

Abstract
Activating mutations in cytokine receptors and transcriptional regulators govern aberrant signal transduction in T‐cell lineage acute lymphoblastic leukemia (T‐ALL). However, the roles played by suppressors of cytokine signaling remain incompletely understood. We examined the regulatory roles of suppressor of cytokine signaling 5 (SOCS5) in T‐ALL cellular signaling networks and leukemia progression. We found that SOCS5 was differentially expressed in primary T‐ALL and its expression levels were lowered in HOXA‐deregulated leukemia harboring KMT2A gene rearrangements. Here, we report that SOCS5 expression is epigenetically regulated by DNA methyltransferase‐3A‐mediated DNA methylation and methyl CpG binding protein‐2‐mediated histone deacetylation. We show that SOCS5 negatively regulates T‐ALL cell growth and cell cycle progression but has no effect on apoptotic cell death. Mechanistically, SOCS5 silencing induces activation of JAK‐STAT signaling, and negatively regulates interleukin‐7 and interleukin‐4 receptors. Using a human T‐ALL murine xenograft model, we show that genetic inactivation of SOCS5 accelerates leukemia engraftment and progression, and leukemia burden. We postulate that SOCS5 is epigenetically deregulated in T‐ALL and serves as an important regulator of T‐ALL cell proliferation and leukemic progression. Our results link aberrant downregulation of SOCS5 expression to the enhanced activation of the JAK‐STAT and cytokine receptor‐signaling cascade in T‐ALL.
Keywords: DNA methylation, histone deacetylation, JAK‐STAT, signal transduction, T‐ALL
Abbreviations
- 5‐AzaC
5‐azacitidine
- DNMT
DNA methyltransferase
- ETP
early T‐cell precursor
- HDAC
histone deacetylase
- IL‐7R
interleukin‐7 receptor
- KMT2A‐R
KMT2A gene rearrangements
- MBD
methyl‐CpG‐binding domain protein
- MeCP2
methyl CpG binding protein‐2
- NSG
NOD.Cg‐PrkdcscidIl2rgtm1Wjl/SzJ
- qRT‐PCR
quantitative real‐time PCR
- SOCS
suppressor of cytokine signaling
- T‐ALL
T‐cell lineage acute lymphoblastic leukemia
- Th
T‐helper
- TSA
Trichostatin A
1. INTRODUCTION
T‐cell lineage acute lymphoblastic leukemia is an aggressive hematopoietic malignancy accounting for 15% of pediatric ALLs.1, 2 Over the past few decades, the cure rate in T‐ALL has significantly increased; however, survival is poor in patients who suffer treatment failure or early relapse.2, 3 Further improvements in survival for T‐ALL will require improved understanding of the mechanism governing leukemogenesis to develop novel treatment approaches. Although much progress has been made in understanding the stage‐specific transformation of T‐cell progenitors in leukemic transformation, the mechanisms of epigenetic dysregulation remain less well understood.4 Genes involved in T‐cell receptor signaling and differentiation, and tumor suppressor genes are commonly differentially methylated genes in T‐ALL.5, 6 Hypermethylation of CpG islands located in the promoter and/or 1st exon/intron region was proposed as an alternative mechanism for tumor suppressor gene inactivation.7, 8, 9
The JAK‐STAT signaling pathway plays an important role in hematopoietic cell growth, differentiation, and survival.10 Similar to other leukemias, dysregulation in JAK‐STAT signaling networks were found in a subset of T‐ALL.1, 10, 11 Studies of JAK‐STAT activating mutations, including IL7R, JAK1, JAK2, JAK3, and TYK2 have been undertaken,11, 12, 13, 14, 15, 16, 17, 18 but the potential roles of negative regulators of signal transduction, including SOCS, remain largely unexplored in the pathogenesis of T‐ALL.
The SOCS family of cytokine‐inducible negative regulators of JAK‐STAT and other signaling pathways includes 8 structurally related family members, SOCS1‐7 and CIS, all of which contain a central Src‐homology 2 domain and a conserved C‐terminal domain termed the SOCS box.19, 20 There is growing evidence implicating SOCS family members in a range of inflammatory diseases and tumors, including hepatocellular carcinoma, colorectal, cervical, and breast cancer.20, 21, 22, 23 Downregulation of SOCS genes was reported in solid tumors with an unfavorable prognosis and hematological malignancies, including AML, and myeloproliferative disorders.21, 22, 24, 25, 26, 27
SOCS5 is expressed in a variety of adult tissues, particularly in primary B and T cells located in the spleen, lymph nodes, thymus, and bone marrow.20, 28 Consistent with its expression in lymphoid organs, SOCS5 has been implicated in Th cell differentiation, particularly in the balance between Th1 and Th2 cells, with SOCS5 preferentially expressed in Th1 cells.28, 29 Growing evidence suggests SOCS5 is tumor suppressor gene, negatively regulating the epidermal growth factor receptor and JAK‐STAT signaling pathways.24, 30, 31, 32 However, little is currently known about the mechanisms by which SOCS5 regulates signal transduction in leukemic cells.
Given the roles of SOCS5 in normal T cell development, we hypothesized that SOCS5 is a critical mediator of JAK‐STAT signaling and T‐ALL progression. Here, we report that SOCS5 is epigenetically regulated by DNA methylation and histone deacetylation. We provide evidence that SOCS5 negatively regulates the activation of the JAK‐STAT signaling pathway and cytokine receptors in T‐ALL. We show that SOCS5 silencing significantly increases T‐ALL proliferation in vitro and leukemia engraftment in a murine model of human leukemia. In summary, we have identified a novel regulator underlying aberrant JAK‐STAT activation in T‐ALL.
2. MATERIALS AND METHODS
2.1. Reagents
All reagents were purchased from Thermo Fisher Scientific (Carlsbad, CA, USA) unless specified otherwise.
2.2. Cells and patient samples
Human T‐ALL cell lines (MOLT4, ALL‐SIL, Jurkat, CCRF‐CEM, KoptK1, and PF382) were cultured in RPMI‐1640 medium supplemented with 10% FBS, 2 mmol/L l‐glutamine, and 100 U/mL penicillin G‐streptomycin in a 5% CO2 incubator at 37°C. The 293‐FT and Phoenix cells were maintained following manufacturer instructions. Murine hematopoietic BaF3 cell line was cultured in RPMI‐1640, 10% FBS, 10 ng/mL mouse IL‐3 (PeproTech, Rocky Hill, NJ, USA), 2 mmol/L L‐glutamine, and 100 U/mL penicillin G‐streptomycin. Human bone marrow CD34+ cells were purchased from Stemcell Technologies (Cambridge, MA, USA). Peripheral blood mononuclear cells were isolated from buffy coats of normal donors (United Blood Services, Albuquerque, NM, USA) by centrifugation in a Ficoll‐Paque (GE Healthcare, Pittsburgh, PA, USA) density gradient. Normal T cells were extracted using a human Pan T‐cell Isolation Kit (Miltenyi Biotec, Auburn, CA, USA). Cryopreserved primary samples were obtained from patients enrolled in Children's Oncology Group T‐ALL study AALL0434.33 All patients or their parent(s)/guardian(s) provided written, informed consent for future research in accordance with the Declaration of Helsinki and local institutional guidelines. The primary cells were cultured as described previously.34
2.3. In vivo leukemia cell transplantation
NSG mice (8‐10 weeks old) were obtained from the University of New Mexico Comprehensive Cancer Center Animal Models Shared Resource (Albuquerque, NM, USA) and housed in a specific pathogen‐free, AAALAC‐accredited facility as described previously.35 Animals were injected through a tail vein with 1 × 106 cells per mouse (T‐ALL cells transduced with SOCS5‐shRNA or with negative control scrambled shRNA). For survival experiments, animals (10 mice per group) were immediately killed when they showed signs of being moribund or had weight loss that exceeded 10%‐15% of their total weight. For leukemia burden analyses (4 mice per group), all mice were killed 25 days post‐engraftment. Leukemic cells were extracted from the bone marrow of femurs, spleen, liver, and brain by centrifugation in a Percoll (GE Healthcare) density gradient. The cells were stained with fluorescent labelled anti‐human APC‐CD45+ and anti‐mouse BV‐421‐CD45+ Ab (BD Biosciences, San Jose, CA, USA) and analyzed by flow on the LSRFortessa flow cytometer. Kaluza Analysis Software (Indianapolis, IN, USA) was used for data analysis.
2.4. Microarray and RNA sequencing datasets
All of the microarray datasets were from publicly available data resources, including GSE7053633 and GSE13159.36 RNA sequencing data was available through the TARGET website, https://ocg.cancer.gov/programs/target. Data were analyzed as described previously.1, 33, 37, 38
2.5. Methylation‐specific PCR and bisulfite sequencing
Genomic DNA was subjected to bisulfite treatment using the DNA Methylation‐Direct Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions. The bisulfite‐treated DNA (150 ng) was amplified using 1× ZymoTaq PreMix and 10 μmol/L primers specific to the methylated or unmethylated DNA sequence. The PCR products were separated on a 2% agarose gel, stained with GelGreen Nucleic Acid Staining Solution (Biotium, Fremont, CA, USA) and visualized using the Bio‐Rad ChemiDoc XRS equipped with Image Lab 5.0 software (Herkules, CA, USA). Primers and the detailed PCR conditions are summarized in Table S1. For sequencing, bisulfite‐treated genomic DNA was amplified using primers listed in Table S2. The PCR products were cloned into the Topo TA cloning kit (Thermo Fisher Scientific). Ten randomly picked clones were sequenced (Eurofins Genomics, Luisville, KY, USA) and aligned using Quantification Tool for Methylation Analysis.39
2.6. Chromatin immunoprecipitation
Chromatin immunoprecipitation was carried out on T‐ALL cells as described.40 DNA was immunoprecipitated with anti‐HDAC1 (sc‐7872X,) anti‐HDAC2 (sc‐7872X), anti‐HDAC3 (sc‐11417X), anti‐MeCP2 (sc‐137070), nonspecific IgG Abs (Santa Cruz Biotechnology, Dallas, TX, USA) or MBD3 (D1B8F) (Cell Signaling Technology, Danvers, MA, USA) and amplified by qRT‐PCR (Table S3) using SYBR Green PCR Master Mix on a StepOnePlus Real‐Time PCR System under standard conditions. Results were quantified by SYBR Green Real‐Time PCR analysis. The fold enrichment of immunoprecipitated samples was normalized on INPUT and expressed relative to the mock‐treated control (IgG). Results were visualized after separating PCR products on 3% agarose gel stained with GelGreen Nucleic Acid Staining.
2.7. Statistical analysis
The statistical analyses were undertaken using GraphPad Prism 7.02 software (GraphPad, La Jolla, CA, USA). The results were considered statistically significant when P < .05. Additional supplementary methods appear in Data S1.
3. RESULTS
3.1. Inactivation of SOCS5 promotes T‐ALL cell proliferation
We first analyzed a panel of 6 T‐ALL cell lines and found that the SOCS5 gene and protein were differentially expressed (Figure 1A). To investigate the biological roles of SOCS5 in T‐ALL, we undertook shRNA‐mediated knockdown of SOCS5 in PF382 and KOPTK1 cells that had higher levels of SOCS5 mRNA and protein. Depletion of SOCS5 mRNA and protein levels was confirmed by qRT‐PCR and immunoblotting, respectively (Figure 1B). Downregulation of SOCS5 expression promoted proliferation of T‐ALL cells as shown by an increase in cell number, increased cell cycle progression in S and G2/M phases, and decreased G1 phase (Figure 1C,D). We next investigated the effects of lentivirus‐induced SOCS5 expression (Figure 1E) on the proliferation of ALL‐SIL and CCRF‐CEM cells. Forced SOCS5 expression suppressed T‐ALL cell growth by inhibiting cell proliferation and reducing the cell cycle in S and G2/M phases, and increased G1 phase (Figure 1F,G). Interestingly, the knockdown and overexpression of SOCS5 had no effect on apoptotic cell death (Figure S1). These results indicate that SOCS5 negatively regulates proliferation of T‐ALL cells in vitro.
Figure 1.
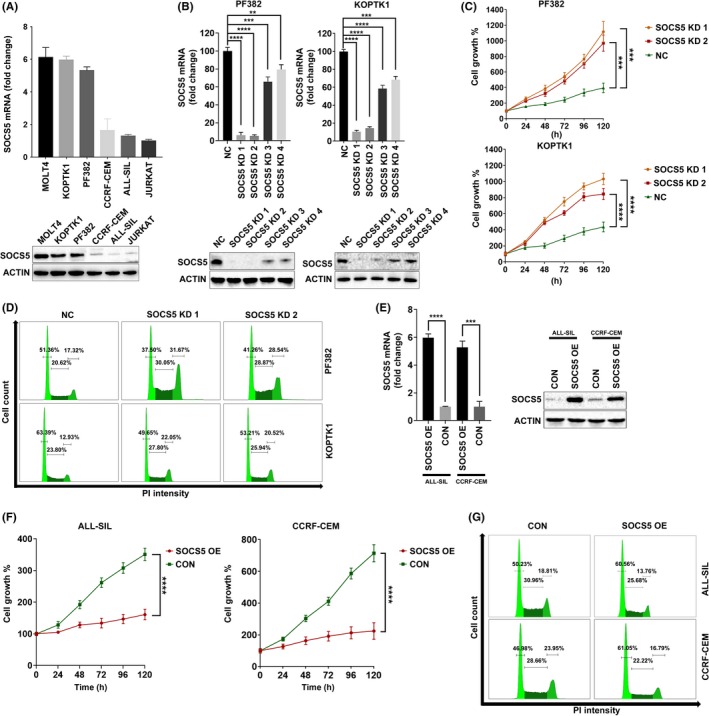
Suppressor of cytokine signaling 5 (SOCS5) negatively regulates T‐cell lineage acute lymphoblastic leukemia (T‐ALL) proliferation and cell cycle progression. A, SOCS5 mRNA and protein levels in T‐ALL cell lines (n = 6). For quantitative real‐time (qRT)‐PCR analyses, data are means ± SD for 3 independent experiments. B, PF382 and KOPTK1 cells were lentivirally transduced with 4 SOCS5 shRNA (SOCS5 KD1, SOCS5 KD2, SOCS5 KD3, and SOCS5 KD4) and scrambled control (NC), and the decrease in SOCS5 transcript and protein levels was confirmed by qRT‐PCR and immunoblotting (**P < .005, *** P < .0005, **** P < .0001). E, Overexpression of SOCS5 mRNA and protein in ALL‐SIL and CCRF‐CEM cells transduced with SOCS5 expressing plasmid (SOCS5 OE) compared to negative control plasmid (CON) was confirmed by qRT‐PCR and immunoblotting (***P < .0005, **** P < .0001). C,F, Growth curves of the transduced T‐ALL cell lines were determined by MTS assay. Data are means ± SD for 2 independent experiments carried out in triplicate (repeated measure ANOVA with Tukey's multiple comparisons test (***P < .0005, **** P < .0001). D,G, Cell cycle distribution was carried out 48 h post transduction by propidium iodide staining followed by flow cytometry analyses. Data are representative for 1 of 3 independent experiments
3.2. SOCS5 negatively regulates MYC and cytokine receptor expression in T‐ALL
With evidence that SOCS5 regulates T‐ALL proliferation, we studied the mechanism by which SOCS5 controls T‐ALL cell growth and signal transduction. We first examined the expression of IL‐7R and IL‐4R, which are involved in T‐ALL progression. Genetic inactivation of SOCS5 upregulated IL‐4R and IL‐7R levels in the tested cells (Figure 2A). In a converse experiment, overexpression of SOCS5 led to receptor downregulation, indicating that SOCS5 negatively regulates the expression of IL‐4R and IL‐7R in T‐ALL (Figure 2B). We also assessed whether the depletion of SOCS5 affects the expression of IL‐4. SOCS5 silencing increased the levels of IL‐4, whereas SOCS5 upregulation reduced its expression (Figure 2). Thus, downregulation of SOCS5 promotes the expression of critical T‐ALL cytokine receptors to enhance T‐ALL proliferation. Because MYC is a transcriptional target of JAK‐STAT signaling, we examined whether SOCS5 affects MYC expression. SOCS5 silencing induced MYC in the tested cells (Figure 2A). Conversely, forced SOCS5 expression reduced MYC protein levels, indicating that SOCS5 could have putative tumor suppressor activity in T‐ALL (Figure 2B).
Figure 2.
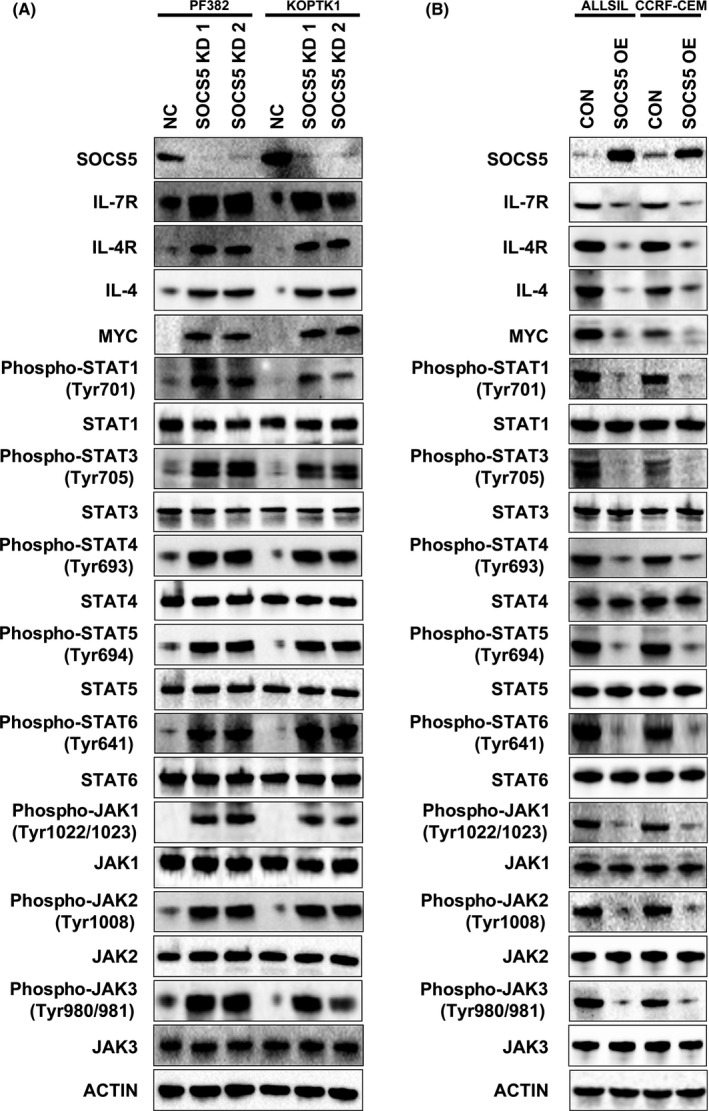
Suppressor of cytokine signaling 5 (SOCS5) negatively regulates cytokine receptors and the JAK‐STAT signaling pathway in T‐cell lineage acute lymphoblastic leukemia cell lines. A, PF382 and KOPTK1 cells were transduced with SOCS5 shRNA (SOCS5 KD1 and SOCS5 KD2) and scrambled negative control (NC). B, ALL‐SIL and CCRF‐CEM cells were transduced with SOCS5 expressing plasmid (SOCS5 OE) or control plasmid (CON). A,B, Cells were lysed and subjected to immunoblotting for the expression of interleukin‐7 receptor (IL‐7R), IL‐4R, IL‐4, and MYC, levels of phosphorylated and total STAT proteins, and activation and total levels of JAK. Western blots were undertaken 3 times and the representative blots are shown
3.3. SOCS5 downregulation induces activation of the JAK‐STAT signaling pathway
To test whether SOCS5 negatively regulates signal transduction in T‐ALL, we examined the JAK‐STAT pathway activation in SOCS5‐depleted cells compared to the negative control. Silencing of SOCS5 expression markedly enhanced the phosphorylation of STAT1, STAT3, STAT4, STAT5, and STAT6 but had no effect on the total STAT levels (Figure 2A). Accordingly, forced SOCS5 expression reduced the activation of the tested STAT proteins, consistent with no changes in their total levels (Figure 2B). We next investigated whether SOCS5 preferentially inhibits JAK1, JAK2, JAK3, and TYK2 in T‐ALL cells. SOCS5 depletion accelerated JAK1, JAK2, and JAK3 phosphorylation but had modest to no effect on TYK2 (Figures 2A and S2). Conversely, overexpressed SOCS5 inhibited activation of all JAK proteins except for TYK2 in the tested cells (Figures 2B and S2). As expected, SOCS5 had no effect on total JAK protein levels. Thus, SOCS5 negatively regulates JAK‐STAT signaling in T‐ALL.
3.4. SOCS5 negatively regulates T‐ALL progression in vivo
Because SOCS5 regulates T‐ALL cell growth in vitro, we next investigated the effects of SOCS5 expression on T‐ALL progression in NSG mice injected with SOCS5‐depleted or control cell lines. SOCS5 depletion significantly reduced survival of the engrafted NSG mice (SOCS5 KD1, n = 10; SOCS5 KD2, n = 10) when compared to the control group (NC, n = 10) (Figure 3A,B). Moreover, tissue analyses from mice killed 25 days post‐injection revealed profound evidence of leukemia progression in mice injected with the SOCS5‐depleted cells (SOCS5 KD1, n = 4; SOCS5 KD2, n = 4) compared to the control group (NC, n = 4). SOCS5 silencing significantly increased leukemia burden in bone marrow, brain, and liver of the tested mice (Figure 3C). PF382 cells did not infiltrate the spleen in all experimental groups, which is consistent with previously published data for PF382 cells.41 These results show that SOCS5 downregulation promotes T‐ALL cell proliferation in vivo, suggesting its suppressive role in leukemia engraftment and progression.
Figure 3.
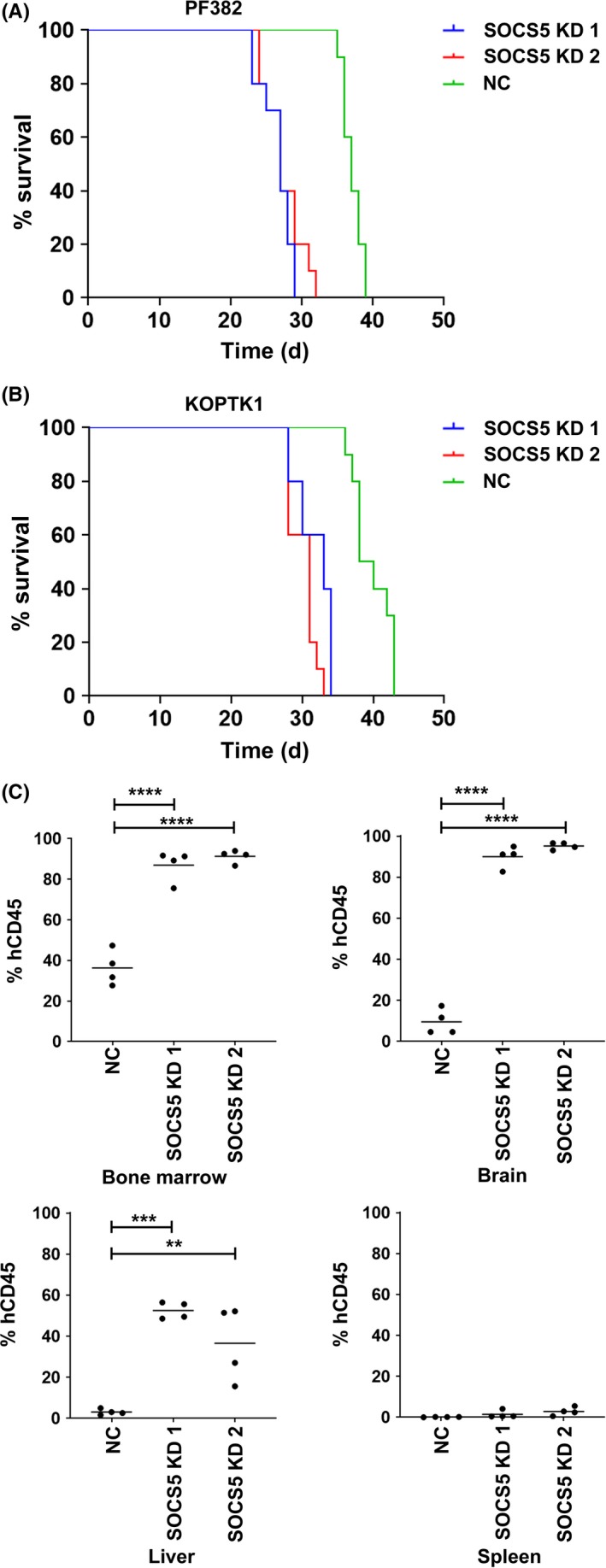
Silencing of suppressor of cytokine signaling 5 (SOCS5) accelerates leukemia progression in murine xenograft model of human T‐cell lineage acute lymphoblastic leukemia. NOD.Cg‐Prkdc scid Il2rg tm1Wjl/SzJ mice were engrafted with 1 × 106 cells (PF382 or KoptK1) transduced with 2 shRNAs targeting SOCS5 (SOCS5 KD1 and SOCS5 KD2) and scrambled control (NC), respectively. A,B, Kaplan‐Meier plot of animal survival in each transplanted group (n = 10 mice per group). For both, PF382 SOCS5 KD1 and SOCS5 KD2 group median survival was 27 d compared to the NC group, in which median survival was 37 d (log‐rank Mantel‐Cox test: SOCS5 KD1 vs NC, P < .0001; SOCS5 KD2 vs NC, P < .0001). For KoptK1 SOCS5 KD1 and SOCS5 KD2 groups, median survival was 33 and 31 d, respectively, compared to the SOCS5 NC group with median survival of 40 d (log‐rank Mantel‐Cox test: SOCS5 KD1 vs NC, P < .0001; SOCS5 KD2 vs NC, P < .0001). C, For leukemia burden analyses, all mice (4 mice per group) were killed 25 d post‐inoculation with transduced PF382 cells and the levels of human and murine CD45+ cells were assessed by flow cytometry (one‐way ANOVA with Dunnett's multiple comparison test: **P < .005; ***P < .0005, ****P < .0001)
3.5. SOCS5 expression is reduced in leukemic patients harboring KMT2A gene rearrangements
To determine SOCS5 expression in primary T‐ALL, we assessed microarray data for 100 T‐ALL samples obtained from children, adolescents, and young adults enrolled in COG AALL0434 (GSE70536),33 and identified different levels of SOCS5 mRNA expression in the tested dataset (Figure 4A). To further identify molecular characteristics associated with distinct SOCS5 expression levels, we analyzed genomic data available for 264 T‐ALL patients reported by Liu et al.1 SOCS5 was differentially expressed between the molecular subtypes of T‐ALL (P < .005) including HOXA‐ and TLX1‐deregulated cases with the lowest, and TLX3‐deregulated cases with the highest levels of SOCS5 mRNA, respectively (Figure 4B). While SOCS5 mRNA levels were higher in immature LYL1/LMO2‐deregulated leukemia, there was no evidence of an association between SOCS5 expression with early T‐cell phenotype or differentiation arrest at distinct stages of T cell development (data not shown). Because HOXA‐deregulated T‐ALL is enriched in alterations involving KMT2A or MLLT10 genes,33, 42, 43 we sought to determine whether SOCS5 mRNA levels were associated with recurrent chromosomal alterations found in T‐ALL. We found that SOCS5 expression was downregulated in T‐ALL patients harboring KMT2A gene rearrangements (KMT2A‐R, P < .005; Figure 4C). The results were consistent with our microarray dataset of 100 T‐ALLs, in which KMT2A‐R samples (n = 12) had lower levels of SOCS5 mRNA compared to the remaining samples (Figure 4D). To further validate our findings, we tested a limited number of primary T‐ALL samples (n = 25; Table S4) and confirmed that both SOCS5 mRNA and protein are differentially expressed in T‐ALL, and that samples harboring KMT2A‐R have lower levels of SOCS5 expression (Figure 4E). SOCS5 mRNA was robustly expressed in PBMC, pan T‐cells, and thymocytes from healthy individuals, but its levels were lower in bone marrow CD34+ cells (Figure 4E). These results show that SOCS5 is deregulated in T‐ALL. Interestingly, SOCS5 downregulation was also identified in B‐ALL and AML primary samples having KMT2A‐R (Figure 4F,G), indicating that SOCS5 inactivation could represent a feature of deregulated signaling networks in KMT2A‐R leukemias. In order to test for the effects of KMT2A rearrangements on SOCS5 expression, BaF3 cells were transduced with KMT2A‐MLLT4 and KMT2A‐MLLT1 constructs, representing the most prevalent KMT2A‐R in T‐ALL (Figure S3, Table S5).1, 33, 44 Forced expression of KMT2A‐MLLT4 and KMT2A‐MLLT1 resulted in the decrease in SOCS5 protein levels and the increased phosphorylation of JAK‐STAT proteins in BaF3 cells (Figure 5A). We next examined IL‐7R expression levels and JAK‐STAT pathway activation in a small set of primary T‐ALL samples with higher (n = 3) and lower/undetectable (n = 3) SOCS5 protein levels. We identified elevated expression of IL‐7R and increased activation of JAK‐STAT proteins in primary T‐ALL samples with lower/undetectable SOCS5 levels (both ALL‐215 and ALL‐216 harbor KMT2A‐R) compared to samples that had higher levels of SOCS5 protein (Figure 5B). We next used lentiviral transduction to induce or silence SOCS5 expression in 2 primary T‐ALL samples, ALL‐214 and ALL‐215 (Table S4). Forced expression of SOCS5 in ALL‐215 primary cells lowered the expression of IL‐7R and decreased the activation of STAT1, STAT3, STAT4, STAT5, and STAT6 as well as JAK1, JAK2, and JAK3 proteins (Figure 5C). Conversely, genetic silencing of SOCS5 in ALL‐214 cells increased the expression of IL‐7R and the activation of the JAK‐STAT signaling pathway (Figure 5C). Together, these results indicate that SOCS5 downregulation is associated with KMT2A gene rearrangements and its lower levels enhance JAK‐STAT and IL‐7R signaling in T‐ALL.
Figure 4.
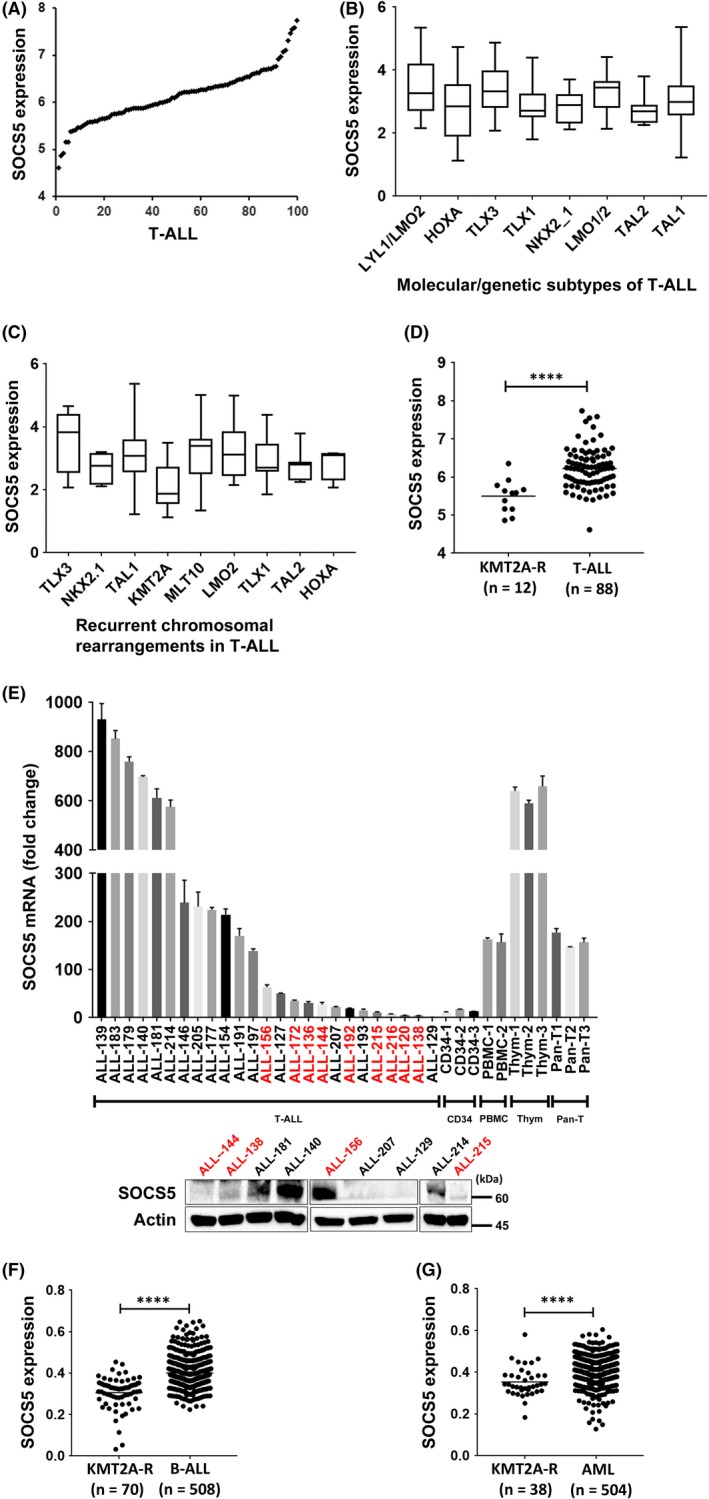
Suppressor of cytokine signaling 5 (SOCS5) expression is lowered in acute leukemias with KMT2A gene rearrangements. A, Microarray data analysis of SOCS5 expression in pediatric and young adult T‐cell lineage acute lymphoblastic leukemia (T‐ALL) patients (n = 100) treated in the COG AALL0434 study (GSE70636).33 Multiple probe sets were tested. The representative probe set for SOCS5 is shown (209647_s_at). B, SOCS5 expression across distinct molecular/genetic subtypes of T‐ALL identified in a cohort of 264 pediatric and young adult T‐ALL patients by RNA sequencing (Kruskal‐Wallis with Dunn's multiple comparison test, P < .005).1 C, SOCS5 expression in 173 T‐ALL cases classified by recurrent chromosomal rearrangements (TLX3, n = 14; NKX2.1, n = 10; TAL1, n = 77; KMT2A, n = 12; MLLT10, n = 12; LMO2, n = 10; TLX1, n = 17; TAL2, n = 7; HOXA, n = 4) (Kruskal‐Wallis with Dunn's multiple comparison test, P < .005).1 D, SOCS5 expression in T‐ALL with KMT2A gene rearrangements (KMT2A‐R) in an independent published cohort of 100 T‐ALL cases tested by microarray (unpaired Mann‐Whitney U test, **** P < .0001).33 E, SOCS5 mRNA levels in primary T‐ALL samples (n = 25), normal bone marrow CD34+ cells (n = 3), normal PBMC (n = 2), pan T‐cells (Pan‐T) (n = 3), and normal thymocytes (Thym, n = 3) by quantitative real‐time PCR. Immunoblotting for SOCS5 protein levels in primary T‐ALL samples (n = 9), for which cellular material was available. Red text indicates the samples harboring KMT2A‐R. F,G, Analysis of SOCS5 mRNA levels in B‐cell (B‐)ALL and AML with KMT2A‐R compared to the remaining cases in a previously reported microarray dataset36 (unpaired Mann‐Whitney U test, ****P < .0001)
Figure 5.
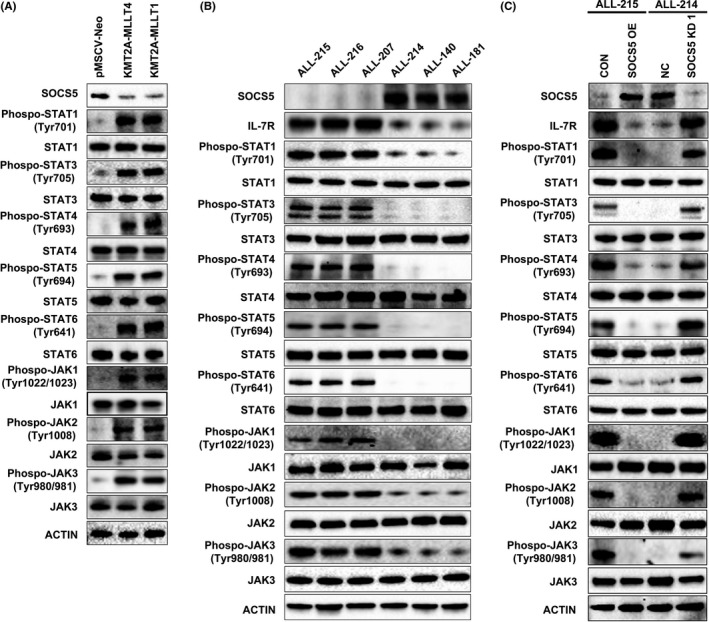
Suppressor of cytokine signaling 5 (SOCS5) negatively regulates interleukin‐7 receptor (IL7R) and JAK‐STAT signaling in BaF3 cells expressing KMT2A‐R and primary T‐cell lineage acute lymphoblastic leukemia (T‐ALL) cells. A, Immunoblotting of BaF3 cells transduced with KMT2A‐MLLT1 or KMT2A‐MLLT4 and negative control (pMSCV‐Neo) for SOCS5 and activated or total levels of JAK‐STAT proteins. B, Immunoblot analyses for IL‐7R and JAK‐STAT activation in primary T‐ALL, including 3 samples with higher and 3 samples with low/undetectable SOCS5 protein levels. C, Primary T‐ALL cells harboring KMT2A gene rearrangements (ALL‐215) were transduced with SOCS5‐expressing plasmid (SOCS5 OE) or negative control plasmid (CON). Primary T‐ALL sample (ALL‐214) was transduced with SOCS5 shRNA (SOCS5 KD1) only (due to insufficient cellular material) and scrambled negative control (NC). The cell lysates were immunoblotted for the expression of SOCS5 and IL‐7R and the levels of phosphorylated and total JAK‐STAT proteins. Representative blots are shown
3.6. SOCS5 downregulation potentiates IL‐7‐induced STAT5 activation
To determine whether SOCS5 inactivation reinforces the JAK‐STAT signaling in response to IL‐7 stimulation, we utilized primary T‐ALL cells (ALL‐214 and ALL‐215) and cell lines (PF382 and ALL‐SIL), which were stimulated with IL‐7 (25 ng/mL; Figure 6). Under basal conditions, SOCS5‐expressing PF382 cells showed the increase in STAT5 phosphorylation after 60 minutes of cytokine stimulation (Figure 6A). Interestingly, genetic inactivation of SOCS5 led to increased STAT5 activation in serum and cytokine‐free environment compared to negative control cells, and increased reactivity of the tested cells to cytokine stimulation. In a converse experiment, low SOCS5 expressing ALL‐SIL cells were hypersensitive to IL‐7 stimulation, leading to high levels of phosphorylated STAT5. The activation of STAT5 was significantly reduced in the cells with forced SOCS5 expression (Figure 6B). To further validate our findings, similar experiments were carried out in primary T‐ALL samples. ALL‐215 cells harboring KMT2A‐R had higher basal levels of STAT5 phosphorylation and showed enhanced STAT5 activation following IL‐7 stimulation compared to T‐ALL cells obtained from an ALL‐214 sample (Figure 6C). Forced SOCS5 expression inhibited STAT5 activation in primary ALL‐215 cells. In contrast, SOCS5 depletion in ALL‐214 cells led to a significant increase in STAT5 activation. Together, these results indicate that SOCS5 silencing leads to hyperactivation of IL‐7‐induced STAT5 signaling.
Figure 6.
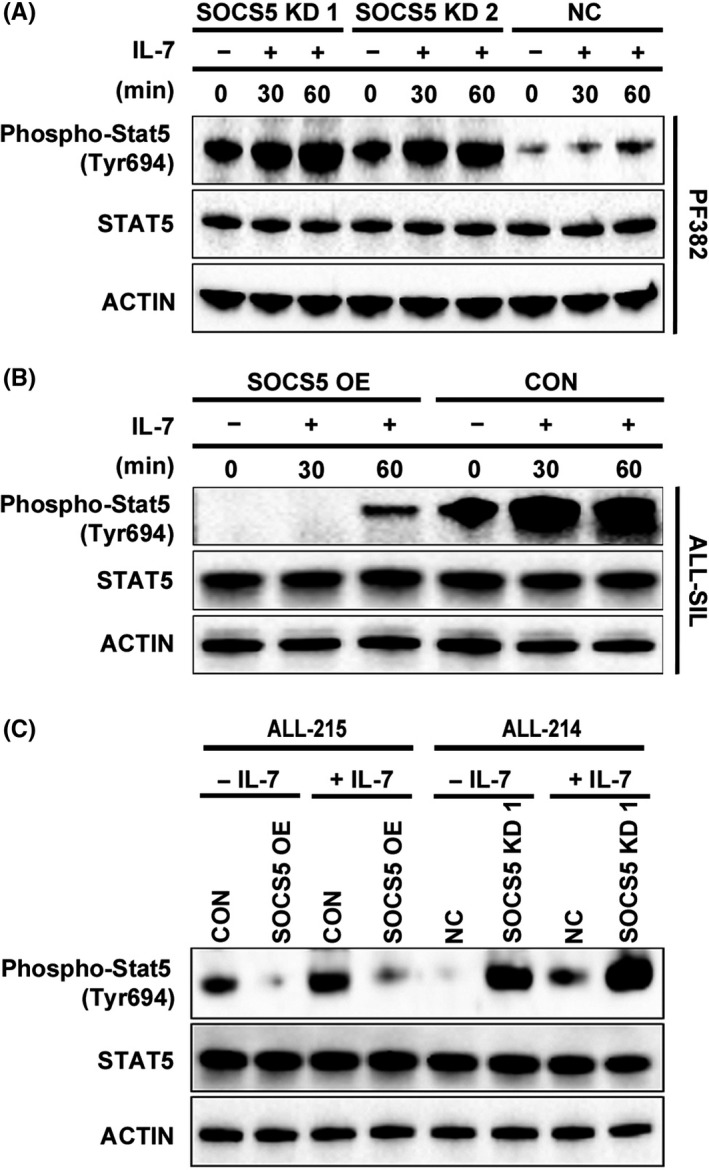
Suppressor of cytokine signaling 5 (SOCS5) downregulation enhances interleukin‐7 (IL‐7)‐induced STAT5 activation. PF382 cell line and primary T‐cell lineage acute lymphoblastic leukemia T‐ALL (ALL‐214) cells were transduced with SOCS5 shRNA (SOCS5 KD1 and SOCS5 KD2) and scrambled control (NC). ALL‐SIL cell line and primary T‐ALL cells (ALL‐215) were transduced with SOCS5‐expressing plasmid (SOCS5 OE) or negative control plasmid (CON). The transduced cell lines (A,B) and primary T‐ALL samples (C) (ALL‐215 and ALL‐214) were serum and cytokine starved (120 min) followed by stimulation with IL‐7 (25 ng/mL). The cell lysates were tested for the levels of phosphorylated and total STAT5. Representative blots are shown
3.7. SOCS5 gene is differentially methylated in T‐ALL
Because we did not identify any mutations in the SOCS5 gene in primary T‐ALL samples (Figure 4E; not shown), we hypothesized that SOCS5 expression is regulated by DNA methylation in T‐ALL. We analyzed the genomic sequence of SOCS5 (Figure 7A) spanning from −60 179 to −59 344 bps and identified densely clustered CpG islands around the promoter and the 1st exon. We performed a methylation‐specific PCR using unmethylated or methylated DNA specific primers (Table S1, Figure S4) to investigate the methylation of the SOCS5 promoter (−60 162 to −59 938 bps) and 1st exon (−59 594 to −59 469 bps) regions in T‐ALL cell lines (n = 4), primary T‐ALL samples (n = 12; Table S4), and normal thymocytes (n = 3). SOCS5 promoter/1st exon were hypermethylated in the cell lines showing lower SOCS5 expression levels (ALL‐SIL and CCRF‐CEM) compared to PF382 and KOPTK1 that had higher levels of SOCS5 mRNA, in which the tested regions were hypomethylated (Figures 1A and 7B). Primary T‐ALL samples with lower levels of SOCS5 expression showed methylation in promoter and/or 1st exon regions (Figures 4E and 7C). By contrast, T‐ALL samples expressing higher levels of SOCS5 mRNA showed absent or partial methylation in the tested regions (Figures 4E and 7C). As expected, normal thymocytes had unmethylated promoter/1st exon regions of the SOCS5 gene (Figures 4E and 7C). To validate our findings, we undertook bisulfite sequencing of the promoter (−60 098 to −59 934 bps) and 1st exon (−59 598 to −59 422 bps) regions of SOCS5. We confirmed the presence of methylated CpG sites (Figure 7D, filled circles) in ALL‐SIL and CCRF‐CEM cells (lower SOCS5 expression) and unmethylated CpG sites (Figure 7D, open circles) in PF383 and KOPTK1 cell lines (higher SOCS5 expression). Bisulfite sequencing of 6 primary T‐ALL samples for which sufficient DNA was available confirmed CpG island methylation in samples with lower levels of SOCS5 expression (ALL‐138, ALL‐192, and ALL‐172) compared to unmethylated CpG islands in ALL‐146, ALL‐205, and ALL‐177, in which SOCS5 was robustly expressed (Figures 4E and 7E). In addition, treatment of T‐ALL cells with the DNA demethylating agent 5‐AzaC led to a decrease in DNA methylation of the SOCS5 promoter/1st exon region and increased expression of SOCS5 mRNA and protein levels (Figures 7F,G and S5). To determine whether SOCS5 is a target for DNA methyltransferase, we analyzed a previously published RNASeq dataset for 264 T‐ALL.1 Among all DNMTs, only DNMT3A expression was inversely correlated with SOCS5 mRNA levels in primary T‐ALL samples (Figure 7H). The role of DNA methylation in regulation of SOCS5 expression was further examined by shRNA‐mediated knockdown of DNMT3A methyltransferase in ALL‐SIL cells. DNMT3A silencing led to increased expression of SOCS5 mRNA and protein levels, corresponding to decreased methylation in promoter and/or 1st exon regions of the SOCS5 gene (Figure 7I‐K). Together, these results indicate that DNA methylation regulates SOCS5 expression in T‐ALL.
Figure 7.
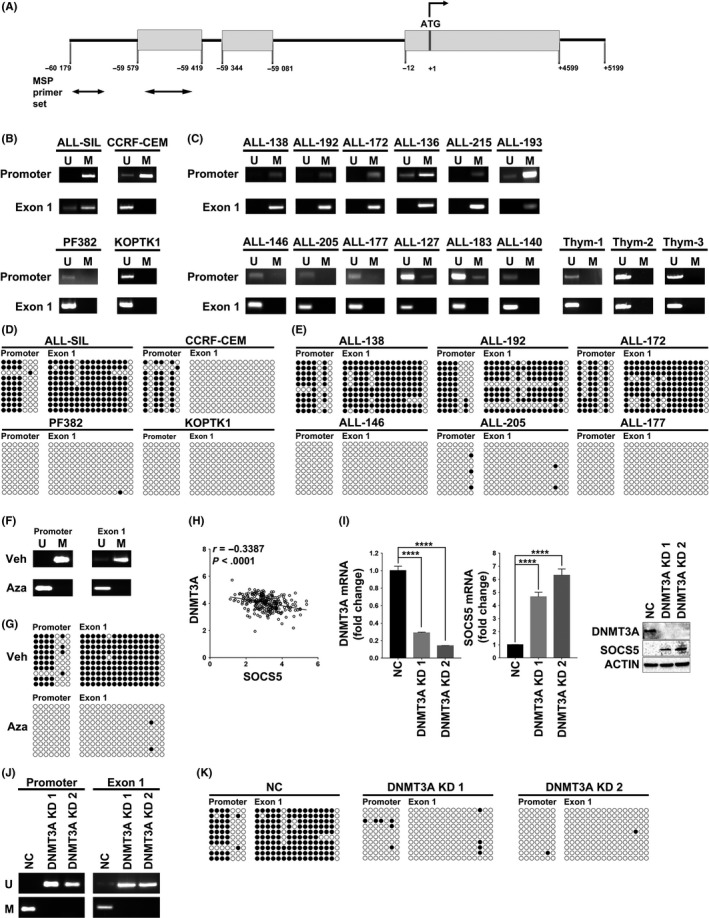
DNA methylation regulates suppressor of cytokine signaling 5 (SOCS5) expression. A, Schematic diagram of the SOCS5 gene. Gray boxes represent exons; the translation start site is at +1, and the arrows indicate direction of translation. The bottom arrows indicate primers used for methylation specific PCR (MS‐PCR). DNA methylation of SOCS5 promoter/1st exon in (B) T‐cell lineage acute lymphoblastic leukemia (T‐ALL) cell lines (n = 4) and (C) primary T‐ALL samples (n = 12) and normal thymocytes (n = 3) were tested by methylation specific (MS)‐PCR. M, methylated; U, unmethylated. D,E, Bisulfite sequencing of SOCS5 promoter/1st exon region in (D) T‐ALL cell lines (n = 4) and (E) primary T‐ALL samples (n = 6) for which sufficient DNA was available. Unmethylated CpG site in the amplified region is shown as an open, white circle and methylated CpG as a closed, black circle. F,G, DNA methylation (F) and bisulfite sequencing (G) of SOCS5 promoter/1st exon in ALL‐SIL cells treated with demethylating agent 5‐azacitidine (Aza) (24 h, 10 μmol/L) or vehicle control (Veh). H, Expression correlation between SOCS5 and DNA methyltransferase‐3A (DNMT3A) in a previously published RNASeq dataset for 264 T‐ALL patients from the COG study (NCT00408005)1 (r, Pearson correlation; P < .0001). I, ALL‐SIL cells were infected with scrambled control (NC) or lentivirus expressing shRNA targeting DNMT3A (DNMT3A KD1 and DNMT3A KD2). Knockdown of DNMT3A and SOCS5 gene expression was examined by quantitative real‐time PCR and immunoblotting. SOCS5 in negative control cells (NC) was normalized to 1. Data are means ± SD for 3 independent experiments (****P < .0001; two‐tailed Student's test). J,K, DNA methylation and bisulfite sequencing analyses of the SOCS5 promoter/1st exon in DNMT3A‐depleted ALL‐SIL cells compared to NC
3.8. Histone deacetylation regulates SOCS5 expression
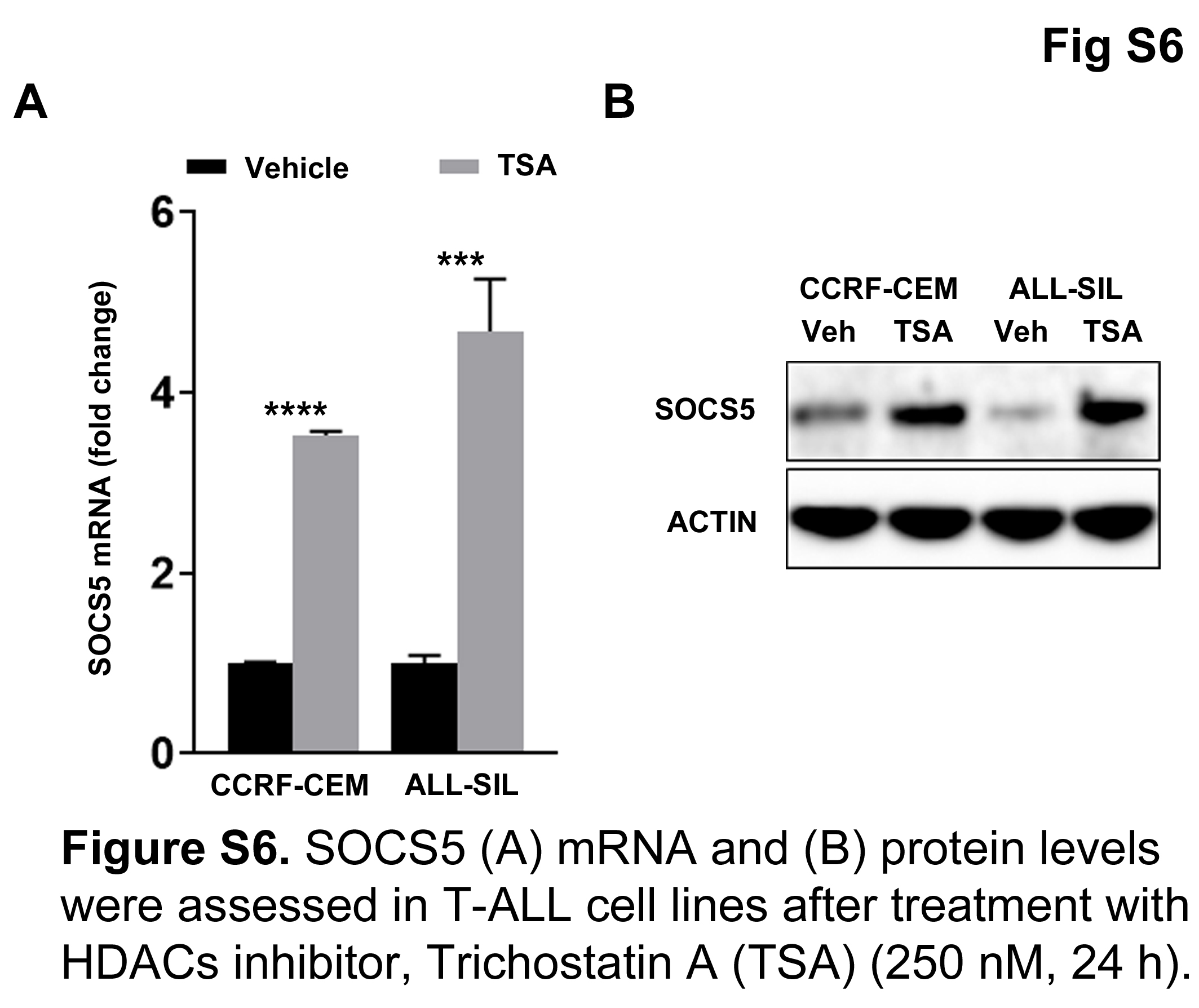
Methylated DNA can be bound by MBDs, which recruit multiprotein co‐repressor complexes carrying HDACs to facilitate transcriptional repression.45, 46, 47 We first tested whether histone deacetylation contributes to SOCS5 silencing by treating T‐ALL cell lines with TSA, a potent inhibitor of class I and II HDACs, and found that TSA treatment induced SOCS5 mRNA and protein expression (Figure S6). To identify specific HDACs involved in the epigenetic regulation of SOCS5 expression, we carried out ChIP using Abs directed against HDAC1, HDAC2, and HDAC3 followed by qRT‐PCR analyses. HDAC1 and HDAC2, but not HDAC3, occupied CpG islands at promoter/1st exon regions of the SOCS5 gene in ALL‐SIL cells and primary T‐ALL samples (n = 2; ALL‐215 and ALL‐216) (Figure 8A). Because HDACs lack DNA binding domains, specific MBDs are required to mediate deacetylation of histone tails. To confirm whether HDAC1 and HDAC2 are recruited to the SOCS5 locus, we carried out ChIP against MeCP2, which is known to recruit SIN3 complex,47, 48 and against MBD3, which is a core subunit of the NuRD complex.46 The SIN3 and NuRD co‐repressor complexes are commonly associated with HDAC1 and HDAC2.46, 47 We found a significant enrichment of MeCP2 at the promoter and 1st exon region of SOCS5 in the tested cells, suggesting that silencing of the SOCS5 expression is regulated by the recruitment of the MeCP2 methyl binding protein and SIN3 co‐repressor complex (Figure 8B). To further investigate whether MeCP2 binding depends on the SOCS5 promoter/1st exon methylation, we treated ALL‐SIL cells with 10 μmol/L demethylating agent, 5‐AzaC. DNA demethylation abrogated MeCP2 binding to the SOCS5 gene (Figure 8C). Thus, MeCP2 regulation of SOCS5 expression is methylation‐dependent. In addition, treatment with 5‐AzaC induced acetylation of histone 3 tails at the SOCS5 locus, which is indicative of chromatin decondensation (Figure 8D). Our findings show that histone deacetylation regulates SOCS5 expression in T‐ALL through the recruitment of the MeCP2‐SIN3 co‐repressor complex.
Figure 8.
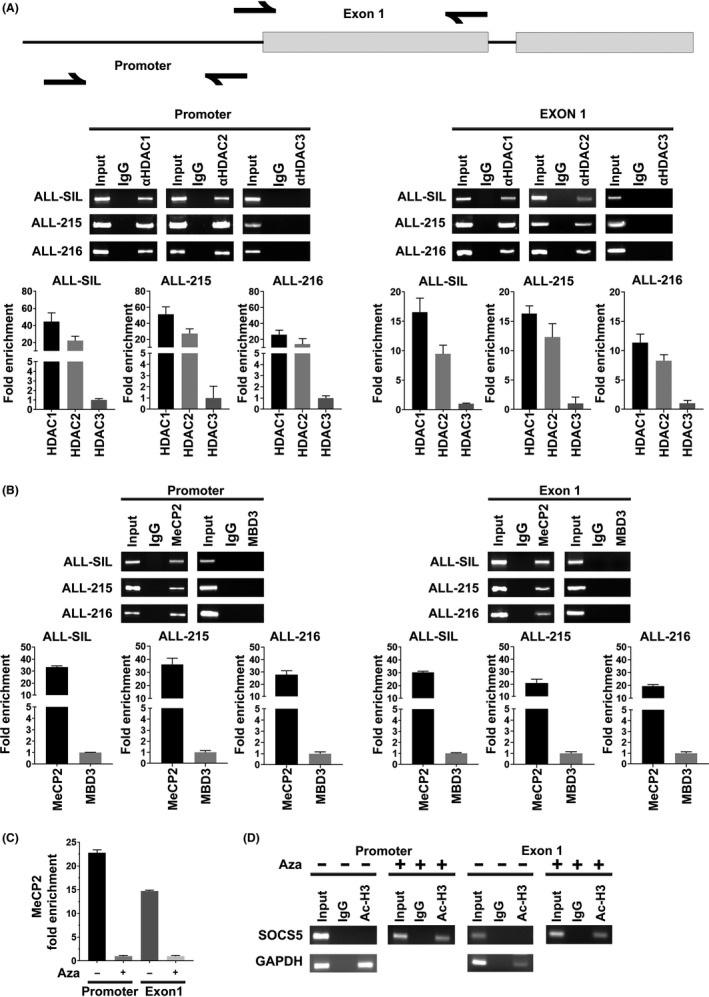
Suppressor of cytokine signaling 5 (SOCS5) expression is regulated through histone deacetylation. A, Identification of specific histone deacetylases (HDAC) and methyl binding proteins at promoter/1st exon regions of the SOCS5 gene. A schematic diagram of the SOCS5 gene (upper) with half arrows indicating the primers used for ChIP analyses. ChIP (lower) with specific (A) α‐HDAC1, α‐HDAC2, α‐HDAC3 and (B) α‐methyl CpG binding protein‐2 (α‐MeCP2) and methyl‐CpG‐binding domain protein‐3 (MBD3) Abs was carried out in ALL‐SIL cells and 2 primary T‐cell lineage acute lymphoblastic leukemia samples, ALL‐215 and ALL‐216. The immunoprecipitates were analyzed using quantitative real‐time (qRT)‐PCR and the amplification product was separated on 3% agarose gel. Data are means ± SD for 3 independent experiments. An IgG Ab was used as negative control. Signals are normalized to input DNA. C, The presence of MeCP2 at promoter/1st regions of SOCS5 was tested in ALL‐SIL cells untreated (−) or treated (+) with 10 μmol/L 5‐azacitidine (Aza, 24 h). The immunoprecipitates were analyzed by qRT‐PCR as described above. D, ChIP analyses for the acetylation status of histone H3 tails at SOCS5 promoter/1st exon regions were done in ALL‐SIL cells using α‐acetylated H3 Ab. The cells were incubated in the absence (−) and in the presence (+) of 10 μmol/L Aza (24 h). Immunoprecipitates (Ac‐H3) were subjected to qRT‐PCR with primer pairs specific for SOCS5 promoter/1st exon region and for GAPDH, as a positive control
4. DISCUSSION
A cytokine‐inducible negative regulator of the JAK‐STAT signaling pathway, SOCS5 is epigenetically deregulated in T‐ALL, leading to a number of previously unappreciated effects on T‐ALL cells. The downstream consequences include inhibition of cell proliferation and leukemia progression, negative regulation of cytokine receptor and JAK‐STAT signaling.
Our analyses show that SOCS5 is differentially expressed in primary T‐ALL samples; however, we and others1 did not identify mutations in the SOCS5 gene that might explain its expression profiles. Although mutations in the SOCS family genes are very rare, aberrant SOCS signaling has been linked to the epigenetic deregulation of their expression.22, 23 For instance, reduced levels of SOCS5 mRNA and its promoter hypermethylation were reported in hepatocellular carcinoma, cervical cancer, and thyroid tumors.21, 24 In this study, we dissected the mechanism of epigenetic deregulation of SOCS5 expression in T‐ALL through aberrant DNA methylation. We showed that SOCS5 mRNA levels were closely associated with its promoter methylation. DNMT3A was identified to negatively regulate the SOCS5 expression levels in T‐ALL cells. Histone acetylation has also been implicated in the epigenetic regulation of the SOCS family genes in solid tumors.21 In addition, early studies identified the CpG binding protein, MeCP2 as a transcriptional silencer.49, 50 Our ChIP analyses provide evidence that MeCP2 binds to the CpG islands of the SOCS5 promoter/1st exon and that MeCP2 regulation of SOCS5 expression is methylation‐dependent. Furthermore, we identified the presence of HDAC1 and HDAC2 at the SOCS5 promoter region, suggesting that MeCP2 recruits HDAC1/2 through SIN3 co‐repressor complexes. Our results indicate that MeCP2 and histone deacetylation are mechanistically linked to the negative regulation of SOCS5 expression in T‐ALL. Other potential mechanisms of epigenetic deregulation of SOCS5 cannot be excluded and might be related to aberrant expression of microRNAs in T‐ALL.51
There is a growing body of evidence indicating that genes involved in T cell differentiation play important roles in T‐ALL pathobiology.1, 52 Our data suggest that SOCS5 is differentially expressed among distinct molecular subtypes of T‐ALL but we found no evidence for the association between SOCS5 mRNA levels and specific stages of T‐cell maturation arrest. Strikingly, we observed that SOCS5 mRNA levels were lower in HOXA‐and TLX1‐deregulated cases but higher in the TLX3‐deregulated subtype, which all represent the Homeobox family members. Moreover, LMO2/LYL1‐deregulated samples, which represent the most immature T‐ALL subsets, had elevated levels of SOCS5 mRNA in contrast to normal CD34+ progenitors, in which SOCS5 expression levels were relatively lower. This interesting observation could be linked to aberrant JAK‐STAT activation commonly found in ETP patients.11, 14 In our studies, SOCS5 was not significantly elevated in ETP samples except for 1 dataset reported by Zhang et al14 (Figure S7). Further studies are required to delineate the roles of ETP phenotype in modulating the activity of SOCS5 in T‐ALL. One of the intriguing observations was that SOCS5 was downregulated in KMT2A‐R T‐ALL. In our study patients, ALL‐144, ALL‐138, and ALL‐215 had KMT2A‐R and almost undetectable SOCS5 protein levels (Figure 4E). We found that forced KMT2A‐R expression lowered SOCS5 protein levels and led to increased activation of JAK‐STAT signaling (Figure 5A). Our previous analyses on deregulated gene expression profiles in KMT2A‐R T‐ALL identified SOCS5 within the most downregulated genes.53 Whether SOCS5 silencing is linked to KMT2A‐R‐driven oncogenesis in T‐ALL and other acute leukemias remains unknown.
In line with previous studies, in which SOCS5 was proposed as a putative tumor suppressor, our results show that forced SOCS5 expression inhibited leukemic cell proliferation and cell cycle progression. Interestingly, apoptosis was not affected, suggesting that SOCS5 specifically regulates proliferation but not cell death in T‐ALL. The results were consistent with our knockdown studies, in which SOCS5 inactivation accelerated cell growth and cell cycle progression in T‐ALL cells. SOCS5 downregulation and its tumor suppressive role were reported in cervical and thyroid cancers.21, 30, 31 However, in chronic lymphocytic leukemia patients, increased SOCS5 expression governed the defective function of dendritic cells.54 Moreover, elevated SOCS5 protein levels were associated with unfavorable prognosis in liver and ovarian cancer (www.proteinatlas.org), strongly suggesting that the roles exerted by SOCS5 are likely tissue‐ and tumor‐specific.
Interleukin‐7 signaling is a critical determinant of normal T cell development and differentiation, and activating mutations in IL‐7R have been shown to drive an oncogenic program in approximately 8%‐10% T‐ALL.1, 14, 16, 55, 56, 57 Here, we show for the first time that SOCS5 negatively regulates the IL‐7R signaling pathway in T‐ALL cells. Because IL‐7R is downregulated in the presence of IL‐7 secreted in the bone marrow microenvironment,55 it will be critical to further assess the roles of SOCS5‐mediated signal transduction in IL‐7 signaling. Our data provide evidence on the roles of SOCS5 downregulation in potentiating STAT5 activation in the presence of IL‐7. In addition, IL‐4 was also shown to stimulate T‐ALL cell growth and proliferation.58 In our study, SOCS5 depletion upregulated both IL‐4R and IL‐4, raising an important question regarding the IL‐4‐dependent autocrine loop, which could self‐induce T‐ALL proliferation. These findings provide an important foundation for further research into the mechanistic link between SOCS5 and cytokine receptor signaling in T‐ALL as autocrine and/or paracrine effects were reported for IL‐2 and IL‐15 in adult T‐cell leukemia.59 In addition to IL‐7R mutations, activating mutations in JAK1, JAK2, JAK3, and STAT5B were associated with aberrant JAK‐STAT activation in T‐ALL cells.1, 17, 18 In this study, we showed that SOCS5 negatively regulates the activation of STAT proteins and selectively regulates phosphorylation of JAK1, JAK2, and JAK3 but not TYK2. These results are of great interest considering that JAK‐STAT activation was also reported in patients lacking mutations in IL‐7R and JAK‐STAT signaling molecules11, 56 suggesting that there are other mechanisms activating this pathway. Our data show that SOCS5 downregulation potentiates JAK‐STAT signal transduction and leukemia progression. Although we did not determine the mutational status of IL‐7R and JAK‐STAT molecules in the tested primary T‐ALL samples (n = 24), our analyses of previously reported 264 T‐ALL patients1 show a lack of correlation between mutations in the JAK‐STAT pathway and the levels of SOCS5 in T‐ALL (Figure S8).
Finally, our work indicates that SOCS5 inactivation accelerated leukemia cell proliferation and engraftment in the T‐ALL xenotransplantation model in vivo, emphasizing its critical role in T‐ALL cell proliferation and leukemia progression. Strikingly, lower levels of SOCS5 facilitated extensive blast dissemination in bone marrow but also in brain and other organs, pointing towards proliferative and possibly migratory roles of SOCS5 in T‐ALL. Previous studies in solid tumors reported the roles of SOCS5 downregulation in promoting cell migration through epidermal growth factor receptor and JAK‐STAT activation.51
In summary, we propose that epigenetic deregulation of SOCS5 expression impacts T‐ALL cell proliferation and leukemic progression. We postulate that downregulation of SOCS5 expression potentiates aberrant JAK‐STAT signal transduction to govern T‐ALL progression. Further studies are warranted to determine whether and how SOCS5 orchestrates with recurrent mutations in the IL‐7R and JAK‐STAT signaling pathway in T‐ALL.
DISCLOSURE
The authors have no conflicts to disclose.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This project was supported by the NIH National Cancer Institute (1R01 CA237165 to KMW). Other grant support: NIH NCATS CTSA (8UL1TR000041), NIH NCI the Cancer Center Support Grant (P30CA118100), dedicated Health Research Funds from the UNM SOM (KMW), 5R01 CA170250 (SAN), Gabrielle's Angel Foundation (HK), U24 CA114766 (COG Specimen Banking), and U10 CA180899 (COG Statistics and Data Center). Material was provided by the COG (AALL15B1‐Q) to KMW. We acknowledge the Analytical and Translational Genomics Shared Resources, Shared Flow Cytometry Resources, Animal Resource Facility, and the Animal Models Shared Resources at the UNM Comprehensive Cancer Center. We kindly thank Drs. Donna Kusewitt and Irina Lagutina for technical assistance.
Sharma ND, Nickl CK, Kang H, et al. Epigenetic silencing of SOCS5 potentiates JAK‐STAT signaling and progression of T‐cell acute lymphoblastic leukemia. Cancer Sci. 2019;110:1931‐1946. 10.1111/cas.14021
REFERENCES
- 1. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T‐lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol. 2012;30(14):1663‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Winter SS, Dunsmore KP, Devidas M, et al. Improved survival for children and young adults with T‐lineage acute lymphoblastic leukemia: results from the Children's Oncology Group AALL0434 methotrexate randomization. J Clin Oncol. 2018;36(29):2926‐2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nordlund J, Backlin CL, Wahlberg P, et al. Genome‐wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013;14(9):r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borssen M, Palmqvist L, Karrman K, et al. Promoter DNA methylation pattern identifies prognostic subgroups in childhood T‐cell acute lymphoblastic leukemia. PLoS ONE. 2013;8(6):e65373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kraszewska MD, Dawidowska M, Larmonie NS, et al. DNA methylation pattern is altered in childhood T‐cell acute lymphoblastic leukemia patients as compared with normal thymic subsets: insights into CpG island methylator phenotype in T‐ALL. Leukemia. 2012;26(2):367‐371. [DOI] [PubMed] [Google Scholar]
- 7. Strathdee G, Davies BR, Vass JK, Siddiqui N, Brown R. Cell type‐specific methylation of an intronic CpG island controls expression of the MCJ gene. Carcinogenesis. 2004;25(5):693‐701. [DOI] [PubMed] [Google Scholar]
- 8. Petak I, Danam RP, Tillman DM, et al. Hypermethylation of the gene promoter and enhancer region can regulate Fas expression and sensitivity in colon carcinoma. Cell Death Differ. 2003;10(2):211‐217. [DOI] [PubMed] [Google Scholar]
- 9. Boultwood J, Wainscoat JS. Gene silencing by DNA methylation in haematological malignancies. Br J Haematol. 2007;138(1):3‐11. [DOI] [PubMed] [Google Scholar]
- 10. Furqan M, Mukhi N, Lee B, Liu D. Dysregulation of JAK‐STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomarker research. 2013;1(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maude SL, Dolai S, Delgado‐Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T‐cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Treanor LM, Zhou S, Janke L, et al. Interleukin‐7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J Exp Med. 2014;211(4):701‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roncero AM, Lopez‐Nieva P, Cobos‐Fernandez MA, et al. Contribution of JAK2 mutations to T‐cell lymphoblastic lymphoma development. Leukemia. 2016;30(1):94‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T‐cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanda T, Tyner JW, Gutierrez A, et al. TYK2‐STAT1‐BCL2 pathway dependence in T‐cell acute lymphoblastic leukemia. Cancer Discov. 2013;3(5):564‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain‐of‐function mutations in childhood T‐cell acute lymphoblastic leukemia. Nat Genet. 2011;43(10):932‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flex E, Petrangeli V, Stella L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205(4):751‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bains T, Heinrich MC, Loriaux MM, et al. Newly described activating JAK3 mutations in T‐cell acute lymphoblastic leukemia. Leukemia. 2012;26(9):2144‐2146. [DOI] [PubMed] [Google Scholar]
- 19. Fujimoto M, Naka T. Regulation of cytokine signaling by SOCS family molecules. Trends Immunol. 2003;24(12):659‐666. [DOI] [PubMed] [Google Scholar]
- 20. Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol. 2013;2(1):1‐29. [PMC free article] [PubMed] [Google Scholar]
- 21. Kim MH, Kim MS, Kim W, et al. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PLoS ONE. 2015;10(4):e0123133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Niwa Y, Kanda H, Shikauchi Y, et al. Methylation silencing of SOCS‐3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24(42):6406‐6417. [DOI] [PubMed] [Google Scholar]
- 23. Evans MK, Yu CR, Lohani A, et al. Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene. 2007;26(13):1941‐1948. [DOI] [PubMed] [Google Scholar]
- 24. Calvisi DF, Ladu S, Gorden A, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Investig. 2007;117(9):2713‐2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen CY, Tsay W, Tang JL, et al. SOCS1 methylation in patients with newly diagnosed acute myeloid leukemia. Genes Chromosom Cancer. 2003;37(3):300‐305. [DOI] [PubMed] [Google Scholar]
- 26. Capello D, Deambrogi C, Rossi D, et al. Epigenetic inactivation of suppressors of cytokine signalling in Philadelphia‐negative chronic myeloproliferative disorders. Br J Haematol. 2008;141(4):504‐511. [DOI] [PubMed] [Google Scholar]
- 27. Weber A, Hengge UR, Bardenheuer W, et al. SOCS‐3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene. 2005;24(44):6699‐6708. [DOI] [PubMed] [Google Scholar]
- 28. Seki Y, Hayashi K, Matsumoto A, et al. Expression of the suppressor of cytokine signaling‐5 (SOCS5) negatively regulates IL‐4‐dependent STAT6 activation and Th2 differentiation. Proc Natl Acad Sci USA. 2002;99(20):13003‐13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brender C, Columbus R, Metcalf D, et al. SOCS5 is expressed in primary B and T lymphoid cells but is dispensable for lymphocyte production and function. Mol Cell Biol. 2004;24(13):6094‐6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kario E, Marmor MD, Adamsky K, et al. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem. 2005;280(8):7038‐7048. [DOI] [PubMed] [Google Scholar]
- 31. Nicholson SE, Metcalf D, Sprigg NS, et al. Suppressor of cytokine signaling (SOCS)‐5 is a potential negative regulator of epidermal growth factor signaling. Proc Natl Acad Sci USA. 2005;102(7):2328‐2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Linossi EM, Chandrashekaran IR, Kolesnik TB, et al. Suppressor of Cytokine Signaling (SOCS) 5 utilises distinct domains for regulation of JAK1 and interaction with the adaptor protein Shc‐1. PLoS ONE. 2013;8(8):e70536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matlawska‐Wasowska K, Kang H, Devidas M, et al. MLL rearrangements impact outcome in HOXA‐deregulated T‐lineage acute lymphoblastic leukemia: a Children's Oncology Group Study. Leukemia. 2016;30:1909‐1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perez DR, Nickl CK, Waller A, et al. High‐throughput flow cytometry identifies small‐molecule inhibitors for drug repurposing in T‐ALL. SLAS Discov. 2018;23:732‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matlawska‐Wasowska K, Ward E, Stevens S, et al. Macrophage and NK‐mediated killing of precursor‐B acute lymphoblastic leukemia cells targeted with a‐fucosylated anti‐CD19 humanized antibodies. Leukemia. 2013;27(6):1263‐1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haferlach T, Kohlmann A, Wieczorek L, et al. Clinical utility of microarray‐based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28(15):2529‐2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang H, Chen IM, Wilson CS, et al. Gene expression classifiers for relapse‐free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B‐precursor acute lymphoblastic leukemia. Blood. 2010;115(7):1394‐1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang H, Wilson CS, Harvey RC, et al. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children's Oncology Group study. Blood. 2012;119(8):1872‐1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36:W170‐W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharma N, Magistroni V, Piazza R, et al. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol Cancer. 2015;14:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cavallo F, Forni M, Riccardi C, Soleti A, Di Pierro F, Forni G. Growth and spread of human malignant T lymphoblasts in immunosuppressed nude mice: a model for meningeal leukemia. Blood. 1992;80(5):1279‐1283. [PubMed] [Google Scholar]
- 42. Ferrando AA, Armstrong SA, Neuberg DS, et al. Gene expression signatures in MLL‐rearranged T‐lineage and B‐precursor acute leukemias: dominance of HOX dysregulation. Blood. 2003;102(1):262‐268. [DOI] [PubMed] [Google Scholar]
- 43. Soulier J, Clappier E, Cayuela JM, et al. HOXA genes are included in genetic and biologic networks defining human acute T‐cell leukemia (T‐ALL). Blood. 2005;106(1):274‐286. [DOI] [PubMed] [Google Scholar]
- 44. Peterson JF, Baughn LB, Pearce KE, et al. KMT2A (MLL) rearrangements observed in pediatric/young adult T‐lymphoblastic leukemia/lymphoma: a 10‐year review from a single cytogenetic laboratory. Genes Chromosom Cancer. 2018;57(11):541‐546. [DOI] [PubMed] [Google Scholar]
- 45. Senese S, Zaragoza K, Minardi S, et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27(13):4784‐4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y, Ng HH, Erdjument‐Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13(15):1924‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 1997;89(3):349‐356. [DOI] [PubMed] [Google Scholar]
- 48. Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19(2):187‐191. [DOI] [PubMed] [Google Scholar]
- 49. Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl‐CpG‐binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386‐389. [DOI] [PubMed] [Google Scholar]
- 50. Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhuang G, Wu X, Jiang Z, et al. Tumour‐secreted miR‐9 promotes endothelial cell migration and angiogenesis by activating the JAK‐STAT pathway. EMBO J. 2012;31(17):3513‐3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Homminga I, Pieters R, Langerak AW, et al. Integrated transcript and genome analyses reveal NKX2‐1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19(4):484‐497. [DOI] [PubMed] [Google Scholar]
- 53. Kang H, Sharma ND, Nickl CK, et al. Dysregulated transcriptional networks in KMT2A‐ and MLLT10‐rearranged T‐ALL. Biomarker Res. 2018;6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Toniolo PA, Liu S, Yeh JE, Ye DQ, Barbuto JA, Frank DA. Deregulation of SOCS5 suppresses dendritic cell function in chronic lymphocytic leukemia. Oncotarget. 2016;7(29):46301‐46314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Silva A, Laranjeira AB, Martins LR, et al. IL‐7 contributes to the progression of human T‐cell acute lymphoblastic leukemias. Can Res. 2011;71(14):4780‐4789. [DOI] [PubMed] [Google Scholar]
- 56. Goossens S, Radaelli E, Blanchet O, et al. ZEB2 drives immature T‐cell lymphoblastic leukaemia development via enhanced tumour‐initiating potential and IL‐7 receptor signalling. Nat Commun. 2015;6:5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ribeiro D, Melao A, van Boxtel R, et al. STAT5 is essential for IL‐7‐mediated viability, growth, and proliferation of T‐cell acute lymphoblastic leukemia cells. Blood Adv. 2018;2(17):2199‐2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cardoso BA, Martins LR, Santos CI, et al. Interleukin‐4 stimulates proliferation and growth of T‐cell acute lymphoblastic leukemia cells by activating mTOR signaling. Leukemia. 2009;23(1):206‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kukita T, Arima N, Matsushita K, et al. Autocrine and/or paracrine growth of adult T‐cell leukaemia tumour cells by interleukin 15. Br J Haematol. 2002;119(2):467‐474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials