

SUMMARY
The melanocortin system is a brain circuit that influences energy balance by regulating energy intake and expenditure. In addition, the brain-melanocortin system controls adipose tissue metabolism to optimize fuel mobilization and storage. Specifically, increased brain-melanocortin signaling or negative energy balance promotes lipid mobilization by increasing sympathetic nervous system input to adipose tissue. In contrast, calorie-independent mechanisms favoring energy storage are less understood. Here, we demonstrate that reduction of brain-melanocortin signaling actively promotes fat mass gain by activating the lipogenic program and adipocyte and endothelial cell proliferation in white fat depots independently of caloric intake via efferent nerve fibers conveyed by the common hepatic branch of the vagus nerve. Those vagally regulated obesogenic signals also contribute to the fat mass gain following chronic high-fat diet feeding. These data reveal a physiological mechanism whereby the brain controls energy stores that may contribute to increased susceptibility to obesity.
Graphical Abstract
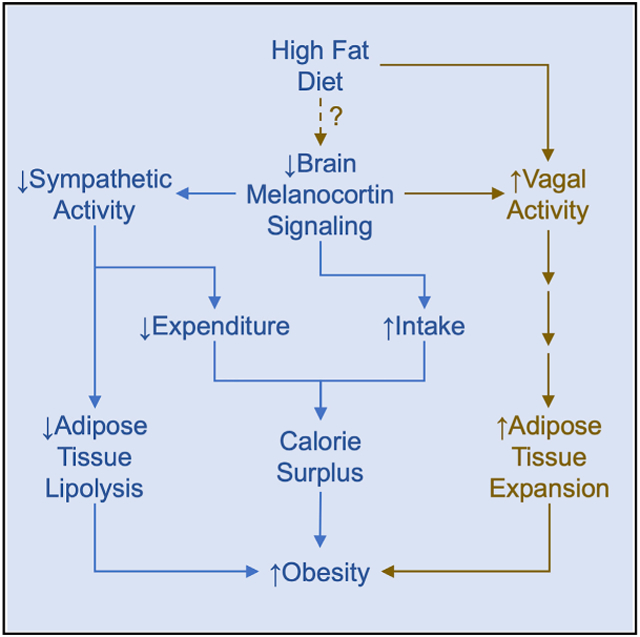
In Brief
Brain-melanocortin signaling controls fat mass indirectly by regulating energy balance and by direct control of lipid mobilization from adipose tissue via sympathetic nervous system activity. Holland et al. show that reduced brain-melanocortin signaling promotes white adipose tissue expansion via signals conveyed by efferent innervation of the vagus nerve.
INTRODUCTION
Energy homeostasis is achieved through the coordinated and integrated activity of numerous processes, including food intake; energy expenditure; the flux of energy-rich fuels among the intestine, blood, and storage organs; and other mechanisms. While numerous brain circuits influence one or several of these processes, of particular note is the brain’s melanocortin system, which receives sensory input from throughout the body indicating current metabolic status, and it, in turn, coordinates many of the processes that influence energy homeostasis. It is well known that the melanocortin system regulates both energy intake and expenditure. Signaling through the melanocortin-4 receptor (Mc4r) directly controls feeding behavior (Garfield et al., 2015). Brain-melanocortin signaling also directly regulates adipose tissue mass independently of caloric intake via direct efferent innervation of peripheral tissues by the sympathetic nervous system (SNS); i.e., we and others have demonstrated that increased brain-melanocortin signaling enhances sympathetic output from the brain (Brito et al., 2007; Nogueiras et al., 2007). A key target of this innervation is adipose tissue, since when sympathetic activity increases, fat cells undergo lipolysis, releasing stored fat during periods of negative energy balance. Ablation of sympathetic innervation to specific fat depots using surgical, chemical, or genetic methods results in an inability to release stored fat and a consequent increase in fat mass, adipocyte size, and cell proliferation (Bartness et al., 2014).
In contrast to its sympathetic innervation, adipose tissue lacks direct innervation by the parasympathetic vagus nerve (Giordano et al., 2006). Nonetheless, there is evidence of parasympathetic involvement in the control of fat mass. Clinical studies have demonstrated that surgical ablation of the abdominal vagus nerve can result in a considerable reduction of body weight (Kral, 1979, 1980; Kral et al., 1993; Smith et al., 1983), and vagal denervation has also been linked to increased weight loss following gastrectomy (Kim et al., 2012; Miyato et al., 2012a, 2012b). These data suggest that increased vagal activity may have a reciprocal role to that of the SNS, promoting a gain of fat mass. Consistent with this, vagal blocking therapy (VBLOC) can provide significant weight loss in obese patients (Apovian et al., 2017; Ikramuddin et al., 2014).
Because of the overall importance of the brain-melanocortin system in energy balance, we investigated its contribution to vagal innervation controlling fat stores. We found that, in opposition to the melanocortin-system-dictated lipolytic role of the SNS, reduced melanocortin signaling mediates adipose tissue expansion due to increased vagal activity and that this brain-vagus axis contributes to diet-induced obesity.
RESULTS
Calorie-Independent Increase in Fat Mass due to Loss of Mc4r Expression or High-Fat Diet Feeding Requires the Integrity of the Vagus Nerve
Subdiaphragmatic vagotomy (sDVX) transiently reduces food intake in Mc4r KO mice, leading to reduced body weight (BW) and fat mass (Dezfuli et al., 2018). To ascertain whether vagal-dependent signals regulate fat mass independently of caloric intake, we pair-fed (PF) wild-type (WT) and Mc4r KO mice daily with a defined low-fat diet (LFD) with the same number of calories eaten by a control group of WT mice fed ad libitum (AL) (Figure 1A). The mice were group-housed (3–4 mice per cage). A group of Mc4r KO mice was maintained on the LFD AL as reference. We also determined whether vagal signals also play a role in the fat mass gain in animals maintained on a high-fat diet (HFD). To this end, an additional group of WT littermates was simultaneously PF with an HFD, but they received only the number of calories eaten by the WT-LFD-AL mice (Figure 1B). First, we tracked fat mass gain over 6 weeks starting at 6–7 weeks of age, and then the mice underwent sDVX or sham surgery (Figures 1A and 1B; Figure S1). To enhance post-operative recovery, all mice were placed on a liquid diet for 3 days before and 7 days after the surgery. Controlled isocaloric feeding resumed afterward. As expected, compared to LFD-AL-WT controls, AL-fed KO mice rapidly gained BW (Figure S2A, left) and fat mass (Figure 1C) but not lean mass (Figure S2A, right), due to overt hyperphagia (Figure 1E). Knockout (KO)-PF mice also gained more BW (Figure S2A, left) due to fat (Figure 1C) but not lean mass gain (Figure S2A, right) than isocalorically fed WT controls (Figures 1C and 1D). sDVX did not reduce fat (Figure 1C) or BW (Figure S2A, left) in LFD-fed WT mice compared to sham LFD-fed WT controls, although it did slightly reduce lean mass (Figure S2A, right). In contrast, sDVX resulted in significantly lower body (Figure S2A, left) and fat mass (Figure 1C) in LFD-PF-KO mice when compared to their sham-operated PF-KO counterparts.
Figure 1. Impact of sDVX on Food-Intake-Independent Gain of Fat Mass Induced by Congenital Loss of Mc4r Expression or by High-Fat Diet Feeding.

(A) Male Mc4r+/+ (WT) and Mc4r−/− (KO) mice (6–7-weeks old) were group-housed (3–4 mice per cage) and fed with a defined LFD. A cage of each genotype was fed ad libitum (AL). Additional WT and KO mice (n=16) were pair-fed (PF) daily to the intake of LFD-AL-WT mice 2 h before the onset of the dark phase, and leftovers in each cage were subtracted. Body composition was assessed by NMR as indicated. After 6 weeks, mice received sham or sDVX surgery and were monitored for 6 additional weeks. During the final week, the mice underwent a glucose tolerance test.
(B) WT mice fed LFD were compared to WT counterparts fed a nutritionally matched HFD (3–4 mice per cage). All PF groups received the same number of calories eaten by LFD-AL-WT mice each day and were monitored as described above.
(C and D) Total fat mass throughout the study comparing WT and KO fed with LFD (C) or WT fed either LFD or HFD (D). *p < 0.05, fat mass at week 12 between PF groups. 2-way ANOVA, Sidak post hoc test. Data represent mean ± SEM. n = 3–4 mice for each AL sham group; n = 16 or 6–8 mice (in presurgery [SX] and post-SX PF groups, respectively).
(E and F) Total caloric intake (normalized per mouse) during the pre-SX and post-SX periods, comparing WT and KO fed with LFD (E) or WT fed either LFD or HFD (F). n = 1 cage for each AL sham group; n = 4 or 2 cages (in pre-SX and post-SX groups, respectively).
Before surgery, HFD-fed WT mice, whether fed AL or PF, gained significantly more fat (Figure 1D) but not lean mass (Figure S2B, right) when compared to their corresponding LFD-AL WT controls, regardless of total caloric intake (Figure 1F). After surgery, both sham-operated HFD-fed groups (AL or PF relative to LFD WT) gained similar amounts of fat. In contrast, sDVX significantly prevented the fat mass gain in HFD-PF fed mice (Figure 1D). Collectively, these data suggest that chronic loss of Mc4r signaling and HFD feeding promote fat mass gain independently of caloric intake via mechanisms that depend on the integrity of the vagus nerve.
AL, sham-operated KO mice were significantly glucose intolerant compared to WT controls, indicating a contribution of obesity (Figure S2C) and hepatic triglycerides (Figure S2E, left) to the glucose intolerance. No differences were detected among the PF groups, regardless of surgery or genotype and despite differential fat mass (Figure S2C). Sham-operated AL and PF HFD mice were also significantly more glucose intolerant than their corresponding LFD controls (Figure S2D), which is also likely related to increased total fat mass (Figure S2D) and hepatic triglycerides (Figure S2F, left). HFD-PF-sDVX mice exhibited improved glucose tolerance compared to HFD-PF-sham mice, which correlated with their reduced fat mass (Figure S2D). Liver glycogen levels in PF-sDVX-KO mice were significantly elevated compared to PF-sham-KO controls (Figure S2E, right). The HFD significantly increased liver glycogen compared to the LFD in the PF groups (p < 0.05), but sDVX had no effect (Figure S2F, right). Muscle triglycerides were significantly elevated in the AL groups due to loss of Mc4r or to HFD feeding (Figures S2G and S2H, respectively). Consistent with the reduced body fat content (Figures 1C and 1D), a 2-way ANOVA detected a significant interaction (p < 0.05) between genotype (Figure S2G) or diet (Figure S2H) and sDVX among the PF groups, although post hoc comparisons failed to detect significant differences. These data suggest that sDVX prevents the calorie-independent fat mass gain due to loss of Mc4r signaling or chronic HFD without promoting ectopic lipid accumulation in tissues important for the control of glucose metabolism.
The Vagus Nerve Mediates Fat Mass Gain that Occurs following Reduced Brain-Melanocortin Signaling
To distinguish whether brain or peripheral Mc4r signaling mediates the control of fat mass by vagal signals, we performed 2-week intracerebroventricular (i.c.v.) infusion of vehicle or the melanocortin-3 receptor and Mc4r blocker SHU9119 (SHU; 1 nmol/day) using osmotic minipumps into the lateral ventricle of sDVX- or sham-operated, chow-fed lean mice (Figure 2A). This SHU dose is ineffective when delivered peripherally in rats (Nogueiras et al., 2007) or mice (data not shown). To avoid potential confounding effects due to the acute impact of vagotomy or post-operative care, we allowed the mice to recover for 4 weeks before the i.c.v. infusion, and we repeated the experiment in a second cohort of mice. During the i.c.v. infusion, all mice received the same average number of calories eaten daily during the weeks prior to the surgery to detect calorie-independent effects (Figures 2B and 2C). Body and lean mass did not differ significantly (Figures 2D and 2E, respectively). In contrast, i.c.v. SHU promoted a significant gain of fat mass in the sham controls but not in the sDVX mice (Figure 2F). sDVX-IcvSHU mice had a slight increase in fat mass compared to sDVX-icvVeh (intracerebral infusion of vehicle) controls, but it did not reach statistical significance. Furthermore, their final fat mass was significantly lower than that of the icvSHU-sham group (Figure 2F). The weights of three visceral fat depots (Figures 2G-2I), as well as subcutaneous inguinal fat (Figure 2J) and interscapular brown adipose tissue (iBAT) depots (Figure 2K), all faithfully recapitulated the effect on whole-body fat mass. Consistent with the previous experiment (Figures S2E and S2F), neither liver weight (Figure 2L) nor liver triglyceride (Figure 2M) or glycogen (Figure 2N) content differed significantly. Likewise, plasma triglycerides (Figure 2O) and free fatty acids (FFAs) (Figure 2P) were similar among groups. These data indicate that sDVX prevents the lipid deposition in different fat depots otherwise promoted by reduced brain-melanocortin signaling.
Figure 2. Impact of sDVX on the Food-Intake-Independent Fat Gain of Mass following Blockade of Brain Melanocortin in Male Mice.

(A) Male C57Bl6 mice (27.1 g) received sDVX or sham surgery 4 weeks prior to receiving icvSHU (1 nmol/day) or vehicle infusion for 14 days in two separate experiments. All mice received the daily average caloric intake consumed by each cohort during the week before the i.c.v. surgery (4.8 and 4.1 g per mouse), and leftovers were subtracted.
(B and C) Daily (B) and cumulative (C) caloric intake.
(D–F) Body (D), lean (E), and fat (F) mass before and after the i.c.v. infusion period.
(G–J) Tissue weight of gonadal (G), mesenteric (H), retroperitoneal (I), and inguinal (J) white adipose tissue.
(K) Tissue weight of interscapular brown adipose tissue.
(L) Liver weight.
(M and N) Hepatic triglyceride (M) and glycogen (N) content.
(O and P) Plasma levels of triglycerides (O) and free fatty acids (P).
*p < 0.05, 3-way ANOVA followed by Tukey’s post hoc test in (F) or 2-way ANOVA followed by Sidak post hoc test in (G)–(K). Data represent mean ± SEM. n = 14–19.
Vagal Efferents Mediate the Control of Fat Mass by Brain-Melanocortin Signaling
To determine whether afferent or efferent vagal nerves mediate the brain-melanocortin control of fat mass, we performed selective vagal subdiaphragmatic deafferentiation (SDA) in lean male Wistar rats. The lack of acute reduction in food intake following cholecystokinin-8 (CCK-8) injection (4 μg/kg, intraperitoneally [i.p.]) confirmed the efficacy of SDA (Figure 3A). Of note, food intake in vehicle-treated SDA rats was significantly reduced during the first hour compared to that of sham controls (Figure 3A). Two weeks after SDA, the rats received either i.c.v. SHU or vehicle for 2 weeks. Feeding was controlled to ensure isocaloric intake (Figure 3B). IcvSHU promoted significant and comparable fat mass gain, despite loss of subdiaphragmatic vagal afferents (Figure 3C). Conversely to the effect on fat mass, a 3-way ANOVA detected a significant effect of icvSHU on lean mass that reached significance only in the sham group when compared to its icvVeh control (Figure 3D). Total body mass did not change significantly (Figure 3E). These data suggest that subdiaphragmatic vagal afferents are not necessary for the increase in fat mass in response to decreased brain-melanocortin signaling.
Figure 3. Impact of Vagal SDA on the Food-Intake-Independent Gain of Fat Mass following Blockade of Brain Melanocortin in Male Rats.

Male Wistar rats (283 g) received SDA or sham surgery 2 weeks before icvSHU or vehicle infusion for 2 weeks.
(A) The anorectic response to i.p.-administered cholecystokinin (CCK; 4 μg/kg) or vehicle in sham and SDA rats.
(B) Chow intake was restricted in rats receiving icvSHU to match the caloric intake of their corresponding controls.
(C–E) Fat (C), lean (D), and body (E) mass before and after the 14-day i.c.v. infusion period.
In (A), C), and (D), *p < 0.05 relative to sham control or as indicated by the bracket, 3-way ANOVA followed by Tukey post hoc test. Data represent mean ± SEM. n = 10–11.
The Common Hepatic Branch of the Vagus Mediates the Control of Fat Mass by Brain-Melanocortin Signaling
We performed selective ablation of the common hepatic branch (hVX) 2 weeks prior to icvSHU. After 1 week of i.c.v. infusion under calorie-controlled conditions to prevent hyperphagia (Figure 4A), icvSHU led to significant fat mass gain in sham, but not in hVX, rats (Figure 4B). Lean (Figure 4C) and total body mass (Figure 4D) did not change significantly. Collectively, these data suggest that reduced brain-melanocortin activity increases fat mass in mice and rats via signals conveyed by efferent nerves in the hVX of the vagus nerve.
Figure 4. Role of the Common Hepatic Branch of the Vagus Nerve on the Food- Intake-Independent Gain of Fat Mass following Blockade of Brain Melanocortin in Male Rats.

Male Wistar (average BW = 296 g) received hVX or sham surgery 1 week prior to icvSHU or vehicle infusion for 1 week.
(A) Chow intake was restricted in rats receiving icvSHU to match the caloric intake of their corresponding controls.
(B–D) Fat (B), lean (C), and body (D) mass before and after the 7-day i.c.v. infusion period.
*p < 0.05, 3-way ANOVA, Tukey post hoc test. Data represent mean ± SEM. n = 8–10.
Reduced Brain-Melanocortin Signaling Promotes Caloric-Intake-Independent Fat Mass Gain under Iso-insulinemic Conditions
Reduced Mc4r signaling leads to hyperinsulinemia (Fan et al., 2000; Martinelli et al., 2011), a critical driver of obesity in mice (Mehran et al., 2012; Frikke-Schmidt et al., 2013). Because the efferent vagus nerve regulates insulin (INS) secretion (Berthoud et al., 1983), we measured baseline INS at the end of studies in icvSHU-treated mice or rats and found no significant differences between vagotomized and sham-operated animals (Figures S3A and S3B). Nonetheless, to unequivocally rule out that fat mass gain due to reduced brain-melanocortin signaling is the result of sustained differences of circulating INS, we induced sustained hyperinsulinemia in lean mice receiving icvSHU or vehicle by administering 20 kDa long-lasting, polyethylene glycol-INS (20 nmol/kg, i.p., twice daily). All groups were meal-fed with pre-i.c.v. surgery caloric amounts to prevent hyperphagia (Figure S4A). INS-treated mice exhibited sustained lower glycemia (Figure S4B). Reduced brain-melanocortin signaling due to icvSHU increased circulating c-peptide and INS levels (Figure S4C). In contrast, all INS-treated mice had nearly undetectable c-peptide, regardless of icv treatment, demonstrating effective suppression of endogenous INS secretion, but they exhibited a near-two-order magnitude increase in INS immunore-activity due to the pharmacological replacement (Figure S4C). IcvSHU promoted similar whole-body fat mass gain in vehicle and INS-treated mice compared to their icvVeh controls (Figure S4D). The increase in total fat mass correlated with an increased mass of discrete fat depots (Figures S4E-S4G). In keeping with previous results (Frikke-Schmidt et al., 2013), high levels of exogenous INS increased fat mass preferentially in the inguinal fat depot, an effect that was only additive to the contribution of icvSHU (Figure S4F). These data suggest that reduced brain-melanocortin signaling increases fat mass via signals, putatively under vagal control, by means other than the sole increase in INS levels.
Reduced Brain-Melanocortin Vagus Axis Signaling Increases Fat Depot Weight despite Unchanged Local Norepinephrine Turnover
To determine the impact of sDVX on the activity of the SNS in specific fat depots, we performed sDVX in lean mice, followed 4 weeks later by icvSHU or vehicle for 2 weeks (Figure S5A). During that period, all mice received daily the same average calories eaten daily during the weeks prior to the i.c.v. surgery (4 g/day). At the end of the i.c.v. infusion, we quantified norepinephrine (NE) turnover (NETO) following the injection of alpha-methyl-p-tyrosine (AMPT) (Vaughan et al., 2014) (Figure S5A) in spleen, iBAT, inguinal white adipose tissue (iWAT), and gonadal white adipose tissue (gWAT). IcvSHU increased the mass of all three fat depots independently of caloric intake, and this increase was significantly blunted by sDVX (Figure S5B). sDVX reduced NETO in the spleen (Figure S5C), consistent with a vagal control of sympathetic tone (Rosas-Ballina et al., 2011) (Figure S5C). Consistent with previous reports (Douris et al., 2015), baseline NETO levels differed substantially among fat depots, from high levels in iBAT to nearly undetectable levels in gonadal fat (Figure S5C). icvSHU significantly reduced NETO in the iBAT of sham-operated mice compared to controls (Figure S5C). Given the role of SNS in the control of uncoupling protein-1 (UCP-1) levels (Bachman et al., 2002), we measured UCP-1 in iBAT samples (Figure 2K) by immunoblot. Consistent with previous reports (Brito et al., 2007; Kooijman et al., 2014), UCP-1 levels were significantly reduced by icvSHU in sham mice (Figure S5D). Intriguingly, UCP-1 was reduced to a similar extent in iBAT of all sDVX mice, and this reduction correlated with the reduced sympathetic tone to iBAT due to sDVX (Figure S5C).
sDVX also reduced NETO in iBAT (main effect of sDVX, p < 0.05; Figure S5C), although within the lower levels exhibited by sDVX mice, NETO was significantly higher in icvSHU mice compared to vehicle controls (Figure S5C). These data suggest a role for signals conveyed by subdiaphragmatic vagal fibers on the maintenance of baseline sympathetic tone to iBAT that intersects with the contribution of the brain-melanocortin signaling. Nonetheless, these data argue against a potential compensatory increase in non-shivering thermogenesis as the main mechanism whereby sDVX prevents icvSHU-induced fat mass gain.
In contrast to what occurred in iBAT, neither sDVX nor chronic icvSHU infusion changed NETO in inguinal or gonadal white fat depots (Figure S5B). Thus, these data suggest that the vagus-dependent control of fat depot mass gain following reduction of brain-melanocortin signaling may not require increased local SNS tone.
The Brain-Melanocortin-Vagus Axis Regulates the Respiratory Exchange Ratio
We housed a cohort of sham or sDVX mice (average BW = 30.3 g) in an indirect calorimetry system for simultaneous measurement of feeding and energy expenditure for the entirety of the 2-week icvSHU infusion. We subtracted total fecal calorie content and any calories due to spillage from total calorie consumption to determine the actual assimilated energy. Neither icvSHU nor sDVX changed fecal calorie density (data not shown), discarding malabsorption as the culprit. Sham-operated icvSHU mice had increased total fecal output that resulted in a modest yet significant increase in fecal caloric loss (Figure S6A). IcvSHU had a small but significant effect (main effect p < 0.05) on total caloric intake (Figure S6B) and on calculated assimilated energy (Figure 5A), but none of the post hoc comparisons among groups identified statistically significant differences. Nonetheless, icvSHU promoted significant fat mass gain in sham-operated mice, and this was significantly mitigated by sDVX (Figure S6C). sDVX did not alter total food intake (Figure S6B), assimilated energy intake (Figure 5A), daily feeding patterns (Figure 5B), or fat mass gain (Figure S6C).
Figure 5. Simultaneous Measurement of Caloric Intake, Energy Expenditure, and RER in Sham and sDVX Mice Receiving icvSHU for 2 Weeks.

Lean male mice (average BW = 30.2 g) received sham or sDVX 4 weeks prior to icvSHU or vehicle infusion for 2 weeks. During that period, the mice were individually housed in sealed chambers. Measurements were taken every 10 min and averaged in bouts of 1 h for energy expenditure (EE) and respiratory exchange ratio (RER). Caloric intake data were accumulated in 2-h intervals.
(A and B) Total (A) and continuous (B) caloric intake.
(C and D) Total (C) and continuous (D) energy expenditure.
(E and F) Average (E) and continuous (F) RER.
Gray vertical bars in (B), (D), and (F) represent the dark phase periods. *p < 0.05, 2-way ANOVA, Sidak post hoc test. Data represent mean ± SEM. n = 8–11.
Locomotor activity did not differ among groups (data not shown). Neither total (Figure 5C) nor daily variations (Figure 5D) on energy expenditure, as measured by indirect calorimetry during the duration of the i.c.v. infusion, differed among groups throughout the experiment. We alternatively calculated the total energy expenditure balance (TEEbal) by considering assimilated energy intake (Figure 5A) and body composition measured by nuclear magnetic resonance (NMR) (Ravussin et al., 2013). This method also failed to identify significant differences in energy expenditure (Figure S6D). In contrast, sham-operated mice receiving icvSHU had an increased respiratory exchange ratio [RER] that was sustained over time and was prevented by sDVX (Figures 5E and 5F).
Collectively, these data imply that reduced brain-melanocortin signaling increases fat mass via the vagus nerve in the face of minimal changes in energy expenditure but plays an integral role in the control of fuel utilization, as suggested by the effect on RER.
The Brain-Melanocortin-Vagus Axis Regulates Gene Expression Favoring Lipogenesis in Adipose Tissue Independently of Caloric Intake
To gain insight into the mechanisms contributing to the control of fat mass by the brain-melanocortin-vagus axis, we performed RNA sequencing for gene expression analysis in gonadal fat. Comparison of sham with sDVX control groups did not identify significantly differentially regulated genes (Figure 6A). In contrast, icvSHU significantly regulated the expression of 228 genes (false discovery rate [FDR] < 0.1; 181 up- and 47 downregulated) in sham-operated mice when compared to vehicle-infused sham controls (Figure 6A; Table S1). Overrepresentation enrichment analysis (ORA) of upregulated genes revealed significant engagement of pathways involved in cholesterol, fatty acid, and glucose metabolism, consistent with the de novo lipogenesis (Figure 6B; Table S2). In contrast, icvSHU in sDVX mice resulted in a significant upregulation of Mest, a regulator of adipocyte size (Takahashi et al., 2005) that was also upregulated by icvSHU in sham mice (Table S1). Comparison between icvSHU-sDVX and icvSHU-sham mice resulted in 43 genes significantly regulated, of which 39 were downregulated (Figure 6A; Table S3). ORA of these significantly downregulated genes identified the blood clotting cascade, the statin pathway, and, consistent with the differences in fat mass, the peroxisome proliferator-activated receptor (PPAR) signaling pathway (Figure 6C; Table S4). These data suggest that reduced brain-melanocortin signaling may facilitate the gain of fat mass by increasing lipogenesis in adipocytes via signals regulated by the vagus nerve.
Figure 6. Impact of sDVX on Gene Expression of Gonadal Fat of Mice Receiving icvSHU for 2 Weeks.

Representative samples (n = 4) from gonadal fat (Figure 2G) were processed for RNA extraction followed by RNaseq for gene expression analysis.
(A) Gonadal depot weight of samples analyzed and genes differentially regulated (FDR < 0.1) between groups indicated by brackets.
(B) Pathways differentially regulated (FDR < 0.1) when considering genes upregulated by icvSHU in sham-operated mice compared to vehicle controls.
(C) Pathways differentially regulated (FDR < 0.1) when considering genes downregulated in sDVX mice receiving icvSHU compared to sham-operated icvSHU counterparts.
The list of individual genes is included as Supplemental Information.
The Brain-Melanocortin-Vagus Axis Regulates Fat Mass Independently of Caloric Intake by Increasing Adipocyte Proliferation in Gonadal Fat
We next asked whether the brain-Mc4r-vagus axis regulates fat mass by promoting adipocyte hyperplasia. We repeated the protocol described earlier (Figure 2A), with the exception of the addition of bromodeoxyuridine (BrdU) in the drinking water between days 3 and 10 of the i.c.v. infusion period (Figure 7A). At the end of the 2-week infusion period, we isolated and immunolabeled stromal vascular cells (SVCs) from gonadal and inguinal fat depots to identify adipocyte progenitors (APs; CD140a+/CD45−/CD31−), endothelial cells (ECs; CD45−/CD31+), and leukocytes (LKs; CD45+) using flow cytometry (Figure S7A). icvSHU promoted significant BW (Figure S7B) and fat mass gain (Figure S7C) in sham-operated mice but not in the sDVX mice. In keeping with this and with the increase in gonadal fat depot weight (Figure 2D), icvSHU increased the percentage of BrdU+ APs and BrdU+ ECs in the gonadal fat depots of sham-operated mice, but not of sDVX mice (Figure 7B), when compared to icvVeh controls. In contrast, icvSHU did not increase BrdU+ APs in the inguinal fat of sham-operated mice but led to a significant increase of BrdU+ ECs compared to sham controls (Figure 7B). The percentage of BrdU+ APs was significantly different between both iWAT sDVX groups, partially due to the low values detected in icvVeh controls (Figure 7B). Neither i.c.v. treatment nor vagotomy impacted the percentage of BrdU+ LKs in gonadal or inguinal fat (Figure 7B). Correlation analysis comparing the percentage of BrdU+ APs from gonadal and inguinal fat revealed a significant positive correlation in sham-operated icvVeh mice (r = 0.91, p < 0.033) and icvSHU mice (r = 0.89, p < 0.016); and linear regression analysis revealed significantly different intercepts (p < 0.01; Figure 7C). sDVX groups did not differ. Collectively, these data indicate that the brain-Mc4r-vagus axis regulates signals that favor AP proliferation, preferentially in gonadal fat, which is consistent with the increased fat mass.
Figure 7. Impact of sDVX on Adipocyte Proliferation in Gonadal and Inguinal Fat Pads of Mice Receiving icvSHU for 2 Weeks.

(A) Sham or sDVX mice (average BW = 27.9 g) were isocalorically fed (4.5 g/day) with chow and received icvSHU or vehicle infusions for 2 weeks. During days 3–10 of i.c.v. infusion, tap water was replaced by BRDU solution (0.8 mg/mL BrdU (Sigma) + 1% D-glucose on tap water) provided fresh daily. Tap water was returned afterward.
(B) Percentage of BrdU+ adipocyte precursors (APs; CD140a+/CD45−/CD31−), endothelial cells (ECs; CD45−/CD31+), and leukocytes (LKs; CD45+) on SVCs of gonadal and inguinal fat, measured using flow cytometry.
(C) Correlation between BrdU+ APs from gonadal and inguinal fat depots.
In (B), *p < 0.05, 2-way ANOVA, Sidak post hoc test. Data represent mean ± SEM. n = 5–7. In (C), correlation analysis detected a significant correlation (p < 0.05) between gWAT and iWAT BrdU+ APs only for Sham-Veh and Sham-SHU; Pearson correlation coefficients (rs) = 0.91 and 0.89, respectively. Linear regression analysis detected a significantly different intercept (p < 0.05) between Sham-Veh (R2 = 0.82) and Sham-SHU (R2 = 0.80).
DISCUSSION
Brain-melanocortin signaling is integral for the control of energy balance. A net reduction of melanocortin receptor activity favors energy intake over expenditure and results in obesity. Here, we provide initial and definitive evidence that a brain-melanocortin-efferent vagal pathway contributes to the control of fat mass gain. This pathway utilizes obesogenic signals to facilitate adipose tissue expansion, both by activation of lipid anabolism and by promoting depot-specific adipocyte proliferation. These data also suggest that components of this axis are regulated during chronic HFD feeding and contribute to diet-induced obesity. These data are consistent with early clinical studies that demonstrate that surgical ablation of the vagus nerve at the abdominal level causes a meaningful reduction of BW (Kral, 1979,1980; Kral et al., 1993). In keeping with this, vagal denervation has been linked to increased weight loss following gastrectomy (Kim et al., 2012; Miyato et al., 2012a, 2012b). These data are all consistent with the concept that increased vagus nerve activity promotes fat mass gain; consistent with this, vagal blocking therapy has been proposed as a treatment of adult patients with obesity (Apovian et al., 2017; Ikramuddin et al., 2014).
The vagus nerve contains afferent and efferent fibers conveying signals between peripheral tissues and the brain. Furthermore, abdominal vagal branches contain nerve fibers other than those of vagal origin. We found that fat mass gain due to reduced brain melanocortin activity can be prevented by (1) ablating the hVX, which contains mainly vagal afferents but also efferents, as well adventitial fibers likely contributed by the SNS (Prechtl and Powley, 1990); and (2) cutting both sub-diaphramatic vagal trunks (i.e., sDVX), which contain both vagal afferents and efferents but lack sympathetic fibers (Prechtl and Powley, 1990); but not by SDA, which eliminates subdiaphragmatic vagal afferents and partially preserves subdiaphragmatic vagal efferents (Norgren and Smith, 1994). Although somewhat surprising, considering the relatively low contribution of efferent fibers to the hVX (Prechtl and Powley, 1990), these data suggest, nonetheless, that the obesogenic activity due to reduced brain melanocortin activity is mediated by vagal efferents which are, at least in the rat, conveyed by that branch.
The peripheral signals whereby the brain-melanocortin-vagus efferent axis regulates fat mass gain remain unknown. A potential direct vagal innervation of adipose tissue seems unlikely, considering the lack of evidence of parasympathetic neurochemistry on white fat depots (Giordano et al., 2006), implying that vagal action elsewhere within the splanchnic compartment regulates fat depot weight. An important shortcoming of surgical approaches as those used here is the limited ability to discriminate the target tissue within the compartment mediating those vagal effects. Novel approaches, including the injection of viral vectors with retrograde properties (Han et al., 2018), should allow probing the selective contribution of specific organs within that compartment responsible of mediating the vagal control on fat mass gain. In the interim, our data suggest other mechanisms critical for the control of fat mass and regulated by the melanocortin system and the vagus nerve. However, neither hyperinsulinemia nor reduced NE turnover at discrete fat depots appears to be sufficient to increase fat mass following the pharmacological blockade of brain melanocortin receptors. Whether those vagally regulated mechanisms play a role in the control of fat mass during conditions favoring lipid mobilization from adipose tissue (i.e., fasting or cold exposure) will require additional examination.
A causal role for hyperinsulinemia mediating the fat mass gain due to reduced brain-melanocortin signaling seemed plausible for multiple reasons. Those include the contribution to fat mass gain of hyperinsulinemia, either due to pharmacological administration (Frikke-Schmidt et al., 2013) or physiologically induced by HFD feeding (Mehran et al., 2012); and the contribution of Mc4r signaling to the control of INS levels. Thus, brain-melanocortin signaling directly regulates circulating INS levels (Fan et al., 2000). Consistently, obese Mc4r-deficient rodents (Girardet and Butler, 2014) and humans (Martinelli et al., 2011) exhibit hyperinsulinemia. On the other hand, vagal efferents, including those contributed by the hVX, have a direct role in the control of INS secretion (Berthoud et al., 1983; Berthoud and Powley, 1987). This raised the possibility that vagotomy could interfere with icvSHU-induced hyperinsulinemia, leading to the differences in fat mass gain. However, blockade of brain-melanocortin signaling with icvSHU promoted similar gains on fat mass, regardless of the levels of circulating (endogenous or exogenous) INS. Thus, although a permissive role of INS signaling is likely required, and hyperinsulinemia might contribute to obesity due to chronic loss of Mc4r signaling, alternative mechanisms under vagal efferent control also play a role in the control of fat mass by brain-melanocortin signaling.
Our data also suggest that the control of fat mass by the brain-melanocortin-vagus axis may not require reduced local sympathetic tone to white adipose tissue. This possibility was plausible, considering that: (1) Mc4rs are expressed throughout multiple neuroanatomical sites projecting to white fat via the SNS (Song et al., 2005); (2) activation of brain Mc4r increases SNS activity in inguinal (Brito et al., 2007) or gonadal fat (Nogueiras et al., 2007); (3) sympathetic postganglionic neurons from the celiac ganglion innervate inguinal fat (Jiang et al., 2017); (4) the vagus nerve innervates the celiac ganglion (Berthoud and Powley, 1993); and (5) this vagus-celiac ganglia interaction is functionally relevant for the control of sympathetic tone in other organs (i.e., spleen) (Rosas-Ballina et al., 2011; Willemze et al., 2015)). In contrast to the contribution of the sympathetic tone to the spleen, sDVX did not impact sympathetic activity in inguinal or gonadal white fat measured as NETO. Indeed, the increase in white fat depot weight following chronic reduction of brain-melanocortin signaling occurred without local reductions of SNS activity to inguinal or gonadal fat depots. In the case of gonadal fat, these data are consistent with our previous experiments in which icvSHU alone did not reduce the firing of sympathetic fibers innervating gonadal fat in rats (Nogueiras et al., 2007). Collectively, these data suggest that, while activation of the brain-melanocortin-SNS axis may contribute to decreased white fat depot weight by promoting lipolysis, vagal-dependent signals are sufficient to mediate its increase in response to a reduction in brain-melanocortin signaling without reductions in local SNS tone.
In contrast to white fat depots, our data confirm a strong positive regulation of the sympathetic tone to iBAT by brain-melanocortin signaling (Song et al., 2008; Voss-Andreae et al., 2007). Surprisingly, sDVX results in a reduction in iBAT sympathetic tone and a corresponding decrease in UCP-1 protein levels. This is consistent with a report indicating that vagus nerve stimulation in humans increases brown adipose tissue activity (Vijgen et al., 2013). Interestingly, recent data suggest that changes in lipogenesis or lipolysis in white adipose tissue regulate SNS activity in iBAT (Guilherme et al., 2018). This cross-talk between white and brown fat occurs through mechanisms that include afferent nerves innervating white fat (Garretson et al., 2016). Our data open the possibility to alternative mechanisms whereby peripheral signals (whether derived or not from white adipose tissue) are conveyed to the brain by vagal afferent fibers at the sub-diaphragmatic level to regulate baseline SNS tone in iBAT.
Our data indicate that signals regulated by the vagus nerve promote significant increases in fat mass following reduction in brain-melanocortin signaling without evident changes in energy intake or expenditure. Notably, the role of Mc4r in the control of energy expenditure seems to parallel the control of SNS activity. Thus, increased melanocortin receptor signaling by use of agonists increases energy expenditure in rats (Jonsson et al., 2001) (Gavini et al., 2016), mice (Hoggard et al., 2004), non-human primates (Kievit et al., 2013), and humans (Chen et al., 2015). In contrast, the reports of possible effects of reduced melanocortin receptor action on energy expenditure are less consistent. Acute i.c.v. injection of the endogenous melanocortin receptor blocker Agrp resulted in a short-term reduction in oxygen consumption in rats (Small et al., 2003). However, i.c.v. administration of the Mc4r blockers HS024 (Jonsson et al., 2001) and SHU9119 (Nogueiras et al., 2007) did not reduce energy expenditure, despite inducing hyperphagia. Notably, these data are consistent with the normal energy expenditure exhibited by Mc4r mutant mice (Sutton et al., 2006; Weideet al., 2003), rats (Almundarij et al., 2016), and human subjects with loss of Mc4r function (Farooqi et al., 2000,2003). In contrast to energy expenditure, reduction in brain-melanocortin signaling consistently results in an increase in the RER in rodents (Almundarij et al., 2016; Cavalcanti-de-Albuquerque et al., 2019; Sutton et al., 2006) and humans (Nogueiras et al., 2007). The RER is an indicator of fuel utilization, such that higher values indicate a reduction of lipid utilization in favor of carbohydrates and often correlate with fat mass gain (Galgani and Ravussin, 2008). Our data depicting differences on RER suggest that signals regulated by the vagus nerve are significant contributors to the control of nutrient partitioning by the brain-melanocortin system.
Consistent with previous findings (Cavalcanti-de-Albuquerque et al., 2019; Nogueiras et al., 2007), gene expression analysis of gonadal fat suggests an increase in de novo lipid synthesis following the reduction in brain-melanocortin signaling. This includes an increase in the local synthesis of fatty acids and triglycerides as well as cholesterol, an indicator of adipocyte size (Le Lay et al., 2001) that can be prevented by sDVX, suggesting specific control of white adipose tissue expansion by the brain-melanocortin-vagal axis. In addition, the expansion of white adipose tissue correlates with an increase in the proliferation of preadipocytes in gonadal fat, as indicated by BrdU labeling. This, as well as the absence of adipocyte proliferation in inguinal fat, despite increased depot weight, is consistent with previous reports (Wang et al., 2013) (Jeffery et al., 2015). On the other hand, both fat depots exhibited an increase in endothelial cell proliferation following reduction of brain-melanocortin signaling that correlated with increased fat depot weight and that was prevented by sDVX. These findings are consistent with an increase in angiogenesis necessary to support the initial steps of adipose tissue expansion (Corvera and Gealekman, 2014; Crewe et al., 2017) and further suggest an active role of the brain-melanocortin-vagus axis in regulating energy stores by actively facilitating adipose tissue expansion.
The current view of the control of energy balance by the brain-melanocortin system includes a bidirectional control of feeding as well as a bidirectional control of the SNS to regulate energy mobilization or storage on white adipose tissue. Our data are consistent with a different model in which sympathetic activation plays a prominent role promoting energy mobilization upon increased melanocortin system signaling, whereas facilitation of energy storage in adipose tissue upon melanocortin system inhibition requires the activation of the parasympathetic nervous system. Our evidence supporting the overlap with the etiology of diet-induced obesity suggests that these mechanisms regulating energy storage are integral to the control of energy balance, and its investigation may provide more efficacious approaches to prevent or to treat obesity.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Diego Perez-Tilve (pereztdo@ucmail.uc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo animal studies
All studies were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice and rats were housed in an AAALAC-approved room with a 12-h light, 12-h dark cycle room held at 22°C and with free access to food and water.
Mice
8-10 week-old male C57BL/6J mice (#000664) were obtained from the Jackson laboratory (Bar harbor, ME). LoxTbMc4r (#006414; Jackson laboratory; KO) mice and wild-type littermate controls (WT) were cross-bred in-house for 5 generations with C57BL/6J mice.
Rats
Male Wistar rats (275 g-290 g) were obtained from Harlan (Indianapolis, IN) and were singly-housed.
Diets
Animals were maintained on pelleted chow (Teklad LM-485, Envigo; 3.1 kcal/g). Some studies utilized a high-fat diet (HFD) (D12331; Research Diets Inc., New Brunswick, NJ; 58% fat, 5.6 kcal/g), and control animals received a matched low-fat diet (LFD, D12329, Research Diets; 11% fat, 4.1 kcal/g).
METHOD DETAILS
Body composition
Body composition was determined by nuclear magnetic resonance (echoMRI, Houston, TX).
Indirect calorimetry
Mice were individually housed at 23°C in chambers for simultaneous measurement of food intake, energy expenditure, respiratory exchange ratio (RER) and locomotor activity via indirect calorimetry (TSE Systems, Chesterfield, MO, USA). Energy expenditure was estimated using the following energy balance equation: TEEbal = Assimilated Energy – (Δ somatic Fat Energy + Δ somatic Fat–Free Energy), assuming 9.4kcal/gm and 1.0 kcal/gm for fat and fat-free mass gained or lost, respectively, as suggested elsewhere (Ravussin et al., 2013).
Fecal caloric content
Fecal energy content was determined in dried fecal homogenate (1g) via a Bomb Calorimeter by the Small Animal Phenotyping sub core at the University of Alabama-Nutrition Obesity Research Center.
Subdiaphragmatic vagotomy (sDVX)
Prior to the surgery, the pelleted diet was replaced by a nutritionally complete liquid diet (Ensure, Abbot) for 3 d. Then, mice were anesthetized using 5% isoflurane in oxygen in an induction chamber, and maintained on 2.5% isoflurane delivered by a nose cone. A laparotomy was performed in the midline. Under a binocular microscope, both vagal trunks attached to the esophagus were identified between the diaphragm and the stomach, isolated and severed. In the sham-operated animals the same procedure was followed except for the scission of the nerve branch. After surgery, the animals received a single sc dose of 5 mg/kg meloxicam (Norbrook, UK) and were maintained on the liquid diet for 3 additional d prior to being returned to the solid diet.
Vagal denervation was confirmed by lack of detection of the retrograde tracer Fluoro-Gold (Flurochrome, CO) at the dorsal motor nucleus of the vagus. Fluoro-Gold was injected (0.8mg, ip) 7-days after sDVX. Mice were euthanized and transcardially perfused with 0.1M PBS followed by 4% paraformaldehyde/0.1M PBS 7 days after Fluoro-Gold injection. Brains were postfixed in 4% paraformaldehyde overnight followed by 30% sucrose in PBS until the brains sank. Brainstems were sectioned (30um) on a freezing stage microtome (Leica Biosystems Inc., Nußloch, Germany) in a series of 4 and stored in cryoprotectant (30% Sucrose, 1% Polyvinyl-pyrolidone (PVP-40), and 30% Ethyleneglycol, in 0.1 M PBS). Sections were rinsed 5 times for 5 minutes (5X5) in 0.1M PBS, washed in a blocking solution (4% goat serum, 0.1% BSA, 0.2% Triton-x-100 in 0.1M PBS) for 1 hr then incubated in anti- Fluoro-Gold (Fluorochrome, rabbit polyclonal) diluted 1:500 in block solution overnight at 4°C. The next day sections were washed 5X5 in 0.1M PBS followed by incubation of Cy3 anti-rabbit (Thermo Scientific, 1:500) secondary. 10x images were acquired under same lighting conditions using a Nikon confocal C2-plus microscope.
Common hepatic branch vagotomy (hVX)
Resection of the common hepatic branch of the vagus nerve was performed as in anesthetized rats via laparotomy as previously described (Perez-Tilve et al., 2010). Briefly, male Wistar rats (250-275 g) were anesthetized with ketamine (86 mg/kg i.p.)/xylazine (5 mg/kg,i.p.) and received buprenorphine analgesia (0.1 mg/kg, s.c.; Buprenex; Reckitt Benckiser Healthcare). A laparotomy was performed in the midline. Under a binocular microscope, hepatic ligaments were severed to deflect the hepatic lobes to the animal right side and to expose the common hepatic vagal branch for transection. In the sham-operated animals the same procedure was followed except the scission of the nerve branch. The animals were allowed to recover for 7 d prior to undergoing chronic intracerebroventricular (icv) infusion.
Subdiaphragmatic vagal deafferentation (SDA)
SDA vagotomy involves left-side intracranial vagal rhizotomy for afferent rootlet resection and transection of the dorsal subdiaphragmatic trunk of the vagus nerve along the esophagus in rats anesthetized with ketamine/xylazineas previously described Klarer et al., 2014). Rats received liquid diet during the initial 3 d and were offered wet chow before returning to solid diet. The recovery period extended for two wk. To test the success of the deafferentation, rats were food deprived overnight and injected intraperitoneally (ip) with 4 μg/kg CCK-8 (Bachem) or vehicle (PBS) 2 h after the onset of the light phase. Food was returned and intake was monitored after 1 and 2 h. Any CCK-treated SDA rat eating < 3.4 g after 2 h (average 6.5 g of the rats left in the experiment) was removed from the study.
Chronic icv infusion in mice
Mice were anesthetized using isoflurane and stereotaxically implanted (David Kopf Instruments, Tujunga, CA) with a cannula (brain infusion kit #3; Alzet Durect, Cupertino, CA) placed in the lateral cerebral ventricle as previously described (Heppner et al., 2014). A polyethylene catheter attached the cannula to a subcutaneously implanted osmotic mini-pump (#1002; Alzet, Cupertino, CA) infusing SHU9119 (SHU, Bachem) at 1 nmol/d or vehicle (saline) for 14 d.
Chronic administration of 20kDa PEG-Insulin (INS)
20-kDa polyethylene glycol-insulin (INS) was synthesized as previously described for lys-pro insulin (Bergenstal et al., 2012) and injected in mice at 20 nmol/kg, ip, twice daily.
Chronic icv infusion in rats
Rats were implanted with a stainless-steel cannula with its tip in the lateral ventricle under ketamine /xylazine anesthesia and buprenorphine analgesia. The cannula was connected to an osmotic minipump (#2001 or #2002, Alzet Durect) subcutaneously placed in the interscapular space, as previously described (Perez-Tilve et al., 2010). The rats received SHU at 2.5 nmol/d or vehicle (saline) for 7 or 14 d as indicated.
Glucose tolerance test
6-h fasted mice received ip glucose (2 g/kg, 20% wt/vol d-glucose [Sigma] in phosphate-buffered solution (pbs)). Blood samples were collected immediately before and 15, 30, 45, 60, 90 and 120 min after injection. Blood glucose was determined with a handheld glucometer (Freestyle, Abbott).
Immunoblot
Frozen brown adipose tissue (BAT) was homogenized in RIPA lysis buffer containing PMSF, sodium orthovanadate, protease inhibitor (Santa Cruz Biotechnology) and phosphatase inhibitor cocktail (Sigma-Aldrich) using a Tissuelyser (QIAGEN). The samples were incubated at 4°C for 30 min and centrifuged for 10 min at 4°C and 10,000 × g, and the supernatants were collected in a fresh tube. Protein concentrations were measured using a Pierce BCA method protein assay kit (Thermo Fisher Scientific). 70 μg were separated by electrophoresis in denaturing conditions using 4%–15% polyacrylamide gels (Bio-Rad Laboratories), and the proteins were transferred for 1.5hours to nitrocellulose membranes (GE Healthcare). The membranes were cut, blocked in 5% nonfat dried milk (20 mm Tris, pH 7.6; 0.9% NaCl; 0.1% Tween 20), and independently incubated overnight at 4°C with rabbit antibodies against UCP-1 (1:1000, Cell Signaling, # 14670) or beta actin (1:2000, Cell Signaling, #4967) in 5% BSA (Sigma-Aldrich). Membranes were then washed and incubated with secondary antibodies (antirabbit-horseradish peroxidase coupled, 1:5,000; Cell Signaling), washed and developed by enhanced chemiluminescence (Western Lightning, Perkin Elmer) and X-ray films (Denville Scientific). Films were scanned and densitometry was assessed using ImageJ 1.48v (https://imagej.nih.gov/ij).
Plasma measurements
Triglycerides (TG) and free fatty acids (FFA) were measured via colorimetric assays using commercially available reagents following manufacturer’s instructions (TR-22421 by Thermo Scientific and 999-34691, 995-34791, 991-34891, 993-35191 by Wako Diagnostics, respectively). Insulin (Crystal Chem #90080) and C-peptide (Crystal Chem, #90050) were analyzed individually by ELISA assay in EDTA-collected plasma.
Tissue triglyceride content
frozen tissue was weighed and homogenized in 50 mM Tris·HCl buffer, pH 7.4, containing 150 mM NaCl, 1 mM EDTA, and 1 μM PMSF. TG concentration was determined by the colorimetric assay listed above.
Tissue Glycogen Content
Frozen liver tissue was homogenized in ddH2O, boiled, and the supernatant collected. Sample concentration was determined by BCA protein assay (Pierce, #23225). Glycogen concentration was determined by a colorimetric glycogen assay (Abcam, ab65620) following manufacturer’s instructions.
Norepinephrine turnover (NETO)
NETO in in spleen, gonadal and inguinal fat depots was determined as described elsewhere (Vaughan et al., 2014) with limited modifications. Briefly, lean, chow pairfed mice, received 2-week icvSHU or vehicle infusion 4 weeks after sham or sDVX. The mice were handled daily during the icv infusion period to reduce stress. Two hours after lights on, food was removed and a group of mice was immediately euthanized by decapitation, the carcasses placed in ice-cold saline solution and tissues extracted, immediately placed in liquid nitrogen and stored at −80°C. A second group received 200mg/kg, ip. of α-Methyl-DL-tyrosine (AMPT; Sigma M3281) followed by another injection 2-hours later. The mice were euthanized 2-hours afterward. Tissues were collected and stored as mentioned above.
The whole organs were homogenized in a Tissuelyser (QIAGEN) for 3 min in a perchloric acid and ascorbic acid (PCA/AA) buffer containing dihydroxybenzylamine (DHBA) (Thermo Scientific, CA) which is used as internal standard. After centrifugation the supernatant (under fat layer in the case of adipose tissues) is added to catecholamine extraction tubes (#NC0205832, Thermo Scientific, CA) and eluted in 250ul of buffer. The buffer (50ul) was loaded in a reverse phase high-performance liquid chromatography (HPLC-ECD) system comprising a Dionex Ultimate 3000 pump (Thermo Fisher) with isocratic flow, a 717plus waters Autosampler with Heater/Cooler Module (Waters, MA) and a Dionex CoulochemIII Electrochemical Detector (ESA, MA). The stationary phase was a commercially available 4.6 × 80 mm column packed with C-18 (3 μm particle size; Dionex HR-80, Fisher) which held at 35°C throughout the measurement. The area under the curve of the peaks for NE (and epinephrine) were calculated using Thermo Scientific Chromeleon 6.8 Chromatography Data System (CDS) software and compared to that from DHBA and normalized to the weight of input tissue. NETO (K) was calculated as K = k*NE0h, where (k) is the rate of NE efflux calculated as k = (log[NE]0h – log [NE]4h)/(0.434 * 4).
Gene expression analysis via RNA-Seq
RNA was extracted using RNAqueous-micro kit (Thermo Fisher from frozen samples homogenized using TissueLyser (QIAGEN). cDNA was synthetized with the iScript after DNase I treatment (Biorad). The RNA quality was determined by Bioanalyzer (Agilent, Santa Clara, CA). To isolate the polyA RNA, NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs, Ipswich, MA) was used with a total of 1 μg of good quality total RNA as input. The NEBNext Ultra Directional RNA Library Prep Kit (New England BioLabs) using QuantStudio 5 Real-Time PCR Systems (Thermo Fisher). To study differential gene expression, individually indexed and compatible libraries were proportionally pooled (≥30 million reads per sample in general) for clustering in cBot system (Illumina, San Diego, CA). Libraries at the final concentration of 16.5 pM were clustered onto a single read (SR) flow cell using Illumina TruSeq SR Cluster kit v3, and sequenced to 51 bp using TruSeq SBS kit on Illumina HiSeq system (Sharma et al., 2018).
Bromodeoxyuridine (BrdU) labeling
Four wk following sham or sDVX surgery, mice received icv infusions of SHU (1nmol/day) or vehicle for 14 d. During the infusion period, all mice received 4 g of standard chow daily 2 h before the onset of the dark phase. On Day 4 of infusion, drinking water was replaced by BRDU solution (0.8mg/ml BrdU (Sigma) + 1% D-Glucose on tap water). The solution was freshly made, light-protected, and provided daily for 7 d at precisely 2 h before lights off. Normal drinking water was returned for 3 additional d. On Day 14 of infusion, mice were euthanized and whole gonadal and inguinal fat depots were collected.
Isolation of stromal vascular cells (SVCs)
Fat depots were collected into 5mL of 1.0 mg/ml collagenase in ADB (w/o glucose and adenosine), minced with scissors and incubated in 37° water bath with shaking for 75 min, shook vigorously by hand and passed through 70um filter into 5mL of KRP buffer. Samples were spun at 300 g for 5min and processed for BRDU labeling using BD FITC BrdU Flow Kit (BD 559619). Briefly, The SVC pellet was resuspended in 200ul staining buffer and incubated with Anti-CD31 PE-Cy7 (BD Biosciences #561410), anti-CD45 PE-CF594 (BD Biosciences #562420) and Anti-CD-140a/PDGFRA, FITC (eBioscience, #11-1401-82) at RT (in dark) for 45min. Then, the samples were washed with 1ml of BD Perm/Wash buffer, spun at 300 g and the pellet resuspended and incubated for 30 min in 100ul in BD Cytofix/Cytoperm buffer. After one additional wash with BD Perm/Wash buffer, cells were resuspended and incubated in 100ul diluted DNase (30ul DNase + 70ul dPBS) at 37°C for 1hr. Then, the samples received BD BRDU antibody (BD 559619) and were incubated at 4°C overnight. Next, the cells were washed with 1ml BD Perm/Wash buffer, spun, resuspended with 250ul of staining buffer, passed through 70um filter and analyzed by flow cytometry, together with unstained and single stained controls.
Flow cytometry
Following preparation of SVC from adipose samples, antigen expression was determined using a BioRad S3e four-color cytometer/cell sorter (Hercules, CA, U.S.A.). BrdU+ leukocytes (LK) were identified as CD45+cells; BrdU+ endothelial cells (EC) as CD31+/CD45− cells; and BrdU+ adipocyte progenitors (AP) as CD45−/CD31−/CD140a+cells. Samples were analyzed using FlowJo software (Ashland, OR, U.S.A.).
QUANTIFICATION AND STATISTICAL ANALYSES
Data are presented as mean ± SEM. Number of replicates is indicated in the figure legends. Analyses were performed using GraphPad Prism, version 8 (GraphPad Software, Inc., San Diego, CA). t tests were used for comparison of two groups. Otherwise, 2 or 3-way ANOVA, followed by Sidak or Tukey’s multiple comparison tests were applied as indicated. Correlation analysis assumed Gaussian distributions and were computed as Pearson correlation coefficients. For linear regression analysis, datasets were fitted using the least square method following the equation (Y = Y Intercept + Slope*X) and compared using the extra sum of squares F-test. p < 0.05 was considered significant.
Bioinformatics RNA-seq data analysis: Sequence reads were aligned to the reference genome using the TopHat aligner (Trapnell et al., 2009), and reads aligning to each known transcript were counted using Bioconductor packages for next-generation sequencing data analysis (Huber et al., 2015). The differential expression analysis between different sample types was performed using the negative binomial statistical model of read counts as implemented in the edgeR Bioconductor package (Anders et al., 2013). Overrepresentation Enrichment Analysis (ORA) of significantly regulated pathways (Wikipathway) was performed using Webgestlat.org (Wang et al., 2017).
DATA SOFTWARE AND AVAILABILITY
The RNA-seq data reported here have been deposited at GEO (https://www.ncbi.nlm.nih.gov/geo/; accession number #GSE128383).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD-140a/PDGFRA, FITC | Thermo Fisher Scientific | Cat# 11-1401-82; RRID:AB_2572476 |
| Anti-CD31 PE-Cy7 | BD Biosciences | Cat# 561410; RRID:AB_10612003 |
| Anti-CD45 PE-CF594 | BD Biosciences | Cat# 562420; RRID:AB_11154401 |
| Anti-FluoroGold | Fluorochrome | N/A |
| Anti-Rabbit IgG | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Goat anti-rabbit IgG, Cy3 | Thermo Fischer Scientific | Cat# A10520; RRID:AB_2534029 |
| Rabbit B-Actin | Cell Signaling Technology | Cat# 4967; RRID:AB_330288 |
| Rabbit monoclonal UCP-1 | Cell Signaling Technology | Cat# 14670; RRID:AB_2687530 |
| Bacterial and Virus Strains | ||
| Biological Samples | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 20kDa-PEG-Insulin | This paper | N/A |
| AMPT | Sigma | M3281 |
| Ascorbic Acid | Sigma | A5960 |
| BRDU | Sigma | B5002 |
| BSA | Sigma | A7030 |
| Buprenex | Reckitt Benckiser | 1249607571 |
| CCK-8 | Bachem | H2080.0005 |
| Collagenase | Sigma | 101035686001 |
| DHBA | Thermo Scientific | NC0415329 |
| Fluoro-Gold | Fluorochrome | N/A |
| Glucose | Sigma | G5767 |
| Isoflourane | Patterson Vet Supply | 07-893-2374 |
| Ketamine | Vedco | VINV-KETA-0VED |
| Meloxicam | Norbrook | 10762061 |
| Perchloric Acid | Sigma | 311421 |
| Phosphatase Inhibitor Cocktail | Sigma | P0044 |
| PMSF | Sigma | P7626 |
| SHU9119 | Bachem | H-3952.0001 |
| Western Lightning (chemiluminescence) | Perkin Elmer | NEL103001EA |
| Xylazine | AKORN | 59399011020 |
| Critical Commercial Assays | ||
| BCA Protein Assay | Pierce | 23225 |
| BD FITC BRDU Flow kit | BD Bioscience | 559619 |
| C-peptide Elisa | Crystal Chem | 90050 |
| Catecholamine Extraction Kit | Thermo Scientific | 450141 |
| Free Fatty Acid Assay | Wako | 999-34691 |
| Free Fatty Acid Assay | Wako | 995-34791 |
| Free Fatty Acid Assay | Wako | 991-34891 |
| Free Fatty Acid Assay | Wako | 993-35191 |
| Glycogen Assay | Abcam | ab65620 |
| Insulin Elisa | Crystal Chem | 90080 |
| RIPA Lysis Buffer System | Santa Cruz | sc-24948 |
| RNAqueous Micro Kit | Ambion | AM1931 |
| Triglyceride Assay | Thermo Scientific | TR-22421 |
| Deposited Data | ||
| RNA sequencing data | GEO | GSE128383 |
| Experimental Models: Organisms/Strains | ||
| Mouse: DIO: C57bl6 | Jackson Laboratories | JAX:000664 |
| Mouse: LoxTbMc4r | Jackson Laboratories | JAX:006414 |
| Rats: Wistar | Harlan | 001 |
| Software and Algorithms | ||
| Chromeleon Chromatography Data System | Thermo Scientific | N/A |
| FlowJo Software | FlowJo | N/A |
| GraphPad Prism 8.0 | Graphpad | N/A |
| ImageJ 1.48v | NIH | N/A |
| Other | ||
| Brain Infusion Kit 3 | Alzet | 0008851 |
| Diet: 11% LFD | Research Diets | D12329 |
| Diet: 58% HFD | Research Diets | D12331 |
| Diet: Chow | Envigo | Teklad LM-485 |
| Diet: Ensure Liquid | Abbott | N/A |
| Freestyle Lite Glucometer | Abbott | N/A |
| Indirect Calorimetry System | TSE Systems | N/A |
| NMR | EchoMRI | N/A |
| Osmotic Minipump 1002 | Alzet | 0004317 |
| Osmotic Minipump 2001 | Alzet | 0000292 |
| Osmotic Minipump 2002 | Alzet | 0000296 |
| S3e Four Color Cytometer/Cell Sorter | Biorad | N/A |
| Stereotax | David Kopf Instr. | N/A |
| Tissuelyser | Quiagen | N/A |
Highlights.
Vagal signals contribute to obesity due to Mc4r deficiency or high-fat feeding
A brain-melanocortin-vagus axis regulates fat mass independently of calorie intake
Vagal signals promote lipogenesis and cell proliferation in white adipose tissue
ACKNOWLEDGMENTS
This work was supported by funds from the University of Cincinnati College of Medicine (F102150) and NIDDK (DK077975) to D.P.-T.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.089.
DECLARATION OF INTERESTS
R.D. is an employee of Novo Nordisk. D.P.-T. maintains research collaborations and receives funding from Novo Nordisk and Cohbar, Inc.
REFERENCES
- Almundarij TI, Smyers ME, Spriggs A, Heemstra LA, Beltz L, Dyne E, Ridenour C, and Novak CM (2016). Physical activity, energy expenditure, and defense of body weight in melanocortin 4 receptor-deficient male rats. Sci. Rep 6, 37435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, and Robinson MD (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- Apovian CM, Shah SN, Wolfe BM, Ikramuddin S, Miller CJ, Tweden KS, Billington CJ, and Shikora SA (2017). Two-year outcomes of vagal nerve blocking (vBloc) for the treatment of obesity in the ReCharge Trial. Obes. Surg 27, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, and Lowell BB (2002). αAR signaling required for diet-induced thermogenesis and obesity resistance. Science 297, 843–845. [DOI] [PubMed] [Google Scholar]
- Bartness TJ, Liu Y, Shrestha YB, and Ryu V (2014). Neural innervation of white adipose tissue and the control of lipolysis. Front. Neuroendocrinol 35, 473–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergenstal RM, Rosenstock J, Arakaki RF, Prince MJ, Qu Y, Sinha VP, Howey DC, and Jacober SJ (2012). A randomized, controlled study of once-daily LY2605541, a novel long-acting basal insulin, versus insulin glargine in basal insulin-treated patients with type 2 diabetes. Diabetes Care 35, 2140–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR, and Powley TL (1987). Characteristics of gastricand pancreatic responses to vagal stimulation with varied frequencies: evidence for different fiber calibers? J. Auton. Nerv. Syst 19, 77–84. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, and Powley TL (1993). Characterization of vagal innervation to the rat celiac, suprarenal and mesenteric ganglia. J. Auton. Nerv. Syst 42, 153–169. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Niijima A, Sauter JF, and Jeanrenaud B (1983). Evidence for a role of the gastric, coeliac and hepatic branches in vagally stimulated insulin secretion in the rat. J. Auton. Nerv. Syst 7, 97–110. [DOI] [PubMed] [Google Scholar]
- Brito MN, Brito NA, Baro DJ, Song CK, and Bartness TJ (2007). Differential activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation. Endocrinology 148, 5339–5347. [DOI] [PubMed] [Google Scholar]
- Cavalcanti-de-Albuquerque JP, Bober J, Zimmer MR, and Dietrich MO (2019). Regulation of substrate utilization and adiposity by Agrp neurons. Nat. Commun 10, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, et al. (2015). RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J. Clin. Endocrinol. Metab 100, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvera S, and Gealekman O (2014). Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim. Biophys. Acta 1842, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crewe C, An YA, and Scherer PE (2017). The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest 127, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezfuli G, Gillis RA, Tatge JE, Duncan KR, Dretchen KL, Jackson PG, Verbalis JG, and Sahibzada N (2018). Subdiaphragmatic vagotomy with pyloroplasty ameliorates the obesity caused by genetic deletion of the melanocortin 4 receptor in the mouse. Front. Neurosci 12, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, Zarebidaki E, Adams AC, Kharitonenkov A, Flier JS, et al. (2015). Central fibroblast growth factor 21 browns white fat via sympathetic action in male mice. Endocrinology 156, 2470–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, and Cone RD (2000). The central melanocortin system can directly regulate serum insulin levels. Endocrinology 141, 3072–3079. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, and O’Rahilly S (2000). Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Invest 106, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, and O’Rahilly S (2003). Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med 348, 1085–1095. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt H, Pedersen TA, Fledelius C, Olsen GS, and Hellerstein M (2013). Adipose weight gain during chronic insulin treatment of mice results from changes in lipid storage without affecting de novo synthesis of palmitate. PLoS ONE 8, e76060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgani J, and Ravussin E (2008). Energy metabolism, fuel selection and body weight regulation. Int. J. Obes 32 (Suppl 7), S109–S119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfield AS, Li C, Madara JC, Shah BP, Webber E, Steger JS, Campbell JN, Gavrilova O, Lee CE, Olson DP, et al. (2015). A neural basis for melanocortin-4 receptor-regulated appetite. Nat. Neurosci 18, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garretson JT, Szymanski LA, Schwartz GJ, Xue B, Ryu V, and Bartness TJ (2016). Lipolysis sensation by white fat afferent nerves triggers brown fat thermogenesis. Mol. Metab 5, 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavini CK, Jones WC 2nd, and Novak CM (2016). Ventromedial hypothalamic melanocortin receptor activation: regulation of activity energy expenditure and skeletal muscle thermogenesis. J. Physiol 594, 5285–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano A, Song CK, Bowers RR, Ehlen JC, Frontini A, Cinti S, and Bartness TJ (2006). White adipose tissue lacks significant vagal innervation and immunohistochemical evidence of parasympathetic innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol 291, R1243–R1255. [DOI] [PubMed] [Google Scholar]
- Girardet C, and Butler AA (2014). Neural melanocortin receptors in obesity and related metabolic disorders. Biochim. Biophys. Acta 1842, 482–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Pedersen DJ, Henriques F, Bedard AH, Henchey E, Kelly M, Morgan DA, Rahmouni K, and Czech MP (2018). Neuronal modulation of brown adipose activity through perturbation of white adipocyte lipogenesis. Mol. Metab 16, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Tellez LA, Perkins MH, Perez IO, Qu T, Ferreira J, Ferreira TL, Quinn D, Liu ZW, Gao XB, et al. (2018). A neural circuit for gutinduced reward. Cell 175, 665–678.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner KM, Piechowski CL, Muller A, Ottaway N, Sisley S, Smiley DL, Habegger KM, Pfluger PT, Dimarchi R, Biebermann H, et al. (2014). Both acyl and des-acyl ghrelin regulate adiposity and glucose metabolism via central nervous system ghrelin receptors. Diabetes 63, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggard N, Rayner DV, Johnston SL, and Speakman JR (2004). Peripherally administered [Nle4,D-Phe7]-alpha-melanocyte stimulating hormone increases resting metabolic rate, while peripheral agouti-related protein has no effect, in wild type C57BL/6 and ob/ob mice. J. Mol. Endocrinol 33, 693–703. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikramuddin S, Blackstone RP, Brancatisano A, Toouli J, Shah SN, Wolfe BM, Fujioka K, Maher JW, Swain J, Que FG, et al. (2014). Effect of reversible intermittent intra-abdominal vagal nerve blockade on morbid obesity: the ReCharge randomized clinical trial. JAMA 312, 915–922. [DOI] [PubMed] [Google Scholar]
- Jeffery E, Church CD, Holtrup B, Colman L, and Rodeheffer MS (2015). Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat. Cell Biol 17, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Ding X, Cao Y, Wang H, and Zeng W (2017). Dense intra-adipose sympathetic arborizations are essential for cold-induced beiging of mouse white adipose tissue. Cell Metab. 26, 686–692.e3. [DOI] [PubMed] [Google Scholar]
- Jonsson L, Skarphedinsson JO, Skuladottir GV, Atlason PT, Eiriksdottir VH, Franzson L, and SchiöD6th HB (2001). Melanocortin receptor agonist transiently increases oxygen consumption in rats. Neuroreport 12, 3703–3708. [DOI] [PubMed] [Google Scholar]
- Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, and Culler MD (2013). Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes 62, 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HH, Park MI, Lee SH, Hwang HY, Kim SE, Park SJ, and Moon W (2012). Effects of vagus nerve preservation and vagotomy on peptide YY and body weight after subtotal gastrectomy. World J. Gastroenterol 18, 4044–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarer M, Arnold M, Günther L, Winter C, Langhans W, and Meyer U (2014). Gut vagal afferents differentially modulate innate anxiety and learned fear. J. Neurosci 34, 7067–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijman S, Boon MR, Parlevliet ET, Geerling JJ, van de Pol V, Romijn JA, Havekes LM, Meurs I, and Rensen PC (2014). Inhibition of the central melanocortin system decreases brown adipose tissue activity. J. Lipid Res 55, 2022–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kral JG (1979). Vagotomy as a treatment for morbid obesity. Surg. Clin. North Am 59, 1131–1138. [DOI] [PubMed] [Google Scholar]
- Kral JG (1980). Effects of truncal vagotomy on body weight and hyperinsulinemia in morbid obesity. Am. J. Clin. Nutr 33 (2, Suppl), 416–419. [DOI] [PubMed] [Google Scholar]
- Kral JG, Görtz L, Hermansson G, and Wallin GS (1993). Gastroplasty for obesity: long-term weight loss improved by vagotomy. World J. Surg 17, 75–78, discussion 79. [DOI] [PubMed] [Google Scholar]
- Le Lay S, Krief S, Farnier C, Lefrère I, Le Liepvre X, Bazin R, Ferré P, and Dugail I (2001). Cholesterol, a cell size-dependent signal that regulates glucose metabolism and gene expression in adipocytes. J. Biol. Chem 276, 16904–16910. [DOI] [PubMed] [Google Scholar]
- Martinelli CE, Keogh JM, Greenfield JR, Henning E, van der Klaauw AA, Blackwood A, O’Rahilly S, Roelfsema F, Camacho-Hübner C, Pijl H, and Farooqi IS (2011). Obesity due to melanocortin 4 receptor (MC4R) deficiency is associated with increased linear growth and final height, fasting hyperinsulinemia, and incompletely suppressed growth hormone secretion. J. Clin. Endocrinol. Metab 96, E181–E188. [DOI] [PubMed] [Google Scholar]
- Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, Botezelli JD, Asadi A, Hoffman BG, Kieffer TJ, et al. (2012). Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 16, 723–737. [DOI] [PubMed] [Google Scholar]
- Miyato H, Kitayama J, Hidemura A, Ishigami H, Kaisaki S, and Nagawa H (2012a). Vagus nerve preservation selectively restores visceral fat volume in patients with early gastric cancer who underwent gastrectomy. J. Surg. Res 173, 60–67. [DOI] [PubMed] [Google Scholar]
- Miyato H, Kitayama J, and Nagawa H (2012b). Vagus nerve preservation results in visceral fat maintenance after distal gastrectomy. Hepatogastroenterology 59, 1299–1301. [DOI] [PubMed] [Google Scholar]
- Nogueiras R, Wiedmer P, Perez-Tilve D, Veyrat-Durebex C, Keogh JM, Sutton GM, Pfluger PT, Castaneda TR, Neschen S, Hofmann SM, et al. (2007). The central melanocortin system directly controls peripheral lipid metabolism. J. Clin. Invest 117, 3475–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgren R, and Smith GP (1994). A method for selective section of vagal afferent or efferent axons in the rat. Am. J. Physiol 267, R1136–R1141. [DOI] [PubMed] [Google Scholar]
- Perez-Tilve D, Hofmann SM, Basford J, Nogueiras R, Pfluger PT, Patterson JT, Grant E, Wilson-Perez HE, Granholm NA, Arnold M, et al. (2010). Melanocortin signaling in the CNS directly regulates circulating cholesterol. Nat. Neurosci 13, 877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prechtl JC, and Powley TL (1990). The fiber composition of the abdominal vagus of the rat. Anat. Embryol. (Berl.) 181, 101–115. [DOI] [PubMed] [Google Scholar]
- Ravussin Y, Gutman R, LeDuc CA, and Leibel RL (2013). Estimating energy expenditure in mice using an energy balance technique. Int. J. Obes 37, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, et al. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Zhang X, and Huang S (2018). Integrate imaging flow cytometry and transcriptomic profiling to evaluate altered endocytic CD1d trafficking. J. Vis. Exp Published online October 29, 2018. 10.3791/57528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small CJ, Liu YL, Stanley SA, Connoley IP, Kennedy A, Stock MJ, and Bloom SR (2003). Chronic CNS administration of Agouti-related protein (Agrp) reduces energy expenditure. Int. J. Obes. Relat. Metab. Disord 27, 530–533. [DOI] [PubMed] [Google Scholar]
- Smith DK, Sarfeh J, and Howard L (1983). Truncal vagotomy in hypothalamic obesity. Lancet 1, 1330–1331. [DOI] [PubMed] [Google Scholar]
- Song CK, Jackson RM, Harris RB, Richard D, and Bartness TJ (2005). Melanocortin-4 receptor mRNA is expressed in sympathetic nervous system outflow neurons to white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol 289, R1467–R1476. [DOI] [PubMed] [Google Scholar]
- Song CK, Vaughan CH, Keen-Rhinehart E, Harris RB, Richard D, and Bartness TJ (2008). Melanocortin-4 receptor mRNA expressed in sympathetic outflow neurons to brown adipose tissue: neuroanatomical and functional evidence. Am. J. Physiol. Regul. Integr. Comp. Physiol 295, R417–R428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton GM, Trevaskis JL, Hulver MW, McMillan RP, Markward NJ, Babin MJ, Meyer EA, and Butler AA (2006). Diet-genotype interactions in the development of the obese, insulin-resistant phenotype of C57BL/6J mice lacking melanocortin-3 or -4 receptors. Endocrinology 147, 2183–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kamei Y, and Ezaki O (2005). Mest/Peg1 imprinted gene enlarges adipocytes and is a marker of adipocyte size. Am. J. Physiol. Endocrinol. Metab 288, E117–E124. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CH, Zarebidaki E, Ehlen JC, and Bartness TJ (2014). Analysis and measurement of the sympathetic and sensory innervation of white and brown adipose tissue. Methods Enzymol. 537, 199–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen GH, Bouvy ND, Leenen L, Rijkers K, Cornips E, Majoie M, Brans B, and van Marken Lichtenbelt WD (2013). Vagus nerve stimulation increases energy expenditure: relation to brown adipose tissue activity. PLoS ONE 8, e77221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss-Andreae A, Murphy JG, Ellacott KL, Stuart RC, Nillni EA, Cone RD, and Fan W (2007). Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology 148, 1550–1560. [DOI] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, and Scherer PE (2013). Tracking adipo-genesis during white adipose tissue development, expansion and regeneration. Nat. Med 19, 1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Vasaikar S, Shi Z, Greer M, and Zhang B (2017). WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45 (W1), W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weide K, Christ N, Moar KM, Arens J, Hinney A, Mercer JG, Eiden S, and Schmidt I (2003). Hyperphagia, not hypometabolism, causes early onset obesity in melanocortin-4 receptor knockout mice. Physiol. Genomics 13, 47–56. [DOI] [PubMed] [Google Scholar]
- Willemze RA, Luyer MD, Buurman WA, and de Jonge WJ (2015). Neural reflex pathways in intestinal inflammation: hypotheses to viable therapy. Nat. Rev. Gastroenterol. Hepatol 12, 353–362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.