

Abstract
Although transforming growth factor beta (TGF‐β) is known to be involved in the pathogenesis and progression of many cancers, its role in renal cancer has not been fully investigated. In the present study, we examined the role of TGF‐β in clear cell renal carcinoma (ccRCC) progression in vitro and in vivo. First, expression levels of TGF‐β signaling pathway components were examined. Microarray and immunohistochemical analyses showed that the expression of c‐Ski, a transcriptional corepressor of Smad‐dependent TGF‐β and bone morphogenetic protein (BMP) signaling, was higher in ccRCC tissues than in normal renal tissues. Next, a functional analysis of c‐Ski effects was carried out. Bioluminescence imaging of renal orthotopic tumor models demonstrated that overexpression of c‐Ski in human ccRCC cells promoted in vivo tumor formation. Enhancement of tumor formation was also reproduced by the introduction of a dominant‐negative mutant TGF‐β type II receptor into ccRCC cells. In contrast, introduction of the BMP signaling inhibitor Noggin failed to accelerate tumor formation, suggesting that the tumor‐promoting effect of c‐Ski depends on the inhibition of TGF‐β signaling rather than of BMP signaling. Finally, the molecular mechanism of the tumor‐suppressive role of TGF‐β was assessed. Although TGF‐β signaling did not affect tumor angiogenesis, apoptosis of ccRCC cells was induced by TGF‐β. Taken together, these findings suggest that c‐Ski suppresses TGF‐β signaling in ccRCC cells, which, in turn, attenuates the tumor‐suppressive effect of TGF‐β.
Keywords: apoptosis, c‐Ski, orthotopic tumor model, renal cell cancer, TGF‐β
1. INTRODUCTION
Renal cell carcinoma (RCC) affects nearly 300 000 people and is the seventh most common form of cancer worldwide and is responsible for over 100 000 deaths annually.1 Widespread use of ultrasonography has increased the number of patients diagnosed with RCC.2 RCC is classified into five histological subtypes: clear cell RCC (ccRCC), papillary RCC, chromophobe RCC, multiocular RCC, and collecting duct carcinoma.3 Among them, ccRCC is the most common histological type of RCC, comprising over 75% of all RCC cases. When RCC is detected early, surgical excision of primary tumors is an effective treatment strategy that results in a 5‐year survival rate of >65%, and even >80% in cases without metastasis. However, there are currently no effective treatments for patients with advanced RCC. Although the combined modality therapy is applied for cases with advanced stage cancer, the efficiency of conventional chemotherapy remains low.
Mutations or deletions in the tumor suppressor von Hippel‐Lindau gene (VHL) have been frequently observed in RCC. The VHL protein (pVHL)/E3 ubiquitin ligase complex targets hypoxia‐inducible factor‐1α (HIF‐1α) for ubiquitin‐proteasome‐mediated degradation.4, 5 Under normoxic conditions, HIF‐1α is hydroxylated on critical proline residues by HIF prolyl hydroxylase (PHD), which requires oxygen, 2‐oxoglutarate, and iron as cosubstrates. pVHL can then recognize and bind hydroxylated HIF‐1α, enabling its ubiquitylation by the pVHL/E3 ubiquitin ligase complex and subsequent degradation by the proteasome. However, under hypoxic conditions, pVHL cannot recognize HIF‐1α because of the dysfunction of PHD, which causes HIF‐1α accumulation. Increased level of HIF‐1α upregulates the expression of its target genes that support tumor growth and vascularization, including vascular endothelial growth factor (VEGF), platelet‐derived growth factor (PDGF), and glucose transporter type 1 (GLUT1). Based on these observations, molecular targeted therapies against VEGF have been recently developed. Although such therapies are used for the treatment of RCC, their effect is limited. Thus, identification of new molecular targets remains important.
Transforming growth factor‐β (TGF‐β) is the prototypic member of the TGF‐β family.6 At the cell surface, TGF‐β binds to two kinase receptors, type II receptor (TβRII) and type I receptor (TβRI, also known as ALK‐5), whereupon they form a heteromeric complex and transduce intracellular signals by phosphorylating receptor‐regulated Smads (R‐Smads), Smad2 and Smad3. These phosphorylated R‐Smads form heteromeric Smad complexes with the common‐partner Smad (co‐Smad), Smad4. Smad complexes associate with various transcription factors and transcriptional coregulators in the nucleus, thereby regulating transcription of the target genes. TGF‐β also activates non‐Smad signaling pathways, including MAPK signaling pathways. TGF‐β regulates a wide variety of biological events, including cell proliferation, apoptosis, cell differentiation, epithelial‐mesenchymal transition (EMT), stem cell maintenance, tissue fibrosis, angiogenesis, and immune responses. During carcinogenesis, cancer progression, and metastasis, TGF‐β has opposite, tumor‐suppressive and tumor‐promotive roles. Because TGF‐β inhibits proliferation and cell survival of epithelial cells, mutations and deletions of TGF‐β signaling pathway components lead to abnormal cell growth during the early stages of cancer.6 In contrast, at the advanced stages of cancer, cancer cells often overcome the inhibitory action of TGF‐β on cell growth and cell survival. EMT induced by TGF‐β enhances mesenchymal characteristics of cancer cells, which stimulates their invasiveness and metastasis. TGF‐β also functions as a potent immunosuppressive cytokine and inhibits antitumor immunity.
Although TGF‐β is thought to act as both tumor‐suppressive and tumor‐promotive factor in a cell type‐dependent method, little is known about the role of TGF‐β in RCC. Thus, in the present study, the effect of TGF‐β signaling on the progression of RCC was investigated by using histopathological examination and in vivo analyses.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
Human ccRCC cells harboring mutant VHL, OS‐RC‐2 (RIKEN Cell Bank, Ibaraki, Japan) and human ccRCC cells harboring wild‐type VHL, Caki‐1 (Cell Resource Center for Biomedical Research, Tohoku University, Miyagi, Japan) were cultured in RPMI‐1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) and minimum essential medium (MEM; Thermo Fisher Scientific) containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, respectively. For the stimulation of TGF‐β or bone morphogenetic protein (BMP), TGF‐β1, TGF‐β3, or BMP‐4 ligands (R&D Systems, Minneapolis, MN, USA) were reconstituted in 4 mmol/L HCl containing 0.1% BSA (Sigma‐Aldrich, St Louis, MO, USA) and used at a concentration of 1 ng/mL (TGF‐β1 and TGF‐β3) or 30 ng/mL (BMP‐4).
2.2. Lentiviral production and infection
We used a lentiviral vector system to overexpress specific genes as previously described.7 For the overexpression of c‐Ski or Noggin, complementary DNAs encoding human SKI or human NOG were inserted into pENTR201 with a multi‐cloning site (pENTR201‐MCS) empty vector and then transferred to a pCSII‐EF‐RfA destination vector by Gateway cloning technology (Thermo Fisher Scientific).8 Introduction of luciferase and HA‐tagged dominant‐negative TGF‐β type II receptor mutant (dnTβRII) was carried out as described previously.9 pCSII‐EF‐GFP was used as positive control for lentiviral infection.
2.3. Immunohistochemistry and TUNEL staining
Immunohistochemistry was carried out as previously described.7 For the immunostaining of human ccRCC tissues, formalin‐fixed, paraffin‐embedded human clinical samples were collected from patients at The University of Tokyo Hospital after informed consent had been obtained. The protocol was approved by the Research Ethics Committee of the Graduate School of Medicine at The University of Tokyo. Sections were subjected to H&E staining or immunostaining using a rabbit anti‐c‐Ski antibody (#19864; Abcam, Cambridge, UK). Stained sections were visualized using a Vectastain Elite ABC kit (PK‐6101; Vector Laboratories, Burlingame, CA, USA). Expression profiles were analyzed by determining the ratio of cells stained by the anti‐c‐Ski antibody in each sample as follows: 80% < ++ ≤ 100%; 50% < + ≤ 80%; 0% < ± ≤ 50%; − = 0%. For the immunostaining of mouse tumor tissues, excised mouse tissue samples were frozen in dry‐iced acetone. The frozen sections were fixed with 4% paraformaldehyde and permeabilized in 0.2% Triton X‐100. Sections were subjected to H&E staining or immunostaining using a rat antimouse CD31 antibody (#550274; BD Biosciences, Franklin Lakes, NJ, USA) and an Alexa Fluor 488‐conjugated antirat IgG antibody (#A‐11006; Life Technologies, Carlsbad, CA, USA). TUNEL staining was carried out using the In situ Cell Death Detection Kit (TMR red; Roche Diagnostics, Basel, Switzerland) and DAPI Fluoromount‐G (Southern Biotech, Birmingham, AL, USA), as previously described.9 Fluorescent images were captured with a BZ‐9000 Fluorescence Microscope (Keyence, Osaka, Japan). CD31‐positive pixels were analyzed with Image J (NIH, Bethesda, MD, USA).
2.4. Immunoblotting
Immunoblotting was carried out as previously described.10 Antibodies against c‐Ski (#A303‐518A; Bethyl Laboratories, Montgomery, TX, USA), Noggin (4C9; Sigma‐Aldrich), and HA (3F10; Sigma‐Aldrich) were used as primary antibodies. Other primary antibodies and secondary antibodies were prepared as previously described.7, 8, 10
2.5. Quantitative real‐time reverse transcription‐PCR analysis
Total RNA was extracted as previously described.7 Complementary DNA was prepared from each cell and subjected to qRT‐PCR analysis as previously described.7 Primer sequences are described in Table S1.
2.6. Mouse renal orthotopic tumor models and bioluminescence imaging
Tumor‐forming ability of ccRCC cells in mice was analyzed using mouse renal orthotopic tumor models and bioluminescence imaging as previously described.11 All protocols were approved by the Animal Ethics Committee of the Graduate School of Medicine at The University of Tokyo. BALB/c‐nu/nu male mice (5‐weeks‐old) were purchased from Sankyo Labo Service Corporation (Tokyo, Japan). Firefly luciferase was introduced into ccRCC cells by infection of lentiviral vectors for bioluminescence imaging.12 The ccRCC cells were resuspended in HBSS (Thermo Fisher Scientific) and then orthotopically injected into mouse kidney (3 × 104 Caki‐1 cells or 3 × 104 OS‐RC‐2 cells in 50 μL per mouse, unless otherwise specified).
2.7. Colony formation assay
Colony formation assay in soft agar was carried out as previously described.12 Colony formation assay in detached culture was done using poly‐2‐hydroxyethyl methacrylate (HEMA; P3932; Sigma‐Aldrich). Cells (1 × 105) were cultured in six‐well plates precoated with poly‐HEMA for 2 days.
2.8. Statistical analysis
Statistical significance of the differences between experimental groups was estimated by using the F‐test followed by the Student's or Welch's t test. All statistical analyses were conducted with a significance level of α = 0.05 (P < 0.05). Scatter diagrams were plotted using GraphPad Prism 6 (GraphPad Software).
3. RESULTS
3.1. Elevated expression of c‐Ski in ccRCC tissues
In order to examine whether TGF‐β signaling pathway components are dysregulated in ccRCC, their expression levels in clinical tissues were analyzed using the Gene Expression Omnibus (GEO) database. There were no significant differences in the expression levels of mRNAs encoding TGF‐β receptors and Smads, including TβRII (TGFBR2), TβRI (TGFBR1), endoglin (ENG), SMAD2, SMAD3, and SMAD4, between ccRCC and corresponding normal renal tissues (Figure 1). However, decreased expression of type III receptor (TGFBR3, also known as betaglycan) in ccRCC tissues was observed (Figure 1), which was in agreement with our previous report.11 In addition, we also found that the expression of mRNA encoding c‐Ski (SKI), a transcriptional corepressor of Smad‐dependent TGF‐β signaling, was significantly increased in ccRCC tissues. Expression of another SKI family member, SKI‐like (SKIL), also known as SnoN, was not increased in ccRCC (Figure 1). Immunohistochemical analysis using human ccRCC tissues (n = 31) showed that c‐Ski was present in the nuclei of cancer cells in many ccRCC cases (Table 1; Figure 1). When compared with its expression in corresponding normal tissues in each case, c‐Ski protein was absent in the nuclei of normal proximal tubule cells (cases with increased c‐Ski expression in ccRCC tissues as compared to that in corresponding normal renal tissues are highlighted in bold in Table 1), suggesting that cancer cells begin expressing c‐Ski during ccRCC progression. In the subsequent experiments, we focused on the role of c‐Ski on tumor formation and TGF‐β signaling in ccRCC cells.
Figure 1.
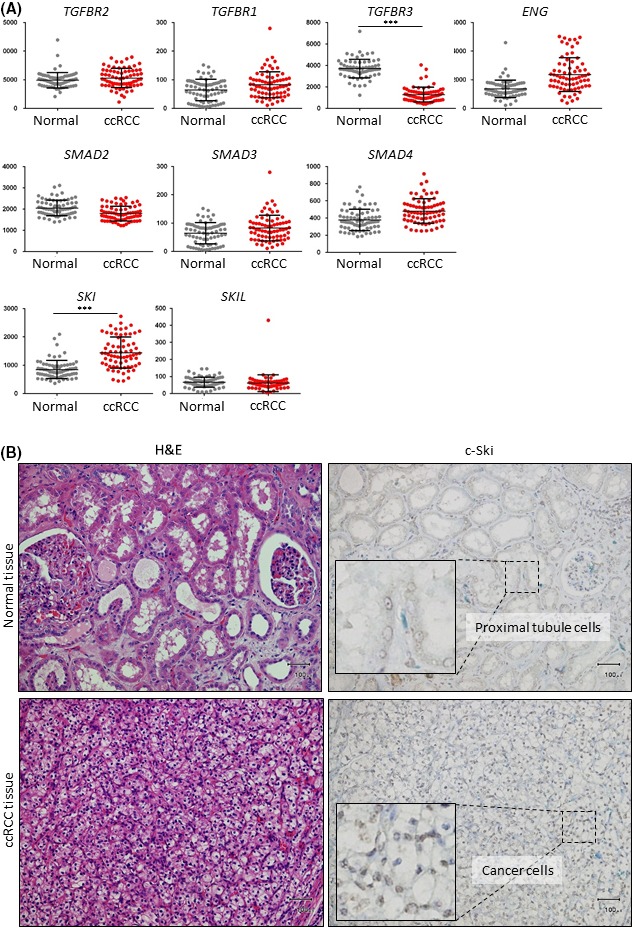
Upregulation of c‐Ski expression in clear cell renal carcinoma (ccRCC) tissues. A, Comprehensive analysis of gene expression data from the NCBI Gene Expression Omnibus (GEO) database (GSE53757) shows expression levels of transforming growth factor beta (TGF‐β) signaling pathway components in ccRCC tissues (n = 72) and matched normal renal tissues (n = 72). Expression values are shown by dot plots. Data represent the mean ± SD. ***P < .001. B, Images of immunohistochemical staining with an anti‐c‐Ski antibody and H&E staining of ccRCC tissues and corresponding normal renal tissues from the same patient. c‐Ski staining in boxed region is shown at high magnification. Scale bars are 100 μm. ENG, endoglin
Table 1.
Expression of c‐Ski in normal renal and clear cell renal carcinoma tissues
| Histological type | Patient | Age | Gender | Grade | Stage | c‐Ski expression | |
|---|---|---|---|---|---|---|---|
| Normal | Cancer | ||||||
| Clear cell renal cell carcinoma | 1 | 59 | F | G1 ≫ G2 | pT2a | − | ++ |
| 2 | 50 | M | G2 > G1 | pT2b | − | + | |
| 3 | 72 | M | G3 | pT3a | − | − | |
| 4 | 64 | M | G3 > G2 | pT3b | − | ++ | |
| 5 | 52 | M | G1 > G2 | pT3a | − | ++ | |
| 6 | 78 | F | G3 > G2 > G1 | pT1b | − | ++ | |
| 7 | 83 | M | G1 > G2 | pT1b | − | ++ | |
| 8 | 57 | M | G2 > G1 | pT1b | − | ++ | |
| 9 | 60 | F | G2 > G3 | pT2a | − | ++ | |
| 10 | 70 | M | G3 > G2 | pT3a | − | −/+ | |
| 11 | 72 | M | G1 > G2 | pT1a | −/+ | ++ | |
| 12 | 53 | M | G3 > G2 | pT3a | − | −/+ | |
| 13 | 58 | M | G2 > G3 | pT1b | − | + | |
| 14 | 78 | M | G2 > G3 > G1 | pT3a | − | + | |
| 15 | 75 | M | G2 > G1 | pT1a | −/+ | ++ | |
| 16 | 43 | F | G2 > G1 | pT1a | − | ++ | |
| 17 | 55 | F | G1 > G2 | pT1a | − | ++ | |
| 18 | 68 | M | G1 > G2 > G3 | pT3a | − | ++ | |
| 19 | 75 | M | G2 > G1 | pT1a | −/+ | −/+ | |
| 20 | 89 | M | G2 > G1 | pT1b | − | + | |
| 21 | 74 | M | G2 > G1 | pT1a | − | + | |
| 22 | 74 | M | G3 > G2 | pT3a | − | + | |
| 23 | 45 | M | G1 > G2 | pT1a | − | ++ | |
| 24 | 64 | M | G2 > G1 | pT1b | − | ++ | |
| 25 | 72 | M | G2 > G3 | pT1b | − | − | |
| 26 | 39 | M | G2 > G3 | pT3a | − | ++ | |
| 27 | 87 | M | G2 > G3 | pT3a | − | −/+ | |
| 28 | 76 | F | G1 > G2 | pT1a | − | ++ | |
| 29 | 43 | M | G1 ≫ G2 | pT1a | − | −/+ | |
| 30 | 66 | M | G3 > G2 | pT2a | − | −/+ | |
| 31 | 74 | M | G1 > G2 | pT3a | − | + | |
3.2. Tumor‐promoting role of c‐Ski during ccRCC progression
To clarify the effect of c‐Ski on ccRCC, we overexpressed c‐Ski in human OS‐RC‐2 ccRCC cells (OS‐RC‐2‐c‐Ski cells) by using lentiviral vectors. Increased expression of SKI mRNA and c‐Ski protein was confirmed by qRT‐PCR and immunoblotting (Figure 2A,B). In OS‐RC‐2‐c‐Ski cells, expression of the TGF‐β target gene, serine peptidase inhibitor, clade E, member 1 (SERPINE1, also known as PAI‐1), was attenuated compared with that in control, GFP‐expressing, OS‐RC‐2‐GFP cells (Figure 2C). This indicated that c‐Ski successfully suppressed Smad‐dependent transcription in OS‐RC‐2 cells. Then, the tumorigenic potentials of OS‐RC‐2‐GFP and OS‐RC‐2‐c‐Ski cells were assessed using a mouse renal orthotopic tumor model. Tumor formation was visualized by bioluminescence imaging after firefly luciferase introduction into cancer cells. Bioluminescence in tumor‐bearing mice and extracted kidneys showed that OS‐RC‐2‐c‐Ski cells formed significantly larger renal tumors than OS‐RC‐2‐GFP cells (Figure 2D,E). We then attempted to reproduce the phenotypes of OS‐RC‐2 cells by injecting another human ccRCC cell line, Caki‐1. Tumors were observed following injections of Caki‐1 cells overexpressing c‐Ski (Caki‐1‐c‐Ski) or GFP (Caki‐1‐GFP) in the mouse renal orthotopic tumor model (Figure 3A‐C). Bioluminescence imaging analyses showed that injection of Caki‐1‐c‐Ski cells also led to the formation of larger tumors than injection of Caki‐1‐GFP cells (Figure 3D,E). These results suggested that c‐Ski enhanced primary tumor formation following injections of ccRCC cells.
Figure 2.
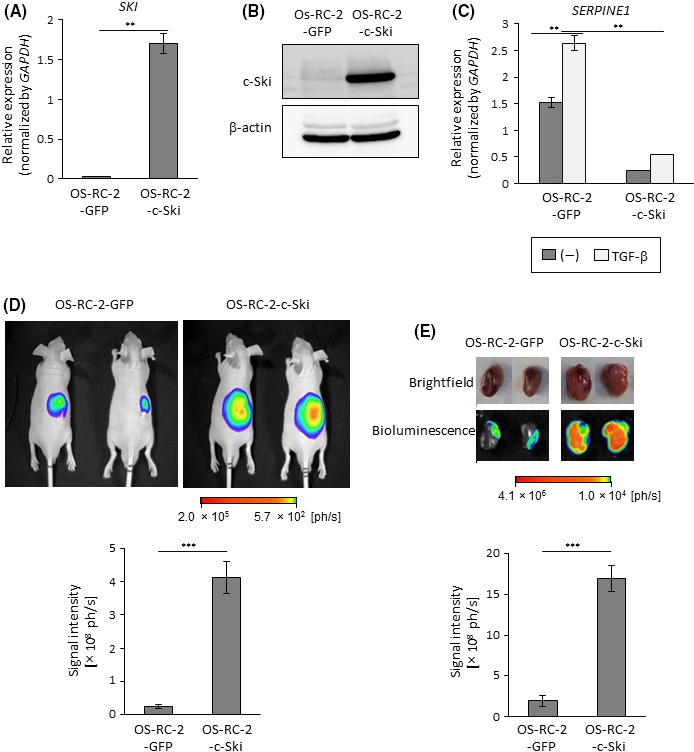
Enhanced tumor formation by c‐Ski in OS‐RC‐2 cells. A, OS‐RC‐2 cells were infected with lentiviral vectors encoding GFP (OS‐RC‐2‐GFP) or c‐Ski (OS‐RC‐2‐c‐Ski) and analyzed by qRT‐PCR for SKI expression. Data represent the mean ± SD. **P < .01. B, Immunoblots of lysates of OS‐RC‐2‐GFP and OS‐RC‐2‐c‐Ski cells with the indicated antibodies. C, qRT‐PCR analysis of SERPINE1 expression. OS‐RC‐2‐GFP and OS‐RC‐2‐c‐Ski cells were stimulated with transforming growth factor beta (TGF‐β) for 2 h and analyzed by qRT‐PCR. Data represent the mean ± SD. **P < .01. D, Tumor‐forming ability of OS‐RC‐2 cells. BALB/c nu/nu male mice received renal orthotopic injection of OS‐RC‐2‐GFP (n = 6) or OS‐RC‐2‐c‐Ski (n = 5) cells (1 × 105 cells per mouse). Upper panels: representative photographs of in vivo bioluminescence imaging of tumor‐bearing mice 13 d after the injection. Lower panel: overall luminescence signal intensity; data represent the mean ± SE. ***P < .001. E, Tumor‐forming ability of OS‐RC‐2 cells. Tumor formation in mice in (D) was examined 3 wks after the injection. Upper panels: representative photographs of brightfield and ex vivo bioluminescence imaging of extracted kidneys. Lower panel: overall luminescence signal intensity; data represent the mean ± SE. ***P < .001
Figure 3.
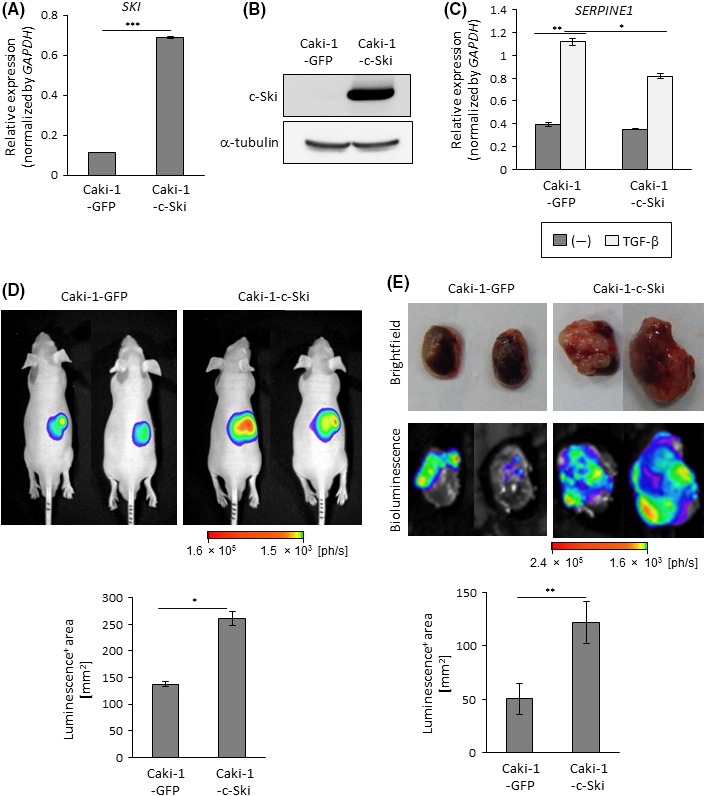
Enhanced tumor formation by c‐Ski in Caci‐1 cells. A, Caki‐1 cells were infected with lentiviral vectors encoding GFP (Caki‐1‐GFP) or c‐Ski (Caki‐1‐c‐Ski) and analyzed by qRT‐PCR for SKI expression. Data represent the mean ± SD. ***P < .001. B, Immunoblots of lysates of Caki‐1‐GFP and Caki‐1‐c‐Ski cells with the indicated antibodies. C, qRT‐PCR analysis of SERPINE1 expression. Caki‐1‐GFP and Caki‐1‐c‐Ski cells were stimulated with transforming growth factor beta (TGF‐β) for 2 h and analyzed by qRT‐PCR. Data represent the mean ± SD. *P < .05, **P < .01. D, Tumor‐forming ability of Caki‐1 cells. BALB/c nu/nu male mice received renal orthotopic injection of Caki‐1‐GFP (n = 6) or Caki‐1‐c‐Ski (n = 5) cells. Upper panels: representative photographs of in vivo bioluminescence imaging of tumor‐bearing mice 3 wks after the injection. Lower panel: luminescence‐positive area in all mice; data represent the mean ± SE. *P < .05. E, Tumor‐forming ability of Caki‐1 cells. Tumor formation in mice in (D) was examined 5 wks after the injection. Upper panels: representative photographs of brightfield and ex vivo bioluminescence imaging of extracted kidneys. Lower panel: luminescence‐positive area; data represent the mean ± SE. **P < .01
3.3. Tumor‐suppressive effect of TGF‐β on ccRCC progression
We hypothesized that c‐Ski may promote tumorigenic activity of RCC cells by inhibiting Smad‐dependent TGF‐β signaling. To test this hypothesis directly, HA‐tagged dnTβRII, the mutant receptor that lacks the intracellular domain, was introduced to OS‐RC‐2 cells. Expression of dnTβRII and attenuation of TGF‐β‐induced phosphorylation of Smad2 were confirmed by immunoblotting (Figure 4A). qRT‐PCR also showed that the induction of SERPINE1 by TGF‐β was impaired by the introduction of dnTβRII (Figure 4B). Then, we determined whether the inhibition of TGF‐β signaling by dnTβRII reproduced the effect of c‐Ski on tumor formation using orthotopic inoculation. In vivo and ex vivo bioluminescence imaging experiments showed that primary tumor formation caused by OS‐RC‐2 cells was accelerated by the introduction of dnTβRII (Figure 4C,D). Moreover, ex vivo bioluminescence imaging showed that OS‐RC‐2‐dnTβRII cells caused metastasis in the lung, which was rarely observed in OS‐RC‐2‐GFP‐xenografted mice (Figure 4E). Similar results were obtained following injection of Caki‐1 cells. TGF‐β signal transduction was inhibited by the introduction of dnTβRII in Caki‐1 cells (Figure 5A,B). Primary tumor formation by Caki‐1 cells was enhanced by dnTβRII (Figure 5C,D). These results suggest that TGF‐β has a tumor‐suppressive role during the formation of primary tumors and lung metastasis in ccRCC. Considering that c‐Ski enhanced primary tumor formation by ccRCC cells (Figures 2, 3), inhibition of TGF‐β signaling by c‐Ski may contribute to tumor formation in ccRCC.
Figure 4.
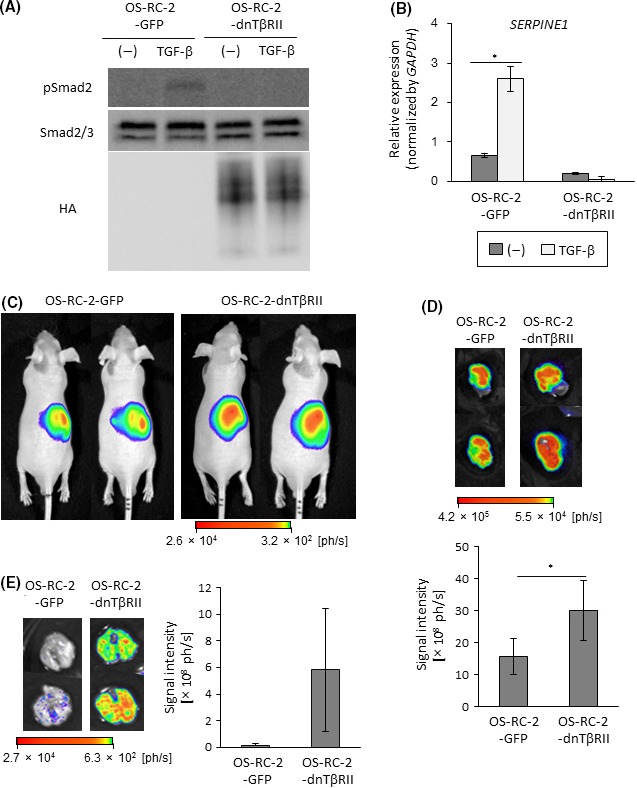
Enhanced tumor formation by dnTβRII in OS‐RC‐2 cells. A, Immunoblots of cell lysates with the indicated antibodies. OS‐RC‐2 cells were infected with lentiviral vectors encoding GFP (OS‐RC‐2‐GFP) or HA‐tagged dnTβRII (OS‐RC‐2‐dnTβRII). Cells were stimulated with transforming growth factor beta (TGF‐β) for 2 h. B, qRT‐PCR analysis of SERPINE1 expression. Cells were stimulated with TGF‐β for 2 h and analyzed by qRT‐PCR. Data represent the mean ± SD. *P < .05. C, Tumor‐forming ability of OS‐RC‐2 cells. BALB/c nu/nu male mice received renal orthotopic injection of OS‐RC‐2‐GFP (n = 8) or OS‐RC‐2‐dnTβRII (n = 8) cells. Representative photographs of in vivo bioluminescence imaging of tumor‐bearing mice 3 wks after the injection. D, Tumor‐forming ability of OS‐RC‐2 cells. Tumor formation in mice in (C) was examined 3 wks after the injection. Upper panels: representative photographs of ex vivo bioluminescence imaging of extracted kidneys. Lower panel: overall luminescence signal intensity; data represent the mean ± SE. *P < .05. E, Metastatic ability of OS‐RC‐2 cells. Metastatic lung tumor in mice shown in (C) was examined 3 wks after the injection. Left panels: representative photographs of ex vivo bioluminescence imaging of extracted lungs. Right panel: overall luminescence signal intensity; data represent the mean ± SE
Figure 5.
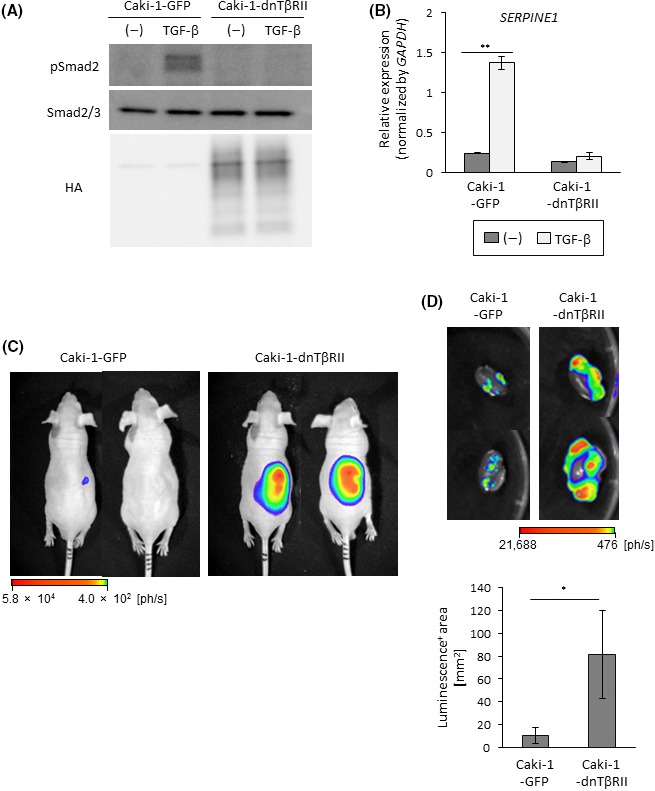
Enhanced tumor formation by dnTβRII in Caci‐1 cells. A, Immunoblots of cell lysates with the indicated antibodies. Caki‐1 cells were infected with lentiviral vectors encoding GFP (Caki‐1‐GFP) or HA‐tagged dnTβRII (Caki‐1‐dnTβRII). Cells were stimulated with transforming growth factor beta (TGF‐β) for 2 h. B, qRT‐PCR analysis of SERPINE1 expression. Cells were stimulated with TGF‐β for 2 h and analyzed by qRT‐PCR. Data represent the mean ± SD. **P < .01. C, Tumor‐forming ability of Caki‐1 cells. BALB/c nu/nu male mice received renal orthotopic injection of Caki‐1‐GFP (n = 5) or Caki‐1‐dnTβRII (n = 4) cells. Representative photographs of in vivo bioluminescence imaging of tumor‐bearing mice 7 wks after the injection. D, Tumor‐forming ability of Caki‐1 cells. Tumor formation in mice shown in (C) was examined 8 wks after the injection. Upper panels: representative photographs of ex vivo bioluminescence imaging of extracted kidney. Lower panel: luminescence‐positive area; data represent the mean ± SE. *P < .05
3.4. Role of bone morphogenetic protein signaling in ccRCC progression
Given that Smad‐dependent BMP and TGF‐β signaling pathways are both inhibited by c‐Ski, it is possible that c‐Ski regulates tumor progression by attenuating BMP signaling. In order to determine whether inhibition of BMP signaling occurs during renal cancer progression, we established renal cancer cells that were not responsive to BMP (Figure S1A,B). Noggin, an extracellular glycoprotein, associates with BMP and acts as a BMP signaling antagonist. qRT‐PCR analysis demonstrated that BMP‐4 induced the expression of inhibitor of DNA binding 1 (ID1) in OS‐RC‐2‐GFP cells, but not in OS‐RC‐2‐Noggin cells (Figure S1B,C), suggesting that Noggin successfully diminished BMP signaling in OS‐RC‐2 cells. In vivo and ex vivo bioluminescence imaging showed that tumor formation caused by OS‐RC‐2 cells was not enhanced by Noggin overexpression (Figure S1D,E), whereas progression of such tumors was accelerated by dnTβRII (Figure 4). Thus, we concluded that the attenuation of BMP signaling was less important for ccRCC than that of TGF‐β signaling, and that c‐Ski enhanced tumor formation caused by ccRCC cells primarily through the inhibition of TGF‐β signaling.
3.5. Lack of the effect of TGF‐β on angiogenesis in ccRCC
Because ccRCC is associated with HIF‐1α hyperactivity, angiogenesis is an important factor for ccRCC development and progression.13 Therefore, we examined whether the tumor‐suppressive effect of TGF‐β was dependent on the attenuation of angiogenesis. Expression of the endothelial marker CD31 (also known as platelet/endothelial cell adhesion molecule 1 or PECAM1) in primary tumor tissues shown in Figure 4D was examined by immunostaining. However, CD31‐positive areas were not significantly different between OS‐RC‐2‐GFP‐ and OS‐RC‐2‐dnTβRII‐derived tumor tissues (Figure S2A). Although ccRCC is usually characterized by increased production of angiogenic factors, qRT‐PCR showed that the upregulation of VEGF‐A (VEGFA) by TGF‐β did not correlate with tumor‐forming ability of OS‐RC‐2 cells (Figure S2B). Expression levels of thrombospondin 1 (THBS1, also known as TSP‐1), an endogenous angiogenic inhibitor, were not altered by TGF‐β in ccRCC cells (Figure S2B). These results suggest that the tumor‐suppressive role of TGF‐β was unlikely mediated by the regulation of angiogenesis in ccRCC cells.
3.6. Effect of TGF‐β on the proliferation of ccRCC cells
Finally, direct effects of TGF‐β on the survival of ccRCC cells were examined. TUNEL staining of tumor tissues showed that the number of apoptotic cells was less frequent in primary tumors derived from OS‐RC‐2‐dnTβRII cells (Figure 6A). Although TGF‐β inhibited colony formation of OS‐RC‐2‐GFP cells in soft agar and detached culture conditions, these effects were cancelled in OS‐RC‐2‐dnTβRII cells (Figure 6B,C). Taken together, these results suggest that TGF‐β inhibited tumor formation of ccRCC cells by inducing apoptosis, which was attenuated by the introduction of dnTβRII.
Figure 6.
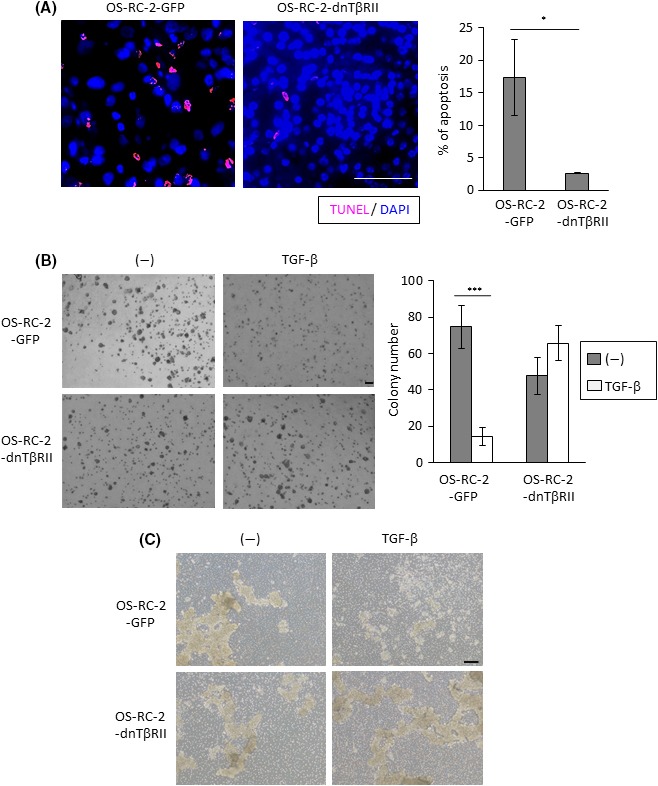
Induction of apoptosis of clear cell renal carcinoma (ccRCC) cells by transforming growth factor beta (TGF‐β). A, Apoptosis in primary tumor tissues dissected from mice shown in Figure 4D, which were killed 3 wks after the transplantation. Left panels: tumor tissues were subjected to TUNEL staining. Representative images were captured for tissue from each mouse. Right panel: apoptosis was quantified by counting the number of TUNEL‐positive cells in three independent fields; data represent the mean ± SD. *P < .05. Scale bar is 50 μm. B, Colony formation of indicated cells in soft agar. Cells were stimulated with or without TGF‐β for 14 d. Left panels: Representative photographs. Right panel: colony number was counted in three independent fields; data represent the mean ± SD. ***P < .001. Scale bar is 200 μm. C, Colony formation of indicated cells in detached culture. Cells were stimulated with or without TGF‐β for 2 d. Representative photographs are shown. Scale bar is 200 μm
4. DISCUSSION
In the present study, the role of TGF‐β signaling on RCC progression was investigated. Histopathological examinations showed that c‐Ski expression was universally elevated in ccRCC tissues, suggesting that TGF‐β signal transduction may be suppressed in ccRCC cells. In vivo experiments showed that c‐Ski overexpression promoted tumor formation, which was also replicated by the introduction of dnTβRII. Thus, we concluded that c‐Ski suppressed TGF‐β signaling in ccRCC cells, which, in turn, attenuated the tumor‐suppressive role of TGF‐β.
Many types of abnormalities in TGF‐β signaling components have been identified in cancers.14 In particular, alterations of Smad4 and TβRII, frequently observed in cancers, strongly attenuate TGF‐β signaling. However, these observations have been mainly restricted to certain types of cancer, such as pancreatic cancer and colorectal cancer. As there have been few similar reports on renal cancers, we examined whether abnormalities in TGF‐β signaling components were observed in tissues affected by ccRCC. Although expression levels of Smad4 and TβRII were not decreased in ccRCC tissues, we herein show that TGFBR3/betaglycan expression was significantly decreased, and c‐Ski expression was significantly increased in ccRCC (Figure 1; Table 1). To the best of our knowledge, this is the first report about increased expression of c‐Ski in ccRCC. We have already reported that the accessory protein TGFBR3/betaglycan promotes binding of TGF‐β to TβRII in ccRCC cells, which enhances TβRI‐dependent signal transduction.11 The decrease in TGFBR3 expression diminishes TGF‐β signaling and negatively regulates the maintenance of aldehyde dehydrogenase (ALDH)‐positive cancer‐initiating cells within ccRCC cells. In addition, increased lamellipodium formation stimulated by focal adhesion kinase (FAK)‐PI3K signaling was observed after TGFBR3 downregulation, which contributed to TGF‐β‐independent migration of ccRCC cells.
The proto‐oncogene c‐Ski was originally identified as a cellular homolog of Sloan‐Kettering retrovirus (v‐Ski).15, 16 Elevated expression levels of c‐Ski correlated with clinical outcome in many types of human tumor tissues, including colorectal cancer, gastric cancer, esophageal cancer, pancreatic cancer, cutaneous melanoma, hemangioma, and acute myeloid leukemia.14, 17, 18, 19, 20 Amplification of SKI and SKIL genes has been demonstrated in several types of cancer cells.20, 21 Given that SKI mRNA was commonly increased in ccRCC tissues in our experiments (Figure 1), gene amplification may account for such an increase in ccRCC cells. c‐Ski protein expression level is also determined by proteasomal degradation regulated by ring finger 111 (RNF111, also known as Arkadia) and cell division cycle 34 (Cdc34)22, 23 proteins. Whereas c‐Ski protein regulates gene transcription in the nucleus of cancer cells, c‐Ski protein in the cytoplasm has also been reported to regulate the proliferation of cancer cells.17, 24, 25 However, according to our immunohistochemical analyses, c‐Ski protein expression was only prominent in the nucleus of ccRCC cells (Figure 1).
Upon TGF‐β stimulation, the Smad complex translocates into the nucleus and regulates transcription of the target genes in collaboration with other transcription factors and coregulators (coactivators and corepressors). Coactivators, such as p300, CREB‐binding protein (CBP), and p300/CBP‐associated factor (P/CAF), promote gene transcription. Corepressors, including the proteins of the Ski family and the Evi‐1 family, inhibit gene transcription. Ski family members c‐Ski and SnoN, which share an overall 50% amino acid similarity with similar domain structures, bind to the C‐terminal MH2 domain in Smad2, Smad3, and Smad4. Binding of c‐Ski prevents Smads from binding to the activators and recruits nuclear receptor corepressor (N‐CoR) and histone deacetylase (HDAC). c‐Ski also disrupts the formation of the Smad complex.26 Eventually, Ski family members act as negative regulators of Smad‐dependent TGF‐β signaling.27, 28, 29 Consistent with their roles as proto‐oncogenes, c‐Ski and SnoN suppress the antiproliferative effect of TGF‐β and promote cancer progression.17, 25, 28 Opposite functions of SnoN (eg, the induction of cellular senescence) have also been reported.30 In the present study, we showed that overexpression of c‐Ski significantly impaired the induction of TGF‐β target genes in ccRCC cells (Figures 2C and 3C). In agreement with these findings, c‐Ski enhanced in vivo tumor formation caused by ccRCC cells (Figures 2D,E and 3D,E), suggesting that c‐Ski antagonized the inhibitory effect of TGF‐β on survival. Thus, c‐Ski showed a tumor‐promotive effect in ccRCC.
Our previous research showed that overexpression of dnTβRII and c‐Ski in human diffuse‐type gastric carcinoma cells inhibited TGF‐β signal transduction, which enhanced tumorigenesis in vivo through the potentiation of angiogenesis.31, 32 Based on these observations, we examined whether a similar molecular mechanism was involved in ccRCC. Although inhibition of TGF‐β signaling in ccRCC cells by dnTβRII accelerated their tumor‐forming ability, histological findings and the number of CD31‐positive cells were not affected (Figure S2A). Likewise, TGF‐β did not significantly influence expression levels of tumor angiogenesis regulators in ccRCC cells, such as THBS1 and VEGFA (Figure S2B), although VEGFA was somewhat increased by TGF‐β in Caki‐1, but not in OS‐RC‐2 cells. Thus, in contrast to the observations in diffuse‐type gastric carcinoma, inhibition of TGF‐β signaling by c‐Ski in ccRCC was important for the survival of cancer cell themselves, rather than for optimization of the tumor microenvironment. Future studies of TGF‐β downstream targets in ccRCC cells may enable identification of novel targets for the treatment of RCC.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGEMENTS
We thank Jun Nishida (The University of Tokyo) for technical advice and assistance and Hiroyuki Miyoshi (Keio University) for providing lentiviral vectors. This work was supported by a KAKENHI Grant‐in‐Aid for Scientific Research on Innovative Areas, Integrative Research on Cancer Microenvironment Network (22112002) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (K. Miyazono); KAKENHI Grants‐in‐Aid for Scientific Research (C) (15K08393; S. Ehata) and (S) (15H05774; K. Miyazono) from the Japan Society for the Promotion of Science; the Yasuda Medical Foundation (K. Miyazono); and the Princess Takamatsu Cancer Research Fund (S. Ehata). This work was also supported in part by a grant for Endowed Department (Department of Medical Genomics) from Eisai Co., Ltd.
Taguchi L, Miyakuni K, Morishita Y, et al. c‐Ski accelerates renal cancer progression by attenuating transforming growth factor β signaling. Cancer Sci. 2019;110:2063‐2074. 10.1111/cas.14018
Luna Taguchi and Kosuke Miyakuni contributed equally to this work.
REFERENCES
- 1. Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373:1119‐1132. [DOI] [PubMed] [Google Scholar]
- 2. Lightfoot N, Conlon M, Kreiger N, et al. Impact of noninvasive imaging on increased incidental detection of renal cell carcinoma. Eur Urol. 2000;37:521‐527. [DOI] [PubMed] [Google Scholar]
- 3. Kovacs G, Akhtar M, Beckwith BJ, et al. The Heidelberg classification of renal cell tumours. J Pathol. 1997;183:131‐133. [DOI] [PubMed] [Google Scholar]
- 4. Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaelin WG Jr. The von Hippel‐Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865‐873. [DOI] [PubMed] [Google Scholar]
- 6. Miyazono K, Katsuno Y, Koinuma D, Ehata S, Morikawa M. TGF‐β signaling in cancer: 2018. Front Med. 2018;12:387‐411. [DOI] [PubMed] [Google Scholar]
- 7. Yokoyama Y, Watanabe T, Tamura Y, Hashizume Y, Miyazono K, Ehata S. Autocrine BMP4 signaling is a therapeutic target in colorectal cancer. Cancer Res. 2017;77:4026‐4038. [DOI] [PubMed] [Google Scholar]
- 8. Watanabe A, Ogiwara H, Ehata S, et al. Homozygously deleted gene DACH1 regulates tumor‐initiating activity of glioma cells. Proc Natl Acad Sci USA. 2011;108:12384‐12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawabata KC, Ehata S, Komuro A, Takeuchi K, Miyazono K. TGF‐β‐induced apoptosis of B‐cell lymphoma Ramos cells through reduction of MS4A1/CD20. Oncogene. 2013;32:2096‐2106. [DOI] [PubMed] [Google Scholar]
- 10. Murai F, Koinuma D, Shinozaki‐Ushiku A, Fukayama M, Miyaozono K, Ehata S. EZH2 promotes progression of small cell lung cancer by suppressing the TGF‐β‐Smad‐ASCL1 pathway. Cell Discov. 2015;1:15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishida J, Miyazono K, Ehata S. Decreased TGFBR3/Betaglycan expression enhances the metastatic abilities of renal cell carcinoma cells through TGF‐β‐dependent and independent mechanisms. Oncogene. 2018;37:2197‐2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takahashi K, Ehata S, Koinuma D, et al. Pancreatic tumor microenvironment confers highly malignant properties on pancreatic cancer cells. Oncogene. 2018;37:2757‐2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lainakis G, Bamias A. Targeting angiogenesis in renal cell carcinoma. Curr Cancer Drug Targets. 2008;8:349‐358. [DOI] [PubMed] [Google Scholar]
- 14. Levy L, Hill CS. Alterations in components of the TGF‐beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41‐58. [DOI] [PubMed] [Google Scholar]
- 15. Li Y, Turck CM, Teumer JK, Stavnezer E. Unique sequence, ski, in Sloan‐Kettering avian retroviruses with properties of a new cell‐derived oncogene. J Virol. 1986;57:1065‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stavnezer E, Barkas AE, Brennan LA, Brodeur D, Li Y. Transforming Sloan‐Kettering viruses generated from the cloned v‐ski oncogene by in vitro and in vivo recombinations. J Virol. 1986;57:1073‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonnon C, Atanasoski S. c‐Ski in health and disease. Cell Tissue Res. 2012;347:51‐64. [DOI] [PubMed] [Google Scholar]
- 18. Makino Y, Yoon JH, Bae E, et al. Repression of Smad3 by Stat3 and c‐Ski/SnoN induces gefitinib resistance in lung adenocarcinoma. Biochem Biophys Res Commun. 2017;484:269‐277. [DOI] [PubMed] [Google Scholar]
- 19. Reed JA, Bales E, Xu W, Okan NA, Bandyopadhyay D, Medrano EE. Cytoplasmic localization of the oncogenic protein Ski in human cutaneous melanomas in vivo: functional implications for transforming growth factor beta signaling. Cancer Res. 2001;61:8074‐8078. [PubMed] [Google Scholar]
- 20. Takahata M, Inoue Y, Tsuda H, et al. SKI and MEL1 cooperate to inhibit transforming growth factor‐beta signal in gastric cancer cells. J Biol Chem. 2009;284:3334‐3344. [DOI] [PubMed] [Google Scholar]
- 21. Buess M, Terracciano L, Reuter J, et al. Amplification of SKI is a prognostic marker in early colorectal cancer. Neoplasia. 2004;6:207‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagano Y, Mavrakis KJ, Lee KL, et al. Arkadia induces degradation of SnoN and c‐Ski to enhance transforming growth factor‐beta signaling. J Biol Chem. 2007;282:20492‐20501. [DOI] [PubMed] [Google Scholar]
- 23. Le Scolan E, Zhu Q, Wang L, et al. Transforming growth factor‐beta suppresses the ability of Ski to inhibit tumor metastasis by inducing its degradation. Cancer Res. 2008;68:3277‐3285. [DOI] [PubMed] [Google Scholar]
- 24. Prunier C, Pessah M, Ferrand N, Seo SR, Howe P, Atfi A. The oncoprotein Ski acts as an antagonist of transforming growth factor‐beta signaling by suppressing Smad2 phosphorylation. J Biol Chem. 2003;278:26249‐26257. [DOI] [PubMed] [Google Scholar]
- 25. Ferrand N, Atfi A, Prunier C. The oncoprotein c‐ski functions as a direct antagonist of the transforming growth factor‐beta type I receptor. Cancer Res. 2010;70:8457‐8466. [DOI] [PubMed] [Google Scholar]
- 26. Luo K, Stroschein SL, Wang W, et al. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signaling. Genes Dev. 1999;13:2196‐2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng XH, Derynck R. Specificity and versatility in TGF‐beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659‐693. [DOI] [PubMed] [Google Scholar]
- 28. Miyazono K, Suzuki H, Imamura T. Regulation of TGF‐beta signaling and its roles in progression of tumors. Cancer Sci. 2003;94:230‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akiyoshi S, Inoue H, Hanai J, et al. c‐Ski acts as a transcriptional co‐repressor in transforming growth factor‐beta signaling through interaction with Smads. J Biol Chem. 1999;274:35269‐35277. [DOI] [PubMed] [Google Scholar]
- 30. Pan D, Zhu Q, Luo K. SnoN functions as a tumour suppressor by inducing premature senescence. EMBO J. 2009;28:3500‐3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kiyono K, Suzuki HI, Morishita Y, et al. c‐Ski overexpression promotes tumor growth and angiogenesis through inhibition of transforming growth factor‐beta signaling in diffuse‐type gastric carcinoma. Cancer Sci. 2009;100:1809‐1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Komuro A, Yashiro M, Iwata C, et al. Diffuse‐type gastric carcinoma: progression, angiogenesis, and transforming growth factor beta signaling. J Natl Cancer Inst. 2009;101:592‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials