues, Pro402 and Pro564, in HIF‐1α (P402A, P564A) in the pTRE‐Tight vector (HIF1αdPA), the QuikChange Site‐Directed Mutagenesis Kit (Stratagene, San Diego, CA, USA) was used according to the manufacturer's recommendations.19 To determine the importance of the oxygen‐dependent degradation (ODD) domain in HIF‐1α,22 HIF‐1α with amino acid 402‐603 deletion was inserted into the pTRE‐Tight vector (HIF1αΔODD).23 Into H358ON cells, HIF1αdPA or HIF1αΔODD was cotransfected with the Linear Hygromycin Marker (Clontech). After selection with hygromycin, single clones were isolated.19 This study was approved by the Nagoya University Center for Gene Research (Approval #14‐106).
2.4. Knockdown analysis using small interfering RNAs
The following double‐stranded 21‐bp RNA oligonucleotides for human HIF‐1α (HIF‐1α: NM_001530) were commercially generated (Invitrogen Life Technologies): HIF‐1α siRNA #1, 5′‐CCUCAGUGUGGGUAUAAGATT‐3′(sense) and 5′‐UCUUAUACCCACACUGAGGTT‐3′(antisense); and HIF‐1α siRNA #2, 5′‐CCAUAUAGAGAUACUCAAATT‐3′(sense) and 5′‐UUUGAGUAUCUCUAUAUGGTG‐3′(antisense). siRNA transfections were carried out according to the manufacturer's instructions.24 For transient knockdown of HIF‐1α or Stealth RNAi negative control, cells were plated at 4 × 105 cells in a 6‐cm dish. After transfection for 24 hours, the culture medium was replaced and incubated for another 96 hours for real‐time PCR and western blotting analysis.
2.5. Real‐time PCR
Total RNA was isolated from treated cells as previously described.25 Real‐time PCR assays were carried out using the GoTaq Probe 1‐Step RTqPCR System (Promega, Madison, WI, USA) with a TaqMan ABI 7300 Sequence Detection System (PE Applied Biosystems, Foster City, CA, USA). The following oligonucleotide primers and probe were used: phosphoglycerate kinase 1 (PGK1) (NM_000291.3) sense (5′‐TGGATGGGCTTGGACTGTGGT‐3′) and antisense (5′‐TGGCAGTGTCTCCACCACCTATGA‐3′), vascular endothelial growth factor A (VEGFA) (NM_001025366.2) sense (5′‐ACATCTTCAAGCCATCCTGTGTG‐3′) and antisense (5′‐TGTGCTGTAGGAAGCTCATCTCT‐3′), HIF‐1α (NM_001243084.1) sense (5′‐ATCCATGTGACCATGAGGAAATG‐3′) and antisense (5′‐TCGGCTAGTTAGGGTACACTTC‐3′), natural antisense HIF‐1α (AS‐HIF) (U85044.1) sense (5′‐AACATGACATTT AGGGACTCAACT‐3′) and antisense (5′‐TGCTTCAACACCTCCAACTC‐3′). These mRNA expression levels were normalized to the 18s rRNA mRNA signals.19
2.6. Western blotting analysis
For whole‐cell extracts, cells were harvested in ice‐cold lysis buffer and cleared by centrifugation.11, 26 The samples were then subjected to SDS‐PAGE and analyzed by immunoblotting. To detect phosphorylation levels of the targeted proteins, PhosSTOP was added to the lysis buffer and Can Get Signal was added to the dilution solution for primary and secondary antibodies.11 β‐Actin was evaluated as a loading control. After scanning the film to create a digital image, the amount of protein on the immunoblots was quantified by image software Quantity One (BioRad Laboratories).11, 27
2.7. Immunofluorescence
Double immunostaining for β‐catenin and E‐cadherin was carried out as previously described.11, 28 Nuclear staining was done by Hoechst 33342. Distribution of β‐catenin and E‐cadherin was determined by confocal laser scanning microscopy (TiEA1R; Nikon Instech Co., Tokyo, Japan) Imaging software (NIS‐Elements AR; Nikon Instech Co.) was used for the fluorescence intensities of β‐catenin, E‐cadherin, and the nucleus.11 To determine cellular distribution of β‐catenin and E‐cadherin in treated cells, fluorescent intensities over a random cross‐section of the cells were plotted.11 Relative levels of intensity at the cell membrane to total plotted intensity in the cells (C/T ratio) were evaluated.
2.8. Statistical analysis
Non‐normally distributed results were analyzed using the Mann‐Whitney test for comparison between any two groups. Furthermore, they were analyzed by nonparametric equivalents of ANOVA for multiple comparisons. P‐value <0.05 was considered statistically significant.
3. RESULTS
3.1. Effects of hypoxia and TGF‐β on EMT phenotypes in H358 cells
First, a kinetic analysis of ECM production and epithelial integrity were carried out in H358 cells cultured under dual stimulation by TGF‐β and persistent hypoxia. Western blotting analysis showed that although combined stimulations for either 6 or 24 hours could not induce fibronectin expression in the cells, de novo expression could be observed in cells treated for 96 hours (Figure 1A‐C). By double immunostaining, we next evaluated distribution of β‐catenin and E‐cadherin in cells treated with dual stimulation by TGF‐β and persistent hypoxia. Double immunostaining showed localization of β‐catenin and E‐cadherin at the cell membrane in cells treated for 6 hours (Figure 1D‐G and Figure S1A‐D,I,J). Meanwhile, the stimulation by TGF‐β and/or persistent hypoxia for 24 hours not only induced translocation of β‐catenin from the cell membrane into the cytoplasm but also internalization of E‐cadherin from the cell surface (Figure 1H‐K and Figure S1E‐H,K,L). We evaluated the longitudinal expression of HIF‐1α and HIF‐2α in the treated cells. Cells incubated under 1% O2 for 6 hours showed a significant increase in stabilized HIF‐1α expression (Figure 1L). Although substantial HIF‐1α expression was observed in the cells, hypoxia for 24 hours attenuated the stabilized HIF‐1α expression (Figure 1L). Furthermore, persistent hypoxia stimulation for 96 hours yielded a significant decrease of approximately 75% in stabilized HIF‐1α expression, compared with that of the cells stimulated for 6 hours (Figure 1L). In contrast, hypoxia stimulation did not induce stabilized HIF‐2α expression in H358 cells during the observational period (Figure 1M). HIF‐1α‐related genes such as PGK1 and VEGFA were induced by hypoxia stimulation (Figure 1N,O). The endogenous level of HIF‐1α transcription was repressed during persistent hypoxia (Figure 1P), accompanied by the finding that de novo natural antisense HIF‐1α (AS‐HIF) expression increased (Figure 1Q).29 TGF‐β also induced an increase in Smad2 phosphorylation (Figure 1R). Scratch assay showed increasing cell migration in the cells with combined stimulation by TGF‐β and/or persistent hypoxia (Figure S1M).
Figure 1.
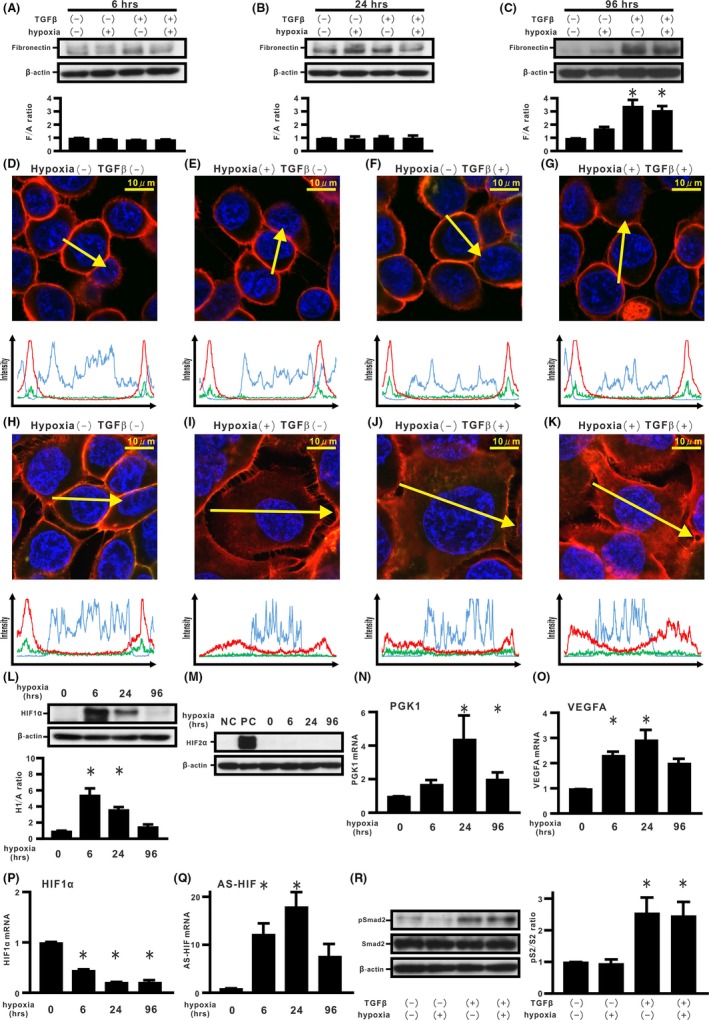
Effects of hypoxia and transforming growth factor β (TGF‐β) on epithelial‐mesenchymal transition (EMT) phenotypes in H358 cells. A‐C, H358 cells were incubated under normoxia or hypoxia (1% O2) in the absence or presence of TGF‐β for the indicated periods (A) 6 h, (B) 24 h, and (C) 96 h. By western blotting analysis, relative expression of fibronectin to β‐actin (F/A ratio) is shown in comparison to that in control cells. *P < 0.05 in comparison with control cells. D‐K, Fluorescence intensities of β‐catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in cells incubated for 6 h (D‐G) and 24 h (H‐K). Upper panels in (D‐K): cells under normoxia or hypoxia in the absence or presence of TGF‐β. Lower panels in (D‐K): fluorescence intensity of β‐catenin (red), E‐cadherin (green), and Hoechst33342 (blue) over a cross‐section of cells along the selected yellow arrows, respectively. Western blotting analyses for hypoxia inducible factor (HIF)‐1α, HIF‐2α, and β‐actin were carried out (L,M). Relative expression of HIF‐1α to β‐actin (H1/A ratio) is shown in comparison to that in control cells (lower panel in L). NC, H358 cells. PC, H358ON cells expressing Dox‐dependent HIF2αdPA treated with Dox. Expression levels of phosphoglycerate kinase 1 (PGK1) (N), VEGFA (O), HIF‐1α (P), and AS‐HIF (Q) mRNA were analyzed by using real‐time PCR and normalized to r18S mRNA. Western blotting analysis for pSmad2 and Smad2 was carried out (left panel in R). Relative expression of pSmad2 to Smad2 (pS2/S2 ratio) is shown in comparison to that in control cells (right panel in R)
3.2. Effect of silencing endogenous HIF‐1α transcription on TGF‐β‐induced EMT phenotypes in H358 cells
In order to determine the role of endogenous levels of HIF‐1α transcription on acquisition of the TGF‐β‐induced EMT phenotypes, siRNAs for HIF‐1α were carried out for H358 cells. siRNAs for HIF‐1α were transfected into H358 cells, resulting in potent HIF‐1α mRNA knockdown (Figure 2A). Induction of PGK1 and VEGFA was not observed in cells treated with TGF‐β and siRNA for HIF‐1α (Figure 2B,C). siRNAs for HIF‐1α did not augment TGF‐β‐induced Smad2 activation (Figure 2D). Silencing of endogenous HIF‐1α mRNA expression did not directly induce de novo fibronectin expression in cells without TGF‐β stimulation 96 hours after transfection (Figure 2E), an observation supported by data showing that no apparent dissociation of the β‐catenin/E‐cadherin complex at the cell membrane took place (Figure 2F,H and Figure S2A,B). TGF‐β‐treated cells with silenced HIF‐1α mRNA expression showed a significant increase in fibronectin expression compared with non‐silenced cells treated with TGF‐β stimulation (Figure 2E). Immunostaining showed that silencing of HIF‐1α mRNA expression did not block dissociation of the β‐catenin/E‐cadherin complex from the cell membrane in cells stimulated with TGF‐β (Figure 2G,I and Figure S2A,B). Other siHIF‐1α experiments showed similar results (data not shown).
Figure 2.
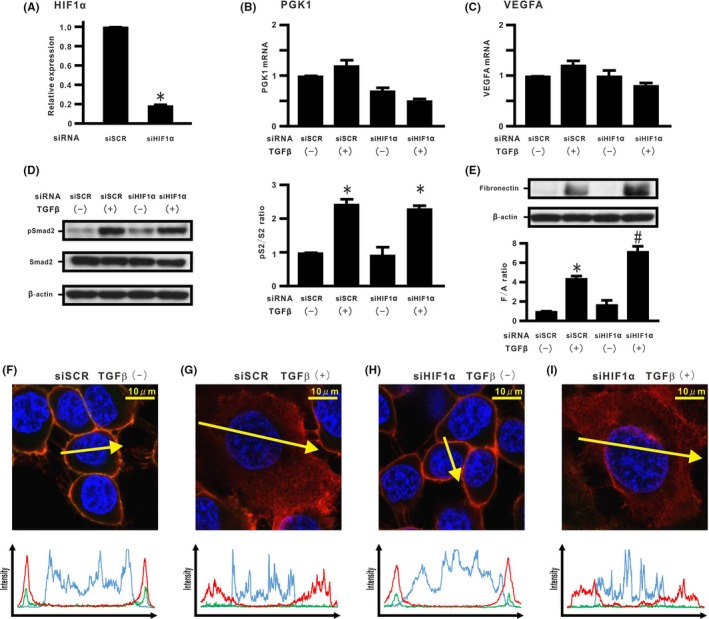
Effect of silencing endogenous hypoxia inducible factor (HIF)‐1α expression on transforming growth factor β (TGF‐β)‐induced epithelial‐mesenchymal transition (EMT) phenotypes in H358 cells. HIF‐1α mRNA was evaluated in H358 cells with transient transduction of control siRNA (siSCR) and HIF‐1α siRNA #1 (siHIF‐1α) by real‐time PCR. A, HIF‐1α mRNA in cells treated with siSCR or siHIF‐1α. B, Phosphoglycerate kinase 1 (PGK1) and (C) vascular endothelial growth factor A (VEGFA) mRNA in cells treated with siSCR or siHIF‐1α in the absence or presence of TGF‐β. D, pSmad2 and Smad2 and (E) fibronectin by western blotting analysis. Right panel in (D): pS2/S2 ratio. Lower panel in (E): F/A ratio. *P < 0.05 in comparison with the control cells. # P < 0.05 in comparison with cells treated with siSCR and TGF‐β. F‐I, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in cells treated with siSCR or siHIF‐1α in the absence or presence of TGF‐β along the selected yellow arrows, respectively
3.3. Effect of stabilized HIF‐1α expression on TGF‐β‐induced EMT phenotypes in H358 cells
Hypoxia inducible factor expression is tightly controlled at the post‐translational level by prolyl hydroxylase domain proteins (PHD).30 By using a Dox‐dependent HIF1αdPA expression system,19 we evaluated whether stabilized HIF‐1α expression could modulate TGF‐β‐induced EMT phenotypes in H358 cells. Cells incubated with Dox for 96 hours retained HIF1αdPA expression (Figure 3A). Induction of HIF‐1α‐related genes, such as PGK1 and VEGFA, were also observed in the cells treated with Dox (Figure 3B,C). Dox‐induced HIF1αdPA expression did not modulate TGF‐β‐induced Smad2 activation (Figure 3D). TGF‐β stimulation did not affect HIF1αdPA expression (Figure 3D,E). When Dox was added, HIF1αdPA alone did not induce fibronectin expression in H358 cells (Figure 3E). In contrast, TGF‐β‐induced fibronectin expression was repressed by de novo HIF1αdPA expression (Figure 3E). Double immunostaining for β‐catenin and E‐cadherin was carried out (Figure 3F‐I and Figure S3A,B). Immunostaining showed that Dox‐induced HIF1αdPA alone did not induce translocation of β‐catenin and internalization of E‐cadherin in the cells (Figure 3G). Although TGF‐β‐treated cells without Dox treatment showed both β‐catenin translocation and E‐cadherin internalization into the cytoplasm (Figure 3H), Dox‐induced HIF1αdPA expression successfully retained the β‐catenin/E‐cadherin complex at the cell membrane in cells cultured with TGF‐β (Figure 3I). When Dox was added, scratch assay did not show repression of cell migration in the TGF‐β‐treated cells (Figure S3C).
Figure 3.
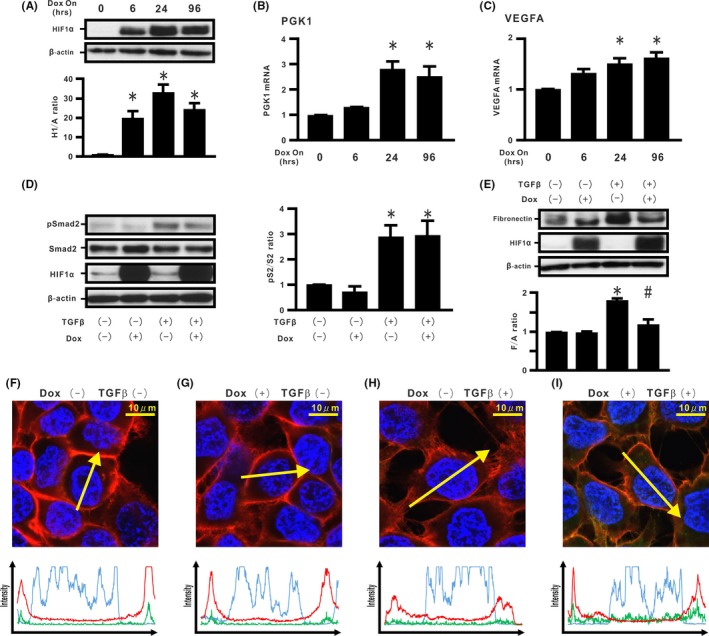
Effect of stabilized hypoxia inducible factor (HIF)‐1α expression on transforming growth factor β (TGF‐β)‐induced epithelial‐mesenchymal transition (EMT) phenotypes in H358 cells. A, Cell extracts from H358ON cells expressing Dox‐dependent HIF1αdPA were harvested at the indicated periods after treatment with Dox. The cell extracts were immunoblotted for HIF1αdPA. Relative expression of HIF1αdPA to β‐actin (H1/A ratio) is shown in comparison to that in control cells. B, Phosphoglycerate kinase 1 (PGK1) and (C) vascular endothelial growth factor A (VEGFA) mRNA in the cells treated with Dox. The cells were incubated with vehicle or Dox for 24 h before TGF‐β treatment. The cells were then treated with vehicle or TGF‐β in the absence or presence of Dox for a further 1 h (Smad2 in D) or 96 h (fibronectin and HIF‐1α in E). D, pSmad2 and Smad2 and (E) fibronectin by western blotting analysis. Right panel in (D): pS2/S2 ratio. Lower panel in (E): F/A ratio. *P < 0.05 in comparison with the control cells. # P < 0.05 in comparison with cells treated with TGF‐β alone. F‐I, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in cells incubated in the absence or presence of Dox and/or TGF‐β along the selected yellow arrows, respectively
3.4. Effect of HIF‐1α lacking an oxygen‐dependent degradation domain on TGF‐β‐induced EMT phenotypes in H358 cells
In H358 cells carrying a Dox‐dependent HIF1αΔODD (Δ402‐603), we evaluated the effect of HIF‐1α lacking the ODD domain on TGF‐β‐induced EMT phenotypes in H358 cells. First, mutant expression was evaluated by western blotting of cells grown in the absence or presence of Dox and TGF‐β. Western blotting verified that the cells successfully expressed protein of the expected size and that its expression was not modulated by TGF‐β stimulation (Figure 4A). Real‐time PCR assay showed that Dox‐treated HIF1αΔODD expression did not induce PGK1 and VEGFA (Figure 4B,C). TGF‐β‐induced Smad2 activation also was not modulated (Figure 4D). Dox‐induced expression of HIF1αΔODD alone did not induce fibronectin production (Figure 4E), and no apparent dissociation of the β‐catenin/E‐cadherin complex was noted (Figure 4F,G and Figure S4A,B). Furthermore, cells with de novo HIF1αΔODD expression did not blunt TGF‐β‐induced fibronectin production (Figure 4E), accompanied by translocation of β‐catenin and internalization of E‐cadherin (Figure 4H,I and Figure S4A,B).
Figure 4.
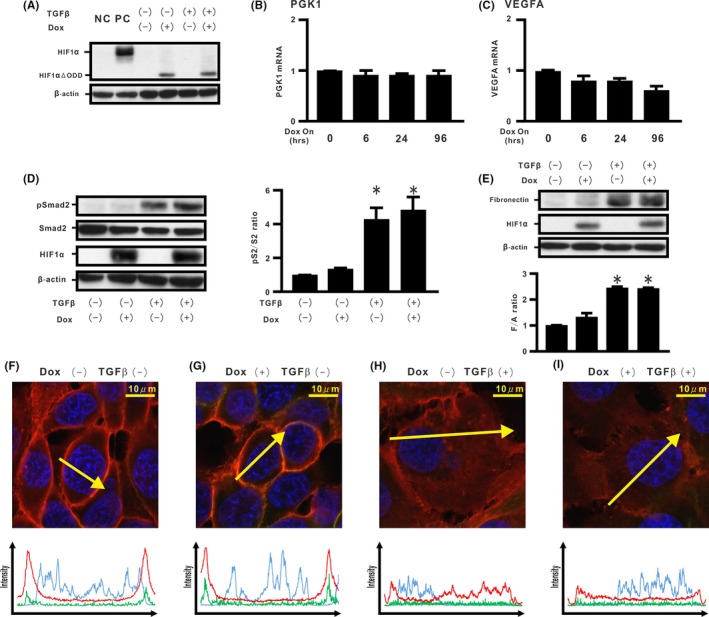
Effect of hypoxia inducible factor (HIF)‐1α lacking oxygen‐dependent degradation domain on transforming growth factor β (TGF‐β)‐induced epithelial‐mesenchymal transition (EMT) phenotypes in H358 cells. We established H358ON cells expressing Dox‐dependent HIF1αΔODD #1. Cells were then treated with vehicle or TGF‐β in the absence or presence of Dox. A representative blot from three independent experiments is shown as (A). NC, H358 cells. PC, H358ON cells expressing Dox‐dependent HIF1αdPA treated with Dox. B, Phosphoglycerate kinase 1 (PGK1) and (C) vascular endothelial growth factor A (VEGFA) mRNA by using real‐time PCR. D, pSmad2 and Smad2 and (E) fibronectin by western blotting analysis. Right panel in (D): pS2/S2 ratio. Lower panel in (E): F/A ratio. *P < 0.05 in comparison with the control cells. F‐I, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in cells incubated in the absence or presence of Dox and/or TGF‐β along the selected yellow arrows, respectively
3.5. Role of induction of endogenous HIF‐1α stabilization on TGF‐β‐induced EMT phenotypes in H358 cells
In order to evaluate the importance of endogenous HIF‐1α stabilization in regulating TGF‐β‐induced EMT phenotypes, HIF‐1α stabilizers were used. H358 cells were treated with cobalt chloride (CoCl2), chelating Fe2+,31 which stabilized HIF‐1α protein expression for 96 hours (Figure 5A). CoCl2 treatment inhibited de novo TGF‐β‐induced fibronectin expression and retained localization of the β‐catenin/E‐cadherin complex (Figure 5B‐F and Figure S5A,B). H358 cells were also treated with FG4592 (FG), a HIF‐1α prolyl hydroxylase inhibitor. FG treatment for 96 hours retained stabilized HIF‐1α protein expression in the cells (Figure 5G). Western blotting analysis showed that FG treatment led to >65% decrease in TGF‐β‐induced fibronectin expression while retaining localization of β‐catenin and E‐cadherin on the cell membrane (Figure 5H‐L and Figure S5C,D).
Figure 5.
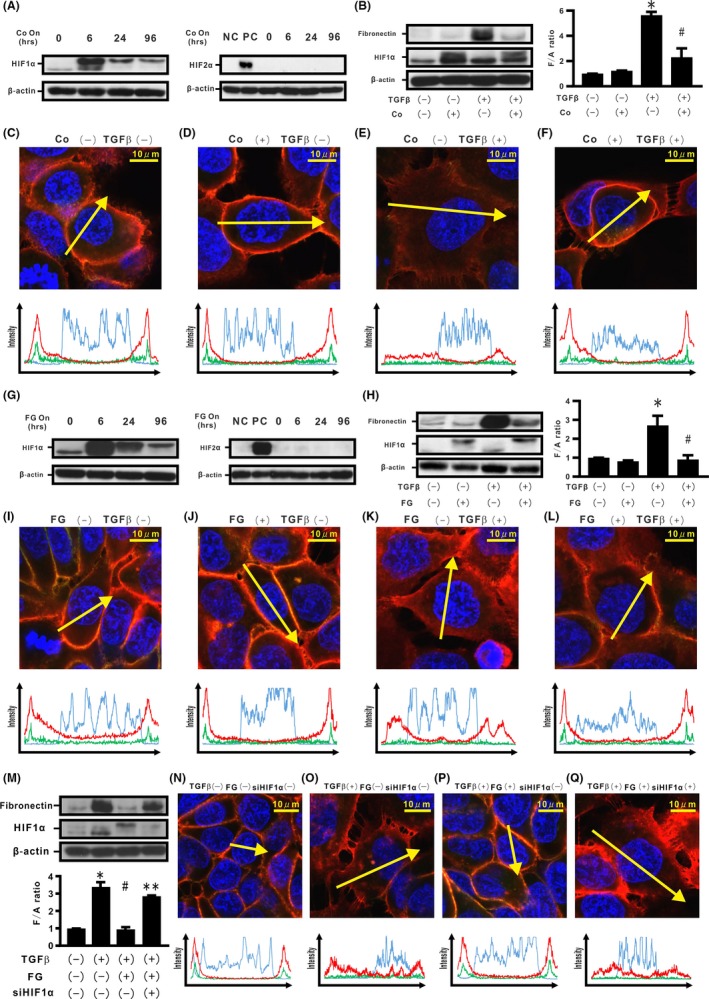
Effects of induction of endogenous hypoxia inducible factor (HIF)‐1α stabilization on transforming growth factor β (TGF‐β)‐induced epithelial‐mesenchymal transition (EMT) phenotypes in H358 cells. H358 cells were treated with CoCl2 (Co) for the indicated time periods (A). Left panel in A: HIF‐1α. Right panel in A: HIF‐2α. NC, negative control; PC, positive control. Cells were also incubated in the absence or presence of Co and/or TGF‐β for 96 h (B). Left panel in B: fibronectin. Right panel in B: F/A ratio. C‐F, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in H358 cells treated in the absence or presence of Co and/or TGF‐β along the selected yellow arrows, respectively. H358 cells were treated with FG4592 (FG) for the indicated time periods (G). Left panel in G: HIF‐1α. Right panel in G: HIF‐2α. Cells were also incubated in the absence or presence of FG and/or TGF‐β for 96 h (H). Left panel in H: fibronectin. Right panel in H: F/A ratio. I‐L, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in H358 cells treated in the absence or presence of FG and/or TGF‐β along the selected yellow arrows, respectively. After transfection of siSCR or siHIF‐1α, H358 cells were incubated in the absence or presence of FG and/or TGF‐β (M). Lower panel in (M): F/A ratio. N‐Q, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in H358 cells treated in the absence or presence of siHIF‐1α, FG, and/or TGF‐β along the selected yellow arrows, respectively. *P < 0.05 in comparison with the control cells. # P < 0.05 in comparison with the cells treated with TGF‐β alone. **P < 0.05 in comparison with cells treated with TGF‐β and FG (M)
To determine whether the inhibitory effect of FG might directly depend on stabilized HIF‐1α protein expression, siRNAs for HIF‐1α were carried out for H358 cells treated with TGF‐β and FG. Silencing of endogenous HIF‐1α mRNA expression abrogated the repression of de novo fibronectin production induced by TGF‐β. When siRNAs for HIF‐1α were added to H358 cells treated with TGF‐β and FG, dissociation of the β‐catenin/E‐cadherin complex from the cell membrane was observed by immunostaining (Figure 5M‐Q and Figure S5E,F). These HIF stabilizers did not show de novo HIF‐2α protein expression in H358 cells by western blotting analysis (Figure 5A,G). When Co or FG was added, scratch assay did not show repression of cell migration in the TGF‐β‐treated cells (Figure S5G,H, respectively).
3.6. Effect of endogenous HIF‐1α stabilization in rat epithelial cells and fibroblasts
In order to determine the effect of endogenous HIF‐1α stabilization in regulating TGF‐β‐induced EMT phenotypes in rat species, we evaluated a rat epithelial cell line, RLE‐6TN. RLE‐6TN cells treated with FG for 96 hours showed HIF‐1α protein expression (Figure 6A). FG treatment repressed TGF‐β‐induced fibronectin production and α‐SMA expression in the cells (Figure 6B,C), an observation supported by immunostaining showing FG‐induced blockade of TGF‐β‐induced translocation of β‐catenin from the cell membrane (Figure 6D‐G and Figure S6A). The function of fibroblasts stimulated by TGF‐β is mainly to increase ECM synthesis and α‐SMA expression with cell contractility.32 Human CC2512 fibroblasts were used to determine whether endogenous HIF‐1α stabilization could regulate the characteristics of TGF‐β‐stimulated fibroblasts. Stabilized HIF‐1α protein expression was observed in cells treated with FG for 96 hours (Figure 6H). FG treatment induced a significant decrease in fibronectin, collagen type I (Col I), and α‐SMA in TGF‐β‐stimulated fibroblasts (Figure 6H,I). Immunostaining showed that FG treatment blocked TGF‐β‐induced translocation of β‐catenin from the cell membrane (Figure 6J‐M and Figure S6B).
Figure 6.
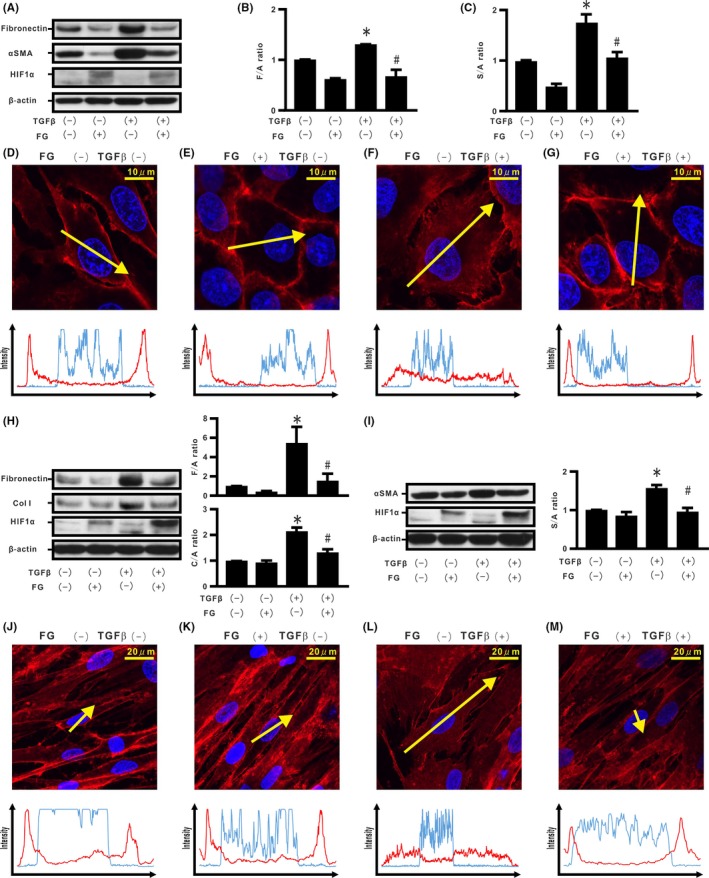
Effect of endogenous hypoxia inducible factor (HIF)‐1α stabilization in rat epithelial cells and fibroblasts. Cells of the rat epithelial cell line (RLE‐6TN) were incubated in the absence or presence of FG4592 (FG) and/or transforming growth factor β (TGF‐β) for 96 h (A). B, F/A ratio. C, α‐Smooth muscle actin (α‐SMA) to β‐actin ratio (S/A ratio). D‐G, β‐Catenin (red) and Hoechst33342 (blue) in the RLE‐6TN‐treated cells in the absence or presence of FG and/or TGF‐β along the selected yellow arrows, respectively. Cells of the human fibroblast cell line (CC2512) were also cultured in the absence or presence of FG and/or TGF‐β for 96 h (H), and cell extracts were evaluated for fibronectin, collagen type I (Col I), α‐SMA, and HIF‐1α, by western blotting (H, I). Upper and right panel in H: F/A ratio. Lower and right panel in H: Col I to β‐actin ratio (C/A) ratio. Right panel in I: S/A ratio. J‐M, β‐Catenin (red) and Hoechst33342 (blue) in CC2512 cells treated in the absence or presence of FG and/or TGF‐β along the selected yellow arrows, respectively. *P < 0.05 in comparison with the control cells. # P < 0.05 in comparison with the cells treated with TGF‐β alone
3.7. Association of endogenous HIF‐1α and protein phosphatases with TGF‐β‐induced EMT phenotypes in H358 cells
Recent studies have suggested a critical role of protein phosphatase in localization of the cadherin/catenin complex at the cell membrane.18, 33 Therefore, H358 cells were treated with okadaic acid (OA), an inhibitor of protein phosphatase 2A activity (PP2A). Although the cells incubated with OA or TGF‐β showed de novo fibronectin production, combined treatment with OA and TGF‐β augmented excessive fibronectin production (Figure 7A,B). Immunostaining showed dissociation of the β‐catenin/E‐cadherin complex from the cell membrane in these cells (Figure 7C‐F and Figure S7A,B). In order to evaluate the role of PP2A on TGF‐β‐induced fibronectin production repressed by stabilized HIF‐1α protein expression, H358 cells carrying Dox‐dependent HIF1αdPA expression were treated with OA and TGF‐β. Although stabilized HIF‐1α induced by Dox repressed de novo fibronectin production induced by TGF‐β, OA treatment abrogated the repression (Figure 7G,H). When OA was added to H358 cells containing stabilized HIF‐1α, dissociation of the β‐catenin/E‐cadherin complex from the cell membrane was observed by immunostaining (Figure 7I‐L and Figure S7C,D).
Figure 7.
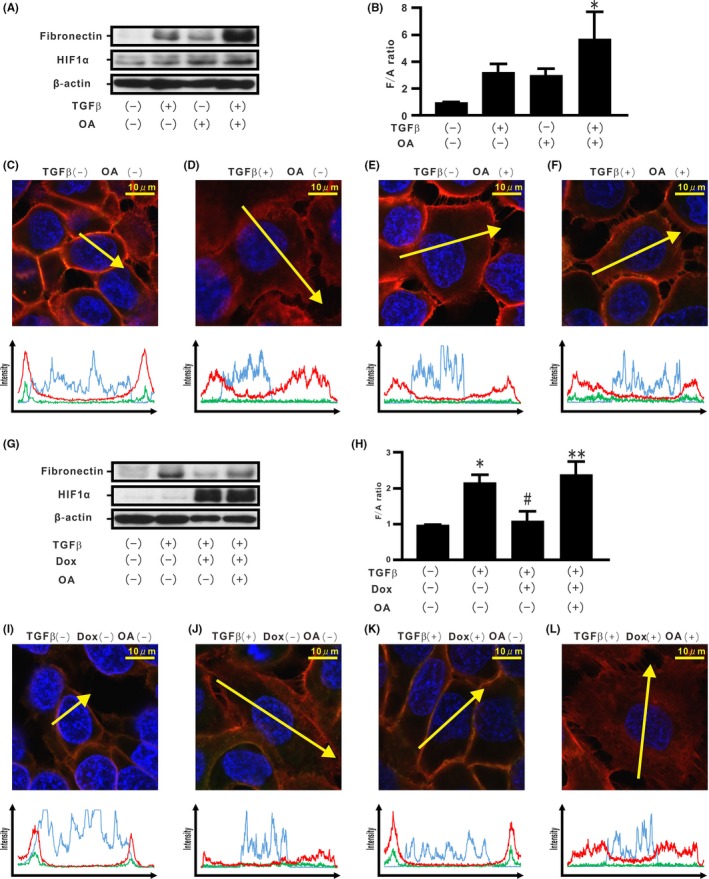
Association of endogenous hypoxia inducible factor (HIF)‐1α and protein phosphatases with transforming growth factor β (TGF‐β)‐induced epithelial‐mesenchymal transition (EMT) phenotypes in H358 cells. H358 cells were incubated in the absence or presence of ocadaic acid (OA) and/or TGF‐β for 96 h (A). B, F/A ratio. C‐F, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in H358 cells treated in the absence or presence of OA and/or TGF‐β along the selected yellow arrows, respectively. H358ON cells carrying HIF1αdPA were treated with TGF‐β in the absence or presence of Dox for 72 h and then incubated in the absence or presence of OA for a further 24 h. Western blotting analysis for fibronectin and HIF‐1α was carried out (G) and the F/A ratio was evaluated (H). I‐L, β‐Catenin (red), E‐cadherin (green), and Hoechst33342 (blue) in H358 cells treated in the absence or presence of TGF‐β and/or Dox and/or OA along the selected yellow arrows, respectively. *P < 0.05 in comparison with the control cells. # P < 0.05 in comparison with the cells treated with TGF‐β alone. **P < 0.05 in comparison with the cells treated with TGF‐β and Dox
4. DISCUSSION
There is increasing awareness of the tissue microenvironment in which acquisition of EMT phenotypes through aberrant crosstalk between cancer cells and CAF affects disease development and progression in lung cancer.34 Therefore, prevention of the development of these EMT phenotypes is one of the most critical targets in treating lung cancer. Our previous studies showed that excessive ECM production is closely associated with β‐catenin translocation into the cytoplasm, stimulated by either TGF‐β or persistent hypoxia in lung cancer cells and lung fibroblasts.10, 11, 20, 27 Although persistent hypoxia also induces acquisition of the EMT phenotypes,20, 35 differential cellular responses to acute and persistent hypoxia were observed in acute lung injury/fibrosis in vitro and in vivo.19, 36 Although combined stimulation by hypoxia and TGF‐β for a very short‐time period (6 hours) did not accelerate TGF‐β‐induced β‐catenin translocation, TGF‐β and/or hypoxia stimulation for short‐time periods (24 hours) induced dissociation of the β‐catenin/E‐cadherin complex in epithelial cells.10, 11, 20
Hypoxia‐inducible factors activate the transcription of genes that might be associated with cancer development.37 Meanwhile, stabilized HIF‐1α protein expression also plays differential roles in the pathogenesis of cancers and injury/fibrosis.37, 38 Several studies have shown that stabilized HIF protein expression might protect tissue from injury.38, 39 In the present study, decreasing levels of HIF‐1α protein expression were associated with disruption of epithelial integrity in H358 cells. Indeed, combined stimulation by TGF‐β and hypoxia for a long‐time period (96 hours) induced loss of HIF‐1α protein expression accompanied by de novo fibronectin production. Decreasing HIF‐1α protein expression in response to prolonged hypoxia might partially be a result of transcriptional suppression of HIF‐1α mRNA by an increase in de novo natural antisense HIF‐1α (AS‐HIF) expression.29
Although silencing of HIF‐1α mRNA expression by siRNA might restore EMT phenotypes in cells experiencing hypoxia,5 the present data suggest that TGF‐β‐induced dissociation of cadherin/catenin complexes and de novo fibronectin expression were exacerbated in H358 cells by silencing of HIF‐1α mRNA expression. Other studies have demonstrated that silencing and deletion of HIF might attenuate barrier functions in injury models through loss of cell‐cell integrity.18, 40 Furthermore, a recent study showed that HIF‐1α‐antisense RNA 2 (HIF1A‐AS2) facilitated the maintenance of mesenchymal phenotypes in glioma cells under hypoxia.41 Thus, loss of endogenous HIF‐1α protein expression by suppression of HIF‐1α transcription under persistent hypoxia might exacerbate aberrant TGF‐β‐induced ECM production.
Oxygen‐dependent degradation of HIF‐1α also involves modification of two proline sites in the HIF‐1α ODD domain by activation of PHD induced by HIF‐1α stabilization.30 The present data showed that stabilized HIF‐1α protein expression by HIF1αdPA blocked dissociation of the β‐catenin/E‐cadherin complex and blunted fibronectin expression in H358 cells treated with TGF‐β. Autocrine VEGF signaling by either TGF‐β or hypoxia stimulation might play a role in the acquisition of aggressive phenotypes in breast cancer.35 Although Dox‐induced stabilized HIF‐1α protein expression induced de novo VEGFA expression, the TGF‐β‐induced EMT phenotypes were depressed. These findings indicate that de novo VEGFA expression did not appear to independently exacerbate the EMT phenotypes in H358 cells. Furthermore, although the Smad signaling pathway, including Smad2 activation, might be partly associated with TGF‐β‐induced ECM expression in epithelial cells,42, 43 Dox‐induced stabilized HIF‐1α protein expression did not attenuate TGF‐β‐induced phosphorylation of Smad2. TGF‐β‐induced transcription of the ECM genes is accelerated by translocation of β‐catenin from E‐cadherin complexes at the cell membrane into the cytoplasm.8, 10, 11, 44 Although TGF‐β‐induced malignant phenotypes involve EMT and aberrant cell motility in lung cancers,1, 2, 10 stabilized HIF‐1α protein expression did not inhibit TGF‐β‐induced aberrant cell motility. These findings indicated that TGF‐β stimulation might involve different mechanisms to induce the acquisition of TGF‐β‐induced malignant phenotypes. Further investigation is warranted. Taken together, tight epithelial integrity induced by stabilized HIF‐1α might inhibit TGF‐β‐induced ECM production, not aberrant cell motility, through blockade of β‐catenin translocation without modification of Smad activation.
Previous studies evaluated the biological effects of HIF‐1α by using an HIF‐1α mutant in which the HIF‐1α ODD domain is deleted.5, 13 Although the HIF‐1α ODD domain involves the N‐terminal transactivation domain (N‐TAD),23 the previous study showed that lack of N‐TAD in HIF‐1α caused a loss of optimal HIF transcriptional activity, possibly as a result of the lack of interaction with additional transcriptional cofactors.13 Indeed, de novo expression of HIF‐1α protein with the deletion of aa 402‐603 did not show repression of TGF‐β‐induced fibronectin expression in H358 cells. In the present study, the role of the HIF‐1α ODD domain, including N‐TAD, in HIF‐1α‐induced repression of aberrant TGF‐β‐induced ECM production, remains elusive.
Enhancement of HIF‐1α through blockade of PHD might induce an increase in transcription of HIF‐1α‐targeting genes, such as VEGFA and PGK1.45 PHD inhibitor (FG) and cobalt chloride (Co), by which HIF‐1α protein expression was stabilized, caused retention of the β‐catenin/E‐cadherin complex at the cell membrane and decreased TGF‐β‐induced ECM expression in lung cancer cells with epithelial characteristics as well as in immortalized rat alveolar type II cells. Aberrant crosstalk between cancer cells and CAF caused excessive ECM production and tissue contractility during development of cancers.12 In human fibroblasts cultured with TGF‐β, treatment with FG inhibited fibronectin and collagen type I production and repressed α‐SMA expression, accompanied by localization of β‐catenin at the cell membrane. Stabilized HIF‐1α expression might regulate aberrant TGF‐β‐stimulated ECM production across cell types and species.
Hypoxia‐inducible factor 2α expression induces vascular endothelial protein tyrosine phosphatase (VE‐PTP) expression, which might be associated with a decrease in vascular endothelial (VE)‐cadherin endocytosis in endothelial cells.18 In the present study, stimulation of TGF‐β and/or hypoxia did not induce stabilized HIF‐2α protein expression in H358 cells. Previous studies showed that PP2A is essential for retaining and stabilizing E‐cadherin/β‐catenin complexes at the plasma membrane.33, 46, 47 Although repression of TGF‐β‐induced EMT phenotypes was achieved by stabilized HIF‐1α protein expression, inhibition of the PP2A function abrogated the repression by stabilized HIF‐1α. Thus, considering the data together, stabilized HIF‐1α protein expression might negatively regulate TGF‐β‐induced ECM production, in part through PP2A activity. A schematic diagram of the repressive role of stabilized HIF‐1α expression on TGF‐β‐induced extracellular matrix production in lung cancer cells is shown in Figure 8.
Figure 8.
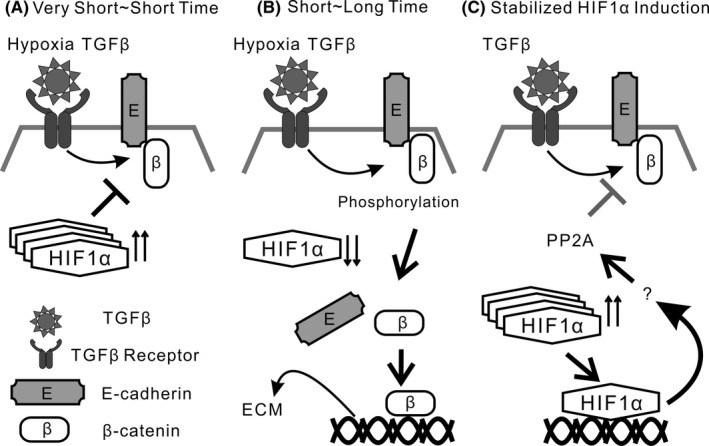
Schematic diagram of repressive role of stabilized hypoxia inducible factor (HIF)‐1α expression on transforming growth factor β (TGF‐β)‐induced extracellular matrix production in lung cancer cells. A, Combined stimulation by hypoxia and TGF‐β for very short‐ or short‐time period does not accelerate β‐catenin translocation, accompanied by stabilized HIF‐1α protein expression. B, TGF‐β and hypoxia stimulation for short‐ or long‐time period induces dissociation of the β‐catenin/E‐cadherin complex in lung cancer cells, accompanied by loss of HIF‐1α protein expression. Persistent stimulation by TGF‐β and hypoxia causes aberrant extracellular matrix (ECM) production. C, Stabilized HIF‐1α protein expression negatively regulates TGF‐β‐induced ECM production in lung cancer cells, in part through protein phosphatase 2 (PP2A) activity
In summary, our study shows, for the first time, that induction of stabilized HIF‐1α protein expression might be a novel therapeutic target to modify the tumor microenvironment in lung cancer cells and CAF.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by Kowa Life Science Foundation and Grant‐in‐Aid for Scientific Research (C) (24591162), and partly supported by a grant to the Diffuse Lung Diseases Research Group from the Ministry of Health, Labour and Welfare, Japan.
Ando A, Hashimoto N, Sakamoto K, et al. Repressive role of stabilized hypoxia inducible factor 1α expression on transforming growth factor β‐induced extracellular matrix production in lung cancer cells. Cancer Sci. 2019;110:1959–1973. 10.1111/cas.14027
REFERENCES
- 1. Gumbiner BM. Regulation of cadherin‐mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6(8):622‐634. [DOI] [PubMed] [Google Scholar]
- 2. Kam Y, Quaranta V. Cadherin‐bound beta‐catenin feeds into the Wnt pathway upon adherens junctions dissociation: evidence for an intersection between beta‐catenin pools. PLoS ONE. 2009;4(2):e4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4(2):165‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang MH, Wu MZ, Chiou SH, et al. Direct regulation of TWIST by HIF‐1alpha promotes metastasis. Nat Cell Biol. 2008;10(3):295‐305. [DOI] [PubMed] [Google Scholar]
- 6. Kojima Y, Acar A, Eaton EN, et al. Autocrine TGF‐beta and stromal cell‐derived factor‐1 (SDF‐1) signaling drives the evolution of tumor‐promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA. 2010;107(46):20009‐20014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liebner S, Cattelino A, Gallini R, et al. Beta‐catenin is required for endothelial‐mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. 2004;166(3):359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial‐mesenchymal transition through beta‐catenin‐T‐cell factor‐4‐dependent expression of transforming growth factor‐beta3. Mol Biol Cell. 2008;19(11):4875‐4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanjore H, Cheng DS, Degryse AL, et al. Alveolar epithelial cells undergo epithelial‐to‐mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286(35):30972‐30980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aoyama D, Hashimoto N, Sakamoto K, et al. Involvement of TGFbeta‐induced phosphorylation of the PTEN C‐terminus on TGFbeta‐induced acquisition of malignant phenotypes in lung cancer cells. PLoS ONE. 2013;8(11):e81133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kusunose M, Hashimoto N, Kimura M, et al. Direct regulation of transforming growth factor beta‐induced epithelial‐mesenchymal transition by the protein phosphatase activity of unphosphorylated PTEN in lung cancer cells. Cancer Sci. 2015;106:1693‐1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nieto MA, Huang RY, Jackson RA, et al. EMT: 2016. Cell. 2016;166(1):21‐45. [DOI] [PubMed] [Google Scholar]
- 13. Hu CJ, Sataur A, Wang L, et al. The N‐terminal transactivation domain confers target gene specificity of hypoxia‐inducible factors HIF‐1alpha and HIF‐2alpha. Mol Biol Cell. 2007;18(11):4528‐4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Page EL, Chan DA, Giaccia AJ, et al. Hypoxia‐inducible factor‐1alpha stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell. 2008;19(1):86‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Generali D, Berruti A, Brizzi MP, et al. Hypoxia‐inducible factor‐1alpha expression predicts a poor response to primary chemoendocrine therapy and disease‐free survival in primary human breast cancer. Clin Cancer Res. 2006;12(15):4562‐4568. [DOI] [PubMed] [Google Scholar]
- 16. Hung JJ, Yang MH, Hsu HS, et al. Prognostic significance of hypoxia‐inducible factor‐1alpha, TWIST1 and Snail expression in resectable non‐small cell lung cancer. Thorax. 2009;64(12):1082‐1089. [DOI] [PubMed] [Google Scholar]
- 17. Baran N, Konopleva M. Molecular pathways: hypoxia‐activated prodrugs in cancer therapy. Clin Cancer Res. 2017;23(10):2382‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gong H, Rehman J, Tang H, et al. HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest. 2015;125(2):652‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sakamoto K, Hashimoto N, Kondoh Y, et al. Differential modulation of surfactant protein D under acute and persistent hypoxia in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;303(1):L43‐L53. [DOI] [PubMed] [Google Scholar]
- 20. Kohnoh T, Hashimoto N, Ando A, et al. Hypoxia‐induced modulation of PTEN activity and EMT phenotypes in lung cancers. Cancer Cell Int. 2016;16:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329‐333. [DOI] [PubMed] [Google Scholar]
- 22. Huang LE, Gu J, Schau M, et al. Regulation of hypoxia‐inducible factor 1alpha is mediated by an O2‐dependent degradation domain via the ubiquitin‐proteasome pathway. Proc Natl Acad Sci USA. 1998;95(14):7987‐7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia‐inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551‐578. [DOI] [PubMed] [Google Scholar]
- 24. Nakashima H, Hashimoto N, Aoyama D, et al. Involvement of the transcription factor twist in phenotype alteration through epithelial‐mesenchymal transition in lung cancer cells. Mol Carcinog. 2012;51(5):400‐410. [DOI] [PubMed] [Google Scholar]
- 25. Hashimoto N, Jin H, Liu T, et al. Bone marrow‐derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113(2):243‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hashimoto N, Kawabe T, Imaizumi K, et al. CD40 plays a crucial role in lipopolysaccharide‐induced acute lung injury. Am J Respir Cell Mol Biol. 2004;30(6):808‐815. [DOI] [PubMed] [Google Scholar]
- 27. Kimura M, Hashimoto N, Kusunose M, et al. Exogenous induction of unphosphorylated PTEN reduces TGFbeta‐induced extracellular matrix expressions in lung fibroblasts. Wound Repair Regen. 2017;25(1):86‐97. [DOI] [PubMed] [Google Scholar]
- 28. Hashimoto N, Phan SH, Imaizumi K, et al. Endothelial‐mesenchymal transition in bleomycin‐induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;43(2):161‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uchida T, Rossignol F, Matthay MA, et al. Prolonged hypoxia differentially regulates hypoxia‐inducible factor (HIF)‐1alpha and HIF‐2alpha expression in lung epithelial cells: implication of natural antisense HIF‐1alpha. J Biol Chem. 2004;279(15):14871‐14878. [DOI] [PubMed] [Google Scholar]
- 30. Semenza GL. Hydroxylation of HIF‐1: oxygen sensing at the molecular level. Physiology (Bethesda). 2004;19:176‐182. [DOI] [PubMed] [Google Scholar]
- 31. Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science. 2001;292(5516):468‐472. [DOI] [PubMed] [Google Scholar]
- 32. Hinz B, Phan SH, Thannickal VJ, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180(4):1340‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gotz J, Probst A, Mistl C, et al. Distinct role of protein phosphatase 2A subunit Calpha in the regulation of E‐cadherin and beta‐catenin during development. Mech Dev. 2000;93(1–2):83‐93. [DOI] [PubMed] [Google Scholar]
- 34. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582‐598. [DOI] [PubMed] [Google Scholar]
- 35. Mak P, Leav I, Pursell B, et al. ERbeta impedes prostate cancer EMT by destabilizing HIF‐1alpha and inhibiting VEGF‐mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;17(4):319‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562‐572. [DOI] [PubMed] [Google Scholar]
- 37. Semenza GL. Hypoxia‐inducible factors in physiology and medicine. Cell. 2012;148(3):399‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. 2014;13(11):852‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Colgan SP, Eltzschig HK. Adenosine and hypoxia‐inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2012;74:153‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mineo M, Ricklefs F, Rooj AK, et al. The long non‐coding RNA HIF1A‐AS2 facilitates the maintenance of mesenchymal glioblastoma stem‐like cells in hypoxic niches. Cell Rep. 2016;15(11):2500‐2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ashcroft GS, Yang X, Glick AB, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1(5):260‐266. [DOI] [PubMed] [Google Scholar]
- 43. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature. 2003;425(6958):577‐584. [DOI] [PubMed] [Google Scholar]
- 44. Kim Y, Kugler MC, Wei Y, et al. Integrin alpha3beta1‐dependent beta‐catenin phosphorylation links epithelial Smad signaling to cell contacts. J Cell Biol. 2009;184(2):309‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Asikainen TM, Schneider BK, Waleh NS, et al. Activation of hypoxia‐inducible factors in hyperoxia through prolyl 4‐hydroxylase blockade in cells and explants of primate lung. Proc Natl Acad Sci USA. 2005;102(29):10212‐10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gotz J, Probst A, Ehler E, et al. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Calpha. Proc Natl Acad Sci USA. 1998;95(21):12370‐12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takahashi K, Nakajima E, Suzuki K. Involvement of protein phosphatase 2A in the maintenance of E‐cadherin‐mediated cell‐cell adhesion through recruitment of IQGAP1. J Cell Physiol. 2006;206(3):814‐820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials