

Abstract
The HIV-1 capsid (CA) protein plays essential roles in both early and late stages of HIV-1 replication and is considered an important, clinically unexploited therapeutic target. As such, small drug-like molecules that inhibit this critical HIV-1 protein have become a priority for several groups. Therefore, in this study we explore small molecule targeting of the CA protein, and in particular a very attractive inter-protomer pocket. We report the design, parallel synthesis, and anti-HIV-1 activity evaluation of a series of novel phenylalanine derivatives as HIV-1 CA protein inhibitors synthesized via Cu(I)-catalyzed alkyne-azide 1,3-dipolar cycloaddition (CuAAC) reaction. We demonstrate robust inhibitory activity over a range of potencies against the HIV-1 NL4-3 reference strain. In particular, compound 13m exhibited the greatest potency and lowest toxicity within this new series with an EC50 value of 4.33 μM and CC50 value of >57.74 μM (SI > 13.33). These values are very similar to the lead compound PF-74 (EC50= 5.95 μM, CC50 > 70.50 μM, SI > 11.85) in our assay, despite significant structural difference. Furthermore, we demonstrate via surface plasmon resonance (SPR) binding assays that 13m interacts robustly with recombinant HIV-1 CA and exhibits antiviral activity in both the early and late stages of HIV-1 replication. Overall, the novel parallel synthesis and structure-activity relationships (SARs) identified within this study set the foundation for further rational optimization and discovery of CA-targeting compounds with improved potency.
Keywords: HIV-1, Phenylalanine derivatives, HIV-1 capsid protein, CuAAC, Surface plasmon resonance, Molecular dynamics simulation
Graphical Abstract
1. Introduction
Acquired immune deficiency syndrome (AIDS), primarily caused by human immunodeficiency virus type 1 (HIV-1), remains a global problem. Although considerable progress has been made with regards to improving the quality and length of life of patients living with HIV with the application of combinatorial antiretroviral therapy (cART) regimen, current therapies still suffer from adverse side effects and the ever-present problems of natural and emergent drug resistance[1, 2]. Accordingly, there is a continuing need for new anti-HIV drugs, especially those acting on novel targets with new scaffolds and mechanisms of action.
The HIV-1 capsid (CA) protein plays crucial roles in both early (uncoating, nuclear entry, reverse transcription, integration, etc.) and late stages (maturation and assembly) of the HIV-1 life cycle [3, 4]. In the early stage, the fusion of the virus with target cell membranes triggers the disassembly or uncoating of CA protein, which is tightly associated with the reverse transcription and synthesis of viral cDNA [5, 6]. Although viral uncoating is not completely understood, the correct spatiotemporal disassembly of the capsid during the initial stage of infection is necessary for the completion of reverse transcription and, hence, infectivity. Moreover, in the late stage of HIV-1 replication, CA protein assembly and maturation are essential for the formation and release of infectious viral particles. Extensive mutational analysis has been performed on CA; analysis that has revealed an extreme genetic fragility, with most changes to the capsid being detrimental for the above processes and the fitness of the virus [7, 8]. Therefore, the HIV-1 CA protein has emerged as an attractive antiviral target for the development of small-molecular inhibitors [9-12].
To date, a series of structurally diverse small-molecular compounds have been identified as HIV-1 CA inhibitors [13-20]. Among them, PF-74 (Fig.1a) is the most widely studied small molecule targeting the HIV-1 CA protein (Fig.1a). PF-74 was developed by Pfizer, USA, and has been demonstrated to inhibit the replication of the majority of isolates at low to sub-micromolar concentrations [11, 13, 21-23]. However, the relatively low inhibitory activity and poor drug-like qualities, most critically extremely poor metabolic stability [13] indicate the urgent needs for further structural optimization of PF-74. Recently, the binding mode of PF-74 in the inter-protomer binding pocket in the CA protein hexamer has been described by several groups in crystallographic studies using native and disulphide stabilized constructs [24-26]. The availability of these structures greatly facilitates the structure-based design of novel compounds based upon the PF-74 scaffold. PF-74 consists of a phenylalanine core (red part, Fig.1a), an indole substituent (pink part, Fig.1a) and a linker between them (blue part, Fig.1a). The interprotomer pocket in which PF-74 binds is defined by four α-helices (H3, H4, H5, and H7) in the NTD interface of one protomer; the phenylalanine core forms interactions with Asn57, with Ile73, Ala105, Tyr130 and Thr107 (Fig.lb). The methylindole ring of PF-74 is oriented towards the CTD interface of the adjacent subunit within the same hexametric ring and interacts with Arg173, Gln63, and Lys182 (Fig.1b) [24-26]. Of particular note, the plasticity of the CTD-NTD interface provides additional space for further modification of the indole substituent and linker of PF-74, as exemplified by compounds I and II with improved potency (Fig.1). Interestingly, in these compounds, the methoxy moiety was frequently observed in the aniline substituent of phenylalanine core (Fig.1) [27, 28].
Fig.1.

The design of novel phenylalanine derivatives as HIV-1 CA inhibitors. (a) Structure of PF-74; (b) The binding mode of PF-74 in the binding site of CA protein hexamer. Red dashed lines indicate H-bond interactions.
Given the attractiveness of the CA protein as a target, and the wealth of structural and mechanistic information available, we chose PF-74 to focus our optimization efforts upon. Specifically, in this study, we chose to explore modification of methylindole moiety as this region has been demonstrated to be quite tolerant to changes [13]. However, unlike previous studies, we demonstrate the successful implementation of a parallel synthesis strategy using the facile Cu(I)-catalyzed alkyne-azide 1,3-dipolar cycloaddition (CuAAC) reaction (Fig.1) [29-32]. This strategy, installs a 1,2,3-triazole unit as a linker in the newly designed compounds, further diversifying the compounds and, most likely, improving solubility (Fig. 1). We chose not to modulate the phenylalanine core region of PF-74 and its reported analogues but chose to include the methoxy-bearing aniline substituent present within the high potency analogues (PF-74).
Therefore, in this novel study, we report the parallel synthesis and antiviral activity of 20 newly designed phenylalanine derivatives. Furthermore, we demonstrate via surface plasmon resonance (SPR) binding assays that representative compounds from this study interact robustly with recombinant HIV-1 CA and exhibit antiviral activity in both the early and late stages of HIV-1 replication. Overall, the novel parallel synthesis and structure-activity relationships (SARs) identified within this study set the foundation for further rational optimization and discovery of CA-targeting compounds with improved potency.
2. Results and discussion
2.1. Chemistry
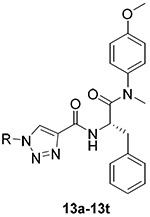
The straightforward synthetic route towards the target compounds was depicted in Scheme 1. (Tert-butoxycarbonyl)-L-phenylalanine (1) was selected as the starting material. It was reacted with 4-methoxy-N-methylaniline, followed by removing the Boc protection to produce the free amine (3). Then, 3 was treated with propiolic acid to yield the key intermediate 4. Meanwhile, the substituted benzyl bromide (5) or chloride (9) were converted to azide 6 or 10 by nucleophilic substitution with NaN3, respectively. Further oxidation of 10 in the presence of 3-chloroperbenzoic acid generated compounds 11 and 12. Substituted phenylboric acids 7 were reacted with NaN3 by the catalytic CuSO4.5H2O to provide the corresponding azides 8. Finally, the target products (13) were obtained by CuAAC reaction between the azide substituents (6, 8, 11 or 12) and key intermediate 4. The newly synthesized compounds were characterized by MS and NMR spectra.
Scheme 1.

The synthetic route of phenylalanine derivatives (13a-13t). Reagents and Conditions. (i) 4-methoxy-N-methylaniline, PyBop, DIEA, CH2Cl2, r.t., 8-9h; (ii): CF3COOH; CH2Cl2, r.t., 6-7h; (iii) propiolic acid, HATU, DIEA, CH2Cl2, r.t., 12h; (iv): NaN3, DMF, r.t., 12-13h; (v): NaN3, CuSO4·5H2O, CH3OH; (vi): 3-Chloroperbenzoic acid, CH2Cl2, 0°C,1h; (vii): 3-chloroperbenzoic acid, CH2Cl2, 15h at r.t.; (viii): CuSO4·5H2O, L-ascorbic acid sodium salt, THF/H2O (V:V = 1:1), 30-60°C, 4-6h.
2.2. Anti-HIV activity evaluation in MT-4 cells
Having demonstrated the utility of our new parallel synthesis scheme, we next sought to test the newly synthesized compounds for in vitro anti-HIV-1 activity in a multicycle assay using fully infectious HIV-1 NL4-3 virus and MT-4 target cells. PF-74 was included as an in-line control to allow for direct comparison with the new compounds. Finally, the toxicity of PF-74 and the new compounds towards the MT-4 cells was also assessed. Table 1 shows the anti-HIV potency (EC50, as measured by a luciferase gene expression assay [33]), cytotoxicity (CC50) as well as selectivity index (SI, the ratio of CC50/EC50) for each of the compounds and control.
Table 1.
Anti-HIV-1 activity and cytotoxicity of the novel phenylalanine derivatives in MT-4 cells infected with the HIV-1 NL4-3 virus.
 |
||||
|---|---|---|---|---|
| Compounds | R | EC50a (μM) | CC50b (μM) | SI |
| 13a | 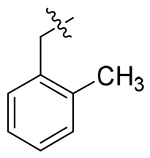 |
8.00 ± 2.37 | > 20.68 | > 2.58 |
| 13b | 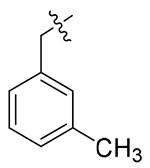 |
NAc | NAc | NDd |
| 13c | 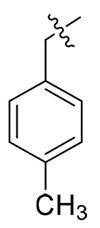 |
NAc | NAc | NDd |
| 13d | 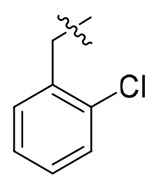 |
> 19.84 | > 19.84 | NDd |
| 13e | 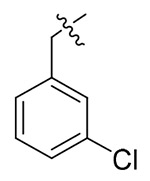 |
NAc | NAc | NDd |
| 13f | 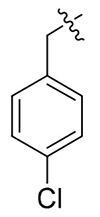 |
> 19.84 | > 19.84 | NDd |
| 13g | 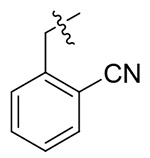 |
> 21.53 | > 21.53 | NDd |
| 13h | 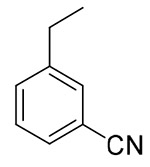 |
9.11 ± 2.88 | > 21.53 | > 2.36 |
| 13i | 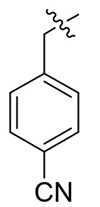 |
8.98 ± 3.10 | > 21.53 | > 2.40 |
| 13j | 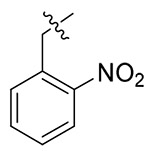 |
10.16 ± 2.22 | > 19.43 | > 1.91 |
| 13k | 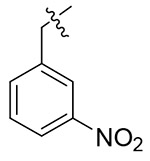 |
NAc | NAc | NDd |
| 13l | 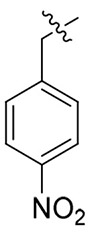 |
> 19.43 | > 19.43 | NDd |
| 13m | 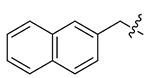 |
4.33 ± 0.83 | > 57.74 | > 13.33 |
| 13n | 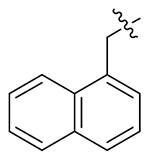 |
> 19.25 | > 19.25 | NDd |
| 13o | 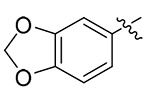 |
14.93 ± 3.94 | > 20.02 | > 1.34 |
| 13p | 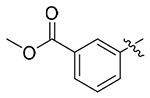 |
6.91 ± 2.43 | > 19.47 | > 2.82 |
| 13q | 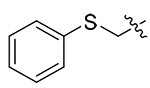 |
NAc | NAc | NDd |
| 13r | 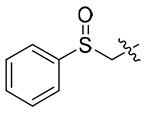 |
6.65 ± 1.47 | > 19.32 | > 2.91 |
| 13s | 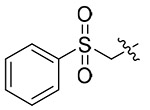 |
NAc | > 18.74 | NDd |
| 13t | 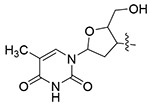 |
10.45 ± 3.08 | > 16.56 | > 1.59 |
| PF-74 | - | 5.95 ± 1.32 | > 70.50 | > 11.85 |
EC50: the concentration of the compound required to achieve 50% protection of MT-4 cells against HIV-1-induced cytopathic effect, determined in at least triplicate against HIV-1 in MT-4 cells; values are the mean ± SD of at least two parallel tests.
CC50: the concentration of the compound required to reduce the viability of uninfected cells by 50%, determined in at least triplicate against HIV-1 in MT-4 cells; values were averaged from at least four independent experiments.
NA: No anti-HIV-1 activity or cytotoxicity at the test concentration.
ND: Not determined.
As shown in the Table 1, it can be observed that some of newly synthesized compounds exhibited from moderate to excellent activity against HIV-1 NL4-3 virus with EC50 values ranging from 14.93 μM to 4.33 μM and SI values between 1.34 and 13.33. Among them, 13m (EC50 = 4.33 ± 0.83 μM, CC50> 57.74 μM), 13p (EC50 = 6.91 ± 2.43 μM, CC50 >19.47 μM) and 13r (EC50 = 6.65 ± 1.47 μM, CC50> 19.32 μM) turned out to be the most potent HIV-1 inhibitors, which were equipotent with PF-74 (EC50 = 5.95 ± 1.32 μM, CC50 > 70.50 μM). In particular, 13m also exhibited an equivalent SI value (13.33) comparable to PF-74 (SI: 11.85).
The exploratory SARs analysis was derived from the results. Firstly, our attention was focused on the SAR of the substituted benzyls (13a-13n). When making a comparison between 13m and 13n with a terminal β- and α-substituted naphthalene group in their structures, respectively, 13m is far more active than 13n in inhibiting HIV-1 NL4-3, indicating that β-substituted naphthalene might form a better interaction with the CTD of CA protein. Similarly, variation of the position of identical substituent in the benzyl (from the ortho-position to the para-position) resulted in different inhibitory activity (13a (2-Me) > 13b (3-Me) and 13c (4-Me); 13i (4-CN) ≈ 13h (3-CN) > 13g (2-CN); 13j (2-NO2) > 13k (3-NO2) and 131 (4-NO2)), with the exception of compounds that were endowed with chlorinated benzyl (13d/13e/13f). These results indicated that benzyl with different positions of the same substituent would have a great influence on the inhibitory activity against HIV-1 NL4-3 virus. In addition, it was noteworthy that different substituents at the same position demonstrated significant effects on the activity results, as suggested by comparison of 13a/13d/13g/13j, 13b/13e/13h/13k and 13c/13f/13i/13l.
Next, we turned our attention to the SAR of synthesized compounds with substituted benzenes (13o/13p). Detailed comparison of the activities of 13o with 13p suggested that one more flexible substituent at the benzene (13p) might strengthen the interaction with the CTD binding site compared to substituent bearing with rigid construction (13o). Further optimization of phenylalanine derivatives was focused on incorporating a group including S atom or its oxidation groups to the linker. Comparing the anti-HIV-1 (NL4-3) activity between 13q, 13r and 13s reveals that had certain influence on the antiviral activity (Table 1). Among compounds above, 13r exhibited the best inhibitory activity with the EC50 value of 6.65 ± 1.47 μM, which is comparable to the reference agent PF-74.
Finally, the commercially available zidovudine unit was introduced as one CTD substituent, resulting in the compound 13t with moderate activity toward HIV-1 NL4-3 (EC50 = 10.45 ±3.08 μM).
On the whole, preliminary SAR studies of these newly designed compounds revealed that the anti-HIV-1(NL4-3) activities of phenylalanine derivatives were not only sensitive to types of CTD substituents but also affected by different linkage positions. The biological evaluation results and the SAR analysis described above will be beneficial to further design of CA protein inhibitors targeting the conformationally dynamic CTD-NTD interface.
2.3. Binding to HIV-1 CA protein
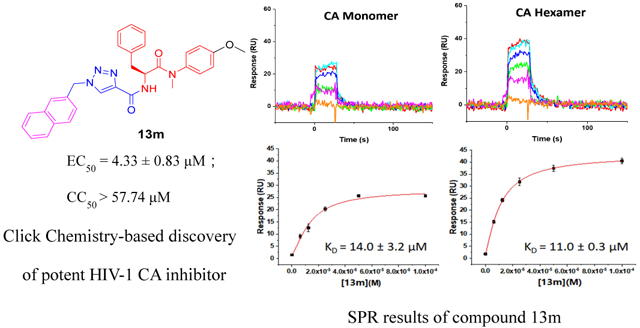
We next sought to demonstrate that the newly synthesized and designed compounds retained target specificity to the HIV-1 CA protein. We chose to perform this analysis on three of our highest potency compounds, 13m, 13p, and 13r. The successful use of surface plasmon resonance (SPR) to quantify the binding interactions of CA-targeting small molecules has been previously reported [13-16]. Therefore, we chose this methodology for our studies.
Moreover, to ascertain whether or not there was any oligomeric preference of the compounds, we chose three different available CA protein constructs; the CA NTD, monomeric CA, and hexameric CA. Again, PF-74 was utilized as the reference compound. Figure 2 shows the results of this analysis.
Fig. 2.


SPR results of compounds 13m (B), 13p (C) and 13r (D) binding to three kinds of CA proteins (NTD, monomer, and hexamer) respectively with PF-74 as the reference (A).
As depicted in Fig. 2A, compounds 13m, 13p, 13r, and PF-74 all bind to the three different CA protein constructs (NTD, monomer and hexamer). PF-74 interacts the tightest overall, with equilibrium dissociation constant (KD) values for the NTD, monomer, and hexamer, of 3.6 μM, 2.7 μM and 64.3 nM, respectively. These values are consistent with the previous affinity studies reported in the literature [13, 24, 26] and demonstrate that PF-74 has a preference for binding to the hexameric form of the CA protein. This result, again, is consistent with the structural information regarding the binding mode of PF-74 in which the molecule makes contact with two adjacent protomers in the CA hexamer. In contrast to the oligomeric preference shown by PF-74, compounds 13m, 13p, and 13r, display comparable affinities to all of the CA protein constructs used, with the exception of 13r that appears to prefer the monomeric form of CA. Moreover, compounds 13m, 13p, and 13r have a markedly different kinetic signatures to PF-74, having rapid on- and off-rates, compared to the rapid on- but relatively slow off-rate of the parental compound. Taken together, this analysis demonstrates that the novel 1,2,3-triazole-containing phenylalanine derivatives retain the target specificity of the parental PF-74 and can be classified as HIV-1 CA inhibitors. Moreover, the use of SPR to determine not only an affinity but a kinetic signature for the compounds may provide a rapid method of screening for higher potency analogues. A recent study has demonstrated that the antiviral potency of a small series of HIV-1 entry inhibitors can be best correlated with the off-rate parameter of their interaction with their protein target [34]. As such, next-generation analogues in this series could be prioritized via a rapid off-rate screening strategy.
2.5. Determination of the action stage of 13m
The observation that all of the new compounds interact with both monomeric and hexameric forms of the HIV-1 CA protein has implications for the stage at which they exert there antiviral effect. PF-74 binds to both CA monomer and hexamer and inhibits HIV-1 replication in the early, infective stages, and the late, assembly stages of the virus lifecycle [11]. We, therefore, sought to see if this dual-stage mode of action was true for our new compounds and chose to focus on 13m as the most potent representative of our derivatives. To achieve this, we utilized the modular nature of the single round infection assay, in which singly-round infective HIV-1 particles are generated recombinantly in HEK293T cells, and then used to infect U87.CD4.CCR5 cells: this allows for the separation of early and late stages of the replication cycle [35].
Effects on post-integration and assembly events were identified by producing Env pseudotyped viruses in the absence or presence of various concentrations of 13m. HIV-1B41 Env pseudotyped virus (which encodes for firefly luciferase as a reporter gene) were diluted ten-fold and used to infect U87.CD4.CCR5 target cells. Compound-induced late-stage effects were demonstrated by a decrease in infectivity in the target cells, compared to the control virus that was generated without the compound present. Compound-induced early-stage effects were observed by using virus produced in the absence of compound to infect U87.CD4.CCR5 target cells in the absence or presence of various concentrations of 13m. As can be seen in Table 2, 13m displays effects in both the post-integration, assembly and pre-integration, infective stages of the HIV-1 life-cycle. The control compound PF-74 also exhibits this dual-stage inhibition profile. These preliminary mechanism-of-action studies set the stage for more rigorous studies with these and higher potency compounds.
Table 2.
Early and late stage anti-HIV-1 activity and cytotoxicity of 13m and PF-74 using HIV-1 Env-pseudotyped virus.
| Therapeutic | ||||
|---|---|---|---|---|
| Compounds | IC50 Early Stage | IC50 Late Stage | CC50 | Index |
| (μM) | (μM) | (μM) | (CC50/IC50) | |
| PF-74 | 0.1 ± 0.02 | 8.3 ± 4.3 | 73 ± 15 | 17.3 |
| 13m | 7.0 ± 0.8 | 31 ± 11 | 54.8 ± 2 | 2.8 |
IC50: the concentration of the compound required to achieve 50% infection of HIV-1 Env-pseudotyped virus in U87.CD4.CCR5 target cells. SD of 3 parallel tests are indicated.
CC50: the concentration of the compound required to reduce the viability of treated cells by 50%. SD of 3 parallel tests are indicated.
Therapeutic Index: defined as the CC50/IC50. The IC50 used was the average of the early and late stage IC50 values.
2.5. Molecular Dynamics (MD) simulation
To better understand the activity results and find the potential binding modes of this series of molecules, the representative compound 13m was analyzed by MD simulation to find its binding to the active site of CA HIV-1 monomer. Figure 3A shows the root mean square deviation (RMSD) of the protein amino acid residues for all heavy atoms during the 1μs MD simulation. It is clear from the figure that the protein structure has deviated from the starting X-ray structure 5HGL. Furthermore, the figure shows that the protein structure could exist in different conformational ensembles due to the different RMSD values. To further investigate the deviation of the protein from the starting X-ray structure, the root mean squares fluctuation (RMSF) of residues was calculated and plotted in Figure 3B. This figure shows that most amino acid residues have deviated from the starting structure, which also indicates the presence of the protein in other conformational forms. The presence of the protein structure in different conformational forms could be accompanied with different binding modes of 13m. Accordingly, RMSD of 13m heavy atoms was calculated in reference to the first frame of the MD simulation and plotted in Figure 3C. Indeed, it is obvious from the figure that 13m is present in different structural clusters, which potentially indicate binding to the active site in different modes.
Fig. 3.


A) RMSD (all heavy atoms) of amino acid residues of CA HIV-1 monomer in reference to the first frame of the 1μs MD simulation. B) RMSF of the backbone Cα atoms for residues of CA HIV-1 monomer during the 1μs MD simulation. C) RMSD (all heavy atoms) of the bound 13m during the 1μs of the MD simulation based on the first frame of the simulation.
According to the RMSD and RMSF results, the entire trajectory has been clustered based on 13m (no fit), after alignment of protein residues, to see the binding modes of 13m in the active site. The clustering procedure yielded nine different structural clusters with two most populated. Figure 4 shows representative structures of the two most populated clusters with their corresponding expanded views for 13m binding to the active site. According to the clustering results, it is clear that CA HIV-1 monomer exists in two different conformational ensembles with RMSD between the representative structures of 6.4 Ǻ. This high RMSD value indicates that they are totally different. Inspection of the expanded views of the first and second clusters in the figure shows that in the most populated cluster 13m binds in similar way to PF-74 structure, where the core scaffold is oriented to the inside of the active site and the substituent is oriented to the outside of the active site. On the other hand, in the second most populated cluster, the core of 13m is oriented to the outside of the active site and the substituent is oriented to the inside of the active site, which is in contrast to the binding of PF-74. According to these results, the binding of 13m in two different binding modes could potentially induce the two different conformations of CA HIV-1 monomer. Furthermore, binding of 13m in two different binding modes could enhance its activity due to the increased chance of binding to the active site.
Fig. 4.

Representative structures of the first (A) and second (C) conformational clusters and their corresponding expanded views of 13m binding to the first (B) and second clusters (D). Protein part of the representative structures was illustrated as surface to show the binding site and conformations.
Interaction of 13m with CA HIV-1 monomer fixed it to the open conformation as shown by Jacques et. al in 5HGL X-ray structure[21], Figures 5 A and C show that GLN50 (in both clusters) is kept far from PRO1, HIS12, THR48 and ASN51 triad, which is important to form hydrogen bond to induce the close conformation. This could fix the hexamer to one conformation and disables its function. In the first conformational cluster 13m binds in similar mode of PF-74 but with different amino acid residues. Where, the phenyl ring of the core region could form hydrophobic interaction with LYS70 similar to PF-74. Furthermore, 13m forms hydrophobic interactions with MET66 and LEU56. It also forms aliphatic hydrogen boning with ASN74 through its methoxy group in the core region, and hydrogen bond with ASN57 at the substituent region. 13m in the other binding mode, which is different from the binding mode of PF-74, forms hydrophobic interaction with LYS70 through its substituent region in contrast to the first binding mode. Also, it forms hydrophobic interactions with MET66 and ALA64 through its core region. Interaction of 13m with CA HIV-1 monomer in different binding modes involves more amino acids, which increases the possibility of inhibiting this protein and could accounts for its high inhibitory activity.
Fig. 5.

Illustration of the open form conformation of CA HIV-1 monomer in first (A) and second (C) clusters. Binding interactions of 13m to the first (B) and second clusters (D).
3. Conclusions
In brief, taking the most studied HIV CA inhibitor PF-74 as lead compound, we designed and expeditiously synthesized a series of 1,2,3-triazole-containing phenylalanine derivatives via CuAAC reaction. Among them, 13m exhibited the best anti-HIV-1 activity (EC50 = 4.33 μM, SI > 13.33), being similar to the lead PF-74 (EC50 = 5.95 μM, SI > 11.85). Direct binding studies using SPR demonstrated that this series of phenylalanine derivatives interact with the HIV-1 CA protein, irrespective of its oligomeric status. In-line with this observation, we have demonstrated the compound 13m inhibits the replication of HIV-1 in both the early and late stages. Finally, through molecular dynamics simulation, we can conclude that 13m potentially has two different binding modes to HIV-1 CA monomer, which has implication for the precise manner of CA protein inhibition in each of the discrete stages of replication. We envisioned that the conformationally dynamic CTD-NTD interface still have ample space for further modification to form potential interaction with nearby hotpot residues. Ongoing studies regarding the further development of these novel capsid inhibitors from our lab will be reported in due course.
4. Experimental section
4.1. Chemistry
All melting points of the compounds were determined on a micro melting point apparatus and were uncorrected. 1H-NMR and 13C-NMR spectra were obtained via a Bruker Avance-400 NMR-spectrometer in DMSO or CDCl3 using TMS as internal reference. Chemical shifts were expressed in δ units (ppm) and J values were presented in hertz (Hz). Related mass spectra dates were determined by a LC Autosampler Device: Standard G1313A instrument. TLC was performed on Silica Gel GF254 for TLC and spots were visualized by irradiation with UV light (λ = 254 nm). Meanwhile, flash column chromatography was performed on column packed with Silica Gel 60 (200-300 mesh). Rotary evaporator under reduced pressure condition was used to concentrate the reaction solutions. Solvents were of reagent grade and purified with standard methods when necessary.
4.1.1. Tert-butyl(S)-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carba mate (2)
A solution of (tert-butoxycarbonyl)-L-phenylalanine (1, 8.75 mmol, 2.3 g) in 15 mL dichloromethane was added PyBop (10.9 mmol, 5.7 g) at 0°C, and the mixture stirred for 0.5 h. Subsequently, DIEA (21.87 mmol, 3.61 mL) and 4-methoxy-N-methylaniline (7.29 mmol, 1.0 g) were added to the mixture and then stirred at room temperature for another 8-9 h (monitored by TLC). The resulting mixture was evaporated under reduced pressure and the residue was initially washed by 1N HCl and extracted with ethyl acetate (3 × 20 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 50 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude intermediate 2 as yellow oil with a yield of 94%. 1H NMR (400 MHz, DMSO-d6δ 7.21 (d, J = 8.4 Hz, 3H), 7.15 (d, J = 7.1 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 6.85 – 6.75 (m, 2H), 4.15 (q, J = 5.4 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.81–2.54 (m, 2H), 1.30 (s, 9H). 13C NMR (100 MHz, DMSO) δ 172.21, 158.98, 155.74, 138.53, 136.13, 129.28, 128.47, 126.70, 115.21, 78.33, 55.94, 53.53, 37.86, 37.10, 28.65. ESI-MS: m/z 385.4 (M + 1)+, C22H28N2O4 (384.2).
4.1.2. (S)-2-amino-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (3)
Trifluoroacetate (34.2 mmol, 5.0 eq) was added dropwise to intermediate 2 (2.63 g, 6.84 mmol, 1.0 eq) in 30 mL dichloromethane and stirred at room temperature for 6-7 h. Then, the resulting mixture solution was alkalized to pH = 9 with saturated sodium bicarbonate solution, and then extracted with dichloromethane (3 × 20 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield the crude product, which was purified by flash column chromatography to afford compound 3 as yellow oil with the yield of 84%. 1H NMR (400 MHz, DMSO-d4) δ 7.34–7.15 (m, 3H), 6.90 (m, 6H), 3.77 (s, 3H), 3.35 (t, J = 6.9 Hz, 1H), 3.06 (s, 3H), 2.76 (dd, J = 12.9, 6.8 Hz, 1H), 2.46 (dd, J = 12.9, 7.1 Hz, 1H). 13C NMR (100 MHz, DMSO) δ 174.96, 158.76, 139.02, 136.36, 129.51, 128.93, 128.46, 126.54, 115.03, 55.83, 53.38, 42.25, 37.45. ESI-MS: m/z 285.3 (M + 1)+, C17>H20N2O2 (284.1).
4.1.3. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)propiolamide (4)
Propiolic acid (4.22 mmol, 0.3 g) and HATU (5.28 mmol, 2.0 g) were mixed in dichloromethane and stirred in an ice bath for 1 h. Then, the intermediate 3 (6.33 mmol, 1.80 g) and DIEA (7.03 mmol, 1.16 mL) were added to the above solution slowly at 0°C. The reaction system was then stirred at room temperature for additional 12 h. The solvent was removed under reduced pressure and then 1N HCl (30 mL) was added, extracted with ethyl acetate (3 × 30 mL). The combined organic layer was washed with saturated sodium bicarbonate (3 × 50 mL). The resulting organic layer was washed with saturated salt water, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude product, which was purified by flash column chromatography to yield compound 4 as yellow oil with the yield of 38%. 1H NMR (400 MHz, DMSO-d6) δ 9.16 (d, J = 7.8 Hz, 1H), 7.17 (td, J = 7.0, 4.5 Hz, 5H), 7.00 (d, J = 8.9 Hz, 2H), 6.85 – 6.82 (m, 2H), 4.45 (ddd, J = 9.7, 7.7, 4.5 Hz, 1H), 3.79 (s, 3H), 3.12 (s, 3H), 2.91 – 2.71 (m, 2H), 2.69 (s, 1H). 13C NMR (100 MHz, DMSO) δ 170.90, 159.10, 151.91, 137.93, 135.87, 129.19, 128.63, 126.96, 115.17, 78.37, 76.80, 55.91, 52.53, 37.85, 36.86. ESI-MS: m/z 337.6 (M + 1)+, C20H20N2O3 (336.1).
4.1.4. General procedure for the synthesis of azide substituents
4.1.4.1. General procedure for the synthesis of azide substituents (6 and 10)
Benzyl bromide (5, 1 g, 1 eq) and sodium azide (NaN3, 1.5 eq) were dissolved in 10 mL N, N-dimethylformamide (DMF). The mixture was added 20 mL water after stirred at room temperature for 12-13 h, and then extracted by ethyl acetate (3 × 20 mL). The organic phase was combined, dried over anhydrous Na2SO4, filtered, and concentrated to yield the crude azide substituent 6, which was used for the next step without further purification. The abovementioned method was applied to the substituent (azidomethyl)(phenyl)sulfane (10, C7H7N3S). 1H NMR (400 MHz, DMSO-d6) δ 7.52–7.47 (m, 2H), 7.42–7.35 (m, 2H), 7.33 – 7.27 (m, 1H), 4.87 (s, 2H).
4.1.4.2. General procedure for the synthesis of azide substituents (8)
Different substituted phenylboronic acid compounds (7, 1 g, 1 eq), sodium azide (NaN3, 1.5 eq) and copper sulfate pentahydrate (CuSO4·5H2, 0.1 eq) were mixed in the 10 mL methanol and then stirred at room temperature for 8-9 h. Subsequently, the reaction solvent was removed under reduced pressure and 20 mL of water was added. Aqueous solution was extracted by ethyl acetate (3 × 20 mL), the combined phase was dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude azide substituents 8 which was used for the next step directly.
4.1.4.3. General procedure for the synthesis of azide substituents (11 and 12)
Under ice cooling, after a solution of 3-chloroperbenzoic acid (mCPBA, 580 mg, 75%, 1.26 mmol) in CH2Cl2 (1.0 mL) was added dropwise to a solution of the intermediate (azidomethyl)(phenyl)sulfane (10, 168 μL, 1.17 mmol) in dry CH2Cl2 (10 mL), the mixture was stirred at room temperature for 15 h. Then saturated sodium bicarbonate solution was poured into the reaction mixture and extracted with ethyl acetate (3 χ 50 mL). The combined organic phase was dried over anhydrous sodium sulphate and then evaporated. The residue was purified using flash chromatography with n-hexane/ethylacetate to afford azide substituent ((azidomethyl)sulfinyl)benzene (12, C7H7N3O2S). 1H NMR (400 MHz, Chloroform-d) δ 8.00 – 7.94 (m, 2H), 7.77 – 7.70 (m, 1H), 7.63 (dd, J = 8.4, 7.1 Hz, 2H), 4.31 (s, 2H, CH2).
This method was also applied to synthesize substituent ((azidomethyl)sulfinyl)benzene (11, C7H7N3OS). Compared to the synthesis of azide substituent ((azidomethyl)sulfinyl)benzene (12), the only difference of ((azidomethyl)sulfinyl)benzene (11) synthesis is that the reaction would be finished after reacting with mCPBA at 0°C merely for 1 h. 1H NMR (400 MHz, Chloroform-d) δ 7.64–7.56 (m, 2H), 7.52–7.35 (m, 3H), 4.21 (d, J = 12.0 Hz, 1H), 3.97 (d, J = 11.9 Hz, 1H)
4.1.5. General procedure for the synthesis of target compounds (13a-13t)
The key intermediate 4 (1.0 eq), azide substituents (6, 8, 10, 11 and 12, 1.1 eq), ascorbic acid sodium (0.6 eq) and CuSO4.5H2O (0.3 eq) were dissolved in the solution of tetrahydrofuran/water (v:v = 1:1). The resulting mixture was stirred at 30-60°C for 4-6 h. Then the reaction mixture was extracted with ethyl acetate (3 × 10 mL), and the combined organic phase was washed with saturated salt water (3 × 10 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give the corresponding crude target product, which was purified by flash column chromatography to afford product 13a-13t. Yield: 30%-80%.
4.1.5.1. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(2-methyl benzyl)-1H-1,2,3-triazole-4-carboxamide (13a).
White solid, yield: 70%. mp: 60-62°C. 1H NMR (400 MHz, DMSO-d6) δ 8.49 (s, 1H), 8.35 (d, J = 8.0 Hz, 1H), 7.31 – 7.12 (m, 8H), 7.08 (d, J = 7.5 Hz, 1H), 7.00 (d, J = 8.4 Hz, 2H), 6.92 – 6.82 (m, 2H), 5.66 (s, 2H), 4.66 (q, J = 7.4 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.81 (m, 2H), 2.30 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.28, 159.56, 159.07, 142.56, 137.98, 136.76, 135.93, 134.20, 130.94, 129.35, 129.24, 129.12, 128.93, 128.60, 127.04, 126.89, 126.79, 115.19, 55.92, 51.76, 51.68, 37.88, 37.27, 19.09. ESI-MS: m/z 484.5 (M + 1)+, 506.4 (M + 23)+, C28H29N5O3 (483.2).
4.1.5.2. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(3-methyl benzyl)-1H-1,2,3-triazole-4-carboxamide (13b).
Yellow solid, yield: 30%. mp: 58-60°C. 1H NMR (400 MHz, DMSO-d6) δ 8.59 (s, 1H), 8.34 (d, J = 8.0 Hz, 1H), 7.23 – 7.12 (m, 8H), 7.01 (d, J = 8.2 Hz, 3H), 6.88 (d, J = 6.8 Hz, 2H), 5.60 (s, 2H), 4.67 (q, J = 6.0 Hz, 1H), 3.81 (s, 3H), 3.12 (d, J = 4.9 Hz, 3H), 2.89 (d, J = 7.1 Hz, 2H), 2.29 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.28, 159.56, 159.08, 142.57, 137.97, 136.77, 135.93, 134.19, 130.94, 129.35, 129.23, 129.13, 128.93, 128.60, 127.03, 126.89, 126.79, 115.19, 55.91, 51.75, 51.69, 37.88, 37.31, 19.09. ESI-MS: m/z 484.5 (M + 1)+, 506.4 (M + 23)+, C28H29N5O3 (483.2).
4.1.5.3. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(4-methyl benzyl)-1H-1,2,3-triazole-4-carboxamide (13c).
Yellow solid, yield: 55%. mp: 63-65°C. 1H NMR (400 MHz, DMSO-d6) δ 8.56 (s, 1H), 8.32 (d, J = 8.0 Hz, 1H), 7.36 – 7.08 (m, 9H), 7.00 (d, J = 8.4 Hz, 2H), 6.92 – 6.83 (m, 2H), 5.58 (s, 2H), 4.66 (q, J = 7.3 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.88 (d, J = 7.1 Hz, 2H), 2.28 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.27, 159.58, 159.07, 142.67, 138.12, 137.98, 135.92, 133.09, 129.80, 129.34, 129.24, 128.60, 128.47, 126.90, 115.19, 55.91, 53.34, 51.73, 37.88, 37.27, 21.17. ESI-MS: m/z 484.5 (M + 1)+, 506.4 (M + 23)+, C28H29N5O3 (483.2).
4.1.5.4. (S)-1-(2-chlorobenzyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropa n-2-yl)-1H-1,2,3-triazole-4-carboxamide (13d).
Yellow solid, yield: 60%. mp: 162-164°C. 1H NMR (400 MHz, DMSO-d6) δ 8.56 (s, 1H), 8.38 (d, J = 8.0 Hz, 1H), 7.54 (dd, J = 7.6, 1.7 Hz, 1H), 7.40 (pd, J = 7.5, 1.7 Hz, 2H), 7.29 – 7.11 (m, 6H), 7.01 (d, J = 8.5 Hz, 2H), 6.94 – 6.75 (m, 2H), 5.76 (s, 2H), 4.67 (q, J = 7.3 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.89 (d, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.28, 159.52, 159.08, 142.50, 137.99, 135.93, 133.30, 133.12, 131.07, 130.87, 130.15, 129.35, 129.24, 128.60, 128.26, 127.45, 126.89, 115.19, 55.92, 51.78, 51.41, 37.88, 37.25. ESI-MS: m/z 504.3 (M + 1)+, 526.4 (M + 23)+, C27H26ClN5O3 (503.1).
4.1.5.5. (S)-1-(3-chlorobenzyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropa n-2-yl)-1H-1,2,3-triazole-4-carboxamide (13e).
White solid, yield: 65%. mp: 288-290°C. 1H NMR (400 MHz, DMSO-d6) δ 8.64 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 7.42 (dd, J = 6.0, 2.9 Hz, 3H), 7.37 – 7.25 (m, 1H), 7.23 – 7.10 (m, 5H), 7.01 (d, J = 8.4 Hz, 2H), 6.93 – 6.80 (m, 2H), 5.67 (s, 2H), 4.67 (q, J = 7.3 Hz, 1H), 3.80 (s, 3H), (s, 3H), 2.89 (d, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.26, 159.54, 159.07, 142.77, 138.44, 138.00, 135.92, 133.78, 131.24, 129.35, 129.25, 128.75, 128.60, 128.37, 127.27, 127.18, 126.89, 115.19, 55.92, 52.75, 51.78, 37.88, 37.24. ESI-MS: m/z 504.3 (M + 1)+, 526.3 (M + 23)+, C27H26ClN5O3 (503.1).
4.1.5.6. (S)-1-(4-chlorobenzyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropa n-2-yl)-1H-1,2,3-triazole-4-carboxamide (13f).
White solid, yield: 50%. mp: 172-174°C. 1H NMR (400 MHz, DMSO-d6) δ 8.61 (s, 1H), 8.34 (d, J = 8.1 Hz, 1H), 7.51 – 7.42 (m, 2H), 7.35 (d, J = 8.3 Hz, 2H), 7.26 – 7.09 (m, 5H), 7.01 (d, J = 8.8 Hz, 2H), 6.93 – 6.81 (m, 2H), 5.65 (s, 2H), 4.67 (q, J = 7.3 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.89 (d, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.26, 159.54, 159.08, 142.75, 137.98, 135.93, 135.08, 133.47, 130.40, 129.34, 129.28, 129.24, 128.60, 127.16, 126.89, 115.20, 55.92, 52.73, 51.75, 37.88, 37.26. ESI-MS: m/z 504.3 (M + 1)+, 526.3 (M + 23)+, C27H26ClN5O3 (503.1).
4.1.5.7. (S)-1-(2-cyanobenzyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1H-1,2,3-triazole-4-carboxamide (13g).
Light yellow solid, yield: 58%. mp: 168-170°C. 1H NMR (400 MHz, DMSO-d6) δ 8.62 (s, 1H), 8.39 (d, J = 8.0 Hz, 1H), 7.93 (dd, J = 7.7, 1.3 Hz, 1H), 7.77 – 7.70 (m, 1H), 7.61 – 7.55 (m, 1H), 7.38 (d, J = Hz, 1H), 7.17 (dt, J = 13.7, 7.5 Hz, 5H), 7.01 (d, J = 8.5 Hz, 2H), 6.91 – 6.84 (m, 2H), 5.87 (s, 2H), 4.67 (q, J = 7.2 Hz, 1H), 3.81 (s, 3H), 3.13 (s, 3H), 2.90 (d, J = 6.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.25, 159.47, 159.08, 142.63, 138.84, 137.99, 135.92, 134.36, 133.91, 129.94, 129.79, 129.35, 129.25, 128.60, 127.65, 126.89, 117.38, 115.20, 111.66, 55.93, 51.79, 51.76, 37.89, 37.24. ESI-MS: m/z 495.4 (M + 1)+, 512.5 (M + 18)+, 517.4 (M + 23)+, C28H26N6O3 (494.2).
4.1.5.8. (S)-1-(3-cyanobenzyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1H-1,2,3-triazole-4-carboxamide (13h).
White solid, yield: 62%. mp: 168-170°C. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (s, 1H), 8.35 (d, J = 8.0 Hz, 1H), – 7.81 (m, 2H), 7.62 (dt, J = 15.3, 7.8 Hz, 2H), 7.17 (dt, J = 13.8, 7.6 Hz, 5H), 7.01 (d, J = 8.5 Hz, 2H), 6.93 – 6.82 (m, 2H), 5.72 (s, 2H), 4.67 (q, J = 7.3 Hz, 1H), 3.81 (s, 3H), 3.12 (s, 3H), 2.89 (d, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.24, 159.50, 159.08, 142.82, 137.97, 137.57, 135.93, 133.45, 132.59, 132.25, 130.60, 129.35, 129.23, 128.60, 127.37, 126.89, 118.85, 115.20, 112.22, 55.92, 52.60, 51.73, 37.88, 37.31. ESI-MS: m/z 495.4 (M + 1)+, 517.5 (M + 23)+, C28H26N6O3 (494.2).
4.1.5.9. (S)-1-(4-cyanobenzyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1H-1,2,3-triazole-4-carboxamide (13i).
White solid, yield: 45%. mp: 190-192°C. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (s, 1H), 8.37 (d, J = 8.0 Hz, 1H), 7.91 – 7.81 (m, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.17 (dt, J = 13.7, 7.6 Hz, 5H), 7.07 – 6.96 (m, 2H), 6.94 – 6.82 (m, 2H), 5.77 (s, 2H), 4.68 (q, J = 7.2 Hz, 1H), 3.81 (s, 3H), 3.13 (s, 3H), 2.90 (d, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.26, 159.53, 159.08, 142.82, 141.51, 138.01, 135.92, 133.25, 129.34, 129.25, 129.20, 128.61, 127.54, 126.89, 118.97, 115.20, 111.51, 55.92, 52.90, 51.80, 37.88, 37.20. ESI-MS: m/z 495.4 (M + 1)+, 517.5 (M + 23)+, C28H26N6O3 (494.2).
4.1.5.10. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(2-nitrobe nzyl)-1H-1,2,3-triazole-4-carboxamide (13j).
Yellow solid, yield: 60%. mp: 63-65°C. 1H NMR (400 MHz, DMSO-d6 δ 8.57 (s, 1H), 8.41 (d, J = 8.0 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.76 (t, J = 7.6 Hz, 1H), 7.66 (t, J = 7.8 Hz, 1H), 7.18 (dt, J = 13.2, 6.8 Hz, 5H), 7.09 (d, J = 7.7 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.94 – 6.83 (m, 2H), 6.02 (s, 2H), 4.68 (q, J = 7.2 Hz, 1H), 3.81 (s, 3H), 3.13 (s, 3H), 2.91 (d, J = 6.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.28, 159.51, 159.09, 148.01, 142.64, 138.00, 135.94, 134.93, 130.82, 130.69, 130.26, 129.35, 129.25, 128.61, 127.89, 126.90, 125.63, 115.20, 55.92, 51.80, 50.80, 37.88, 37.25. ESI-MS: m/z 515.5 (M + 1)+, 537.4 (M + 23)+, C27H26N6O5 (514.2).
4.1.5.11. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(3-nitrobe nzyl)-1H-1,2,3-triazole-4-carboxamide (13k).
Light yellow solid, yield: 67%. mp: 61-63 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.37 (d, J = 8.0 Hz, 1H), 8.25 (t, J = 2.0 Hz, 1H), 8.22 (d, J = 8.3 Hz, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.69 (t, J = 7.9 Hz, 1H), 7.18 (dd, J = 18.4, 7.9 Hz, 5H), 7.01 (d, J = 8.4 Hz, 2H), 6.92 – 6.83 (m, 2H), 5.83 (s, 2H), 4.67 (q, J = 7.2 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.89 (d, J = 6.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.24, 159.50, 159.08, 148.37, 142.83, 138.11, 137.98, 135.93, 135.23, 130.94, 129.34, 129.24, 128.59, 127.44, 126.89, 123.73, 123.38, 115.20, 55.92, 52.51, 51.77, 37.88, 37.25. ESI-MS: m/z 515.5 (M + 1)+, 537.4 (M + 23)+, C27H26N6O5 (514.2).
4.1.5.12. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(4-nitrobe nzyl)-1H-1,2,3-triazole-4-carboxamide (13l).
Light yellow solid, yield: 57%. mp: 65-67°C. 1H NMR (400 MHz, DMSO-d6) δ 8.67 (s, 1H), 8.38 (d, J = 8.0 Hz, 1H), 8.29 – 8.20 (m, 2H), 7.54 (d, J = 8.6 Hz, 2H), 7.17 (dt, J = 13.8, 7.7 Hz, 5H), 7.01 (d, J = 8.8 Hz, 2H), 6.93 – 6.84 (m, 2H), 5.84 (s, 2H), 4.68 (q, J = 12 Hz, 1H), 3.81 (s, 3H), 3.13 (s, 3H), 2.90 (d, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.25, 159.51, 159.09, 147.77, 143.44, 142.85, 137.99, 135.93, 129.53, 129.34, 129.24, 128.61, 127.60, 126.89, 124.42, 115.21, 55.93, 52.64, 51.78, 37.88, 37.25. ESI-MS: m/z 515.4 (M + 1)+, C27H26N6O5 (514.2).
4.1.5.13. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(naphthal en-2-ylmethyl)-1H-1,2,3-triazole-4-carboxamide (13m).
White solid, yield: 65%. mp: 72-74°C. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1H), 8.34 (d, J = 8.0 Hz, 1H), 7.93 (dd, J = 8.9, 4.9 Hz, 3H), 7.87 (s, 1H), 7.54 (dd, J = 6.5, 3.1 Hz, 2H), 7.49 – 7.41 (m, 1H), 7.24 – 7.12 (m, 5H), 7.00 (d, J = 8.4 Hz, 2H), 6.94 – 6.82 (m, 2H), 5.82 (s, 2H), 4.67 (q, J = 7.4 Hz, 1H), 3.80 (s, 3H), 3.12 (s, 3H), 2.88 (d, J = 7.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.26, 159.57, 159.08, 142.74, 137.97, 135.93, 133.57, 133.21, 133.01, 129.35, 129.23, 129.05, 128.60, 128.31, 128.09, 127.46, 127.19, 127.07, 126.98, 126.88, 126.11, 115.20, 55.91, 53.73, 51.73, 37.88, 37.31. ESI-MS: m/z 520.4 (M + 1)+, 542.4 (M + 23)+, C31H29N5O3 (519.2).
4.1.5.14. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-(naphthal en-l-ylmethyl)-1H-1,2,3-triazole-4-carboxamide (13n).
White solid, yield: 68%. mp: 78-80°C. NMR (400 MHz, DMSO-d6) δ 8.54 (s, 1H), 8.33 (d, J = 8.0 Hz, 1H), (d, J = 8.1 Hz, 1H), 7.99 (t, J = 8.5 Hz, 2H), 7.57 (ddt, J = 20.7, 14.9, 7.3 Hz, 3H), 7.43 (d, J = 7.1 Hz, 1H), 7.24 – 7.09 (m, 5H), 6.99 (d, J = 8.4 Hz, 2H), 6.91 – 6.80 (m, 2H), 6.15 (s, 2H), 4.64 (q, J = 7.6 Hz, 1H), 3.79 (s, 3H), 3.10 (s, 3H), 2.87 (d, J = 5.3 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.25, 159.50, 159.06, 142.56, 137.95, 135.91, 133.84, 131.54, 130.99, 129.67, 129.33, 129.21, 128.58, 127.85, 127.35, 127.12, 126.88, 126.69, 126.05, 123.59, 115.17, 55.91, 51.72, 51.54, 37.86, 37.29. ESI-MS: m/z 520.4 (M + 1)+, 542.4 (M + 23)+, C31H29N5O3 (519.2).
4.1.5.15. (S)-1-(benzo[d][1,3]dioxol-5-yl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1H-1,2,3-triazole-4-carboxamide (13o).
White solid, yield: 65%. mp: 136-138°C. 1H NMR (400 MHz, DMSO-d6 δ 9.10 (s, 1H), 8.42 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 2.2 Hz, 1H), 7.41 (dd, J = 8.4, 2.2 Hz, 1H), 7.25 – 7.10 (m, 6H), 7.06 – 7.00 (m, 2H), 6.95 – 6.89 (m, 2H), 6.16 (s, 2H), 4.72 (q, J = 7.2 Hz, 1H), 3.81 (s, 3H), 3.14 (s, 3H), 2.94 (d, J = 6.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.20, 159.36, 159.10, 148.64, 148.29, 143.20, 137.94, 135.94, 131.05, 129.40, 129.26, 128.62, 126.92, 125.40, 115.22, 114.89, 109.11, 102.71, 55.93, 51.79, 37.92, 37.34. ESI-MS: m/z 500.3 (M + 1)+, 522.4 (M + 23)+, C27H25N5O5 (499.1).
4.1.5.16. Methyl(S)-3-(4-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamoyl)-1H-1,2,3-triazol-1-yl)benzoate (13p).
White solid, yield: 80%. mp: 170-172°C. 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 8.52 (d, J = 8.0 Hz, 1H), 8.50 – 8.43 (m, 1H), 8.27 – 8.20 (m, 1H), 8.09 (dt, J = 7.8, 1.3 Hz, 1H), 7.78 (t, J = 8.0 Hz, 1H), 7.29 – 7.13 (m, 5H), 7.02 (d, J = 8.4 Hz, 2H), 6.99 – 6.88 (m, 2H), 4.73 (q, J = 7.2 Hz, 1H), 3.92 (s, 3H), 3.82 (s, 3H), 3.15 (s, 3H), 2.95 (d, J = 6.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.19, 165.67, 159.23, 159.11, 143.57, 137.96, 136.98, 136.65, 135.94, 131.09, 130.00, 129.40, 129.26, 128.63, 126.92, 125.59, 125.49, 121.25, 115.22, 55.93, 53.07, 51.86, 37.93, 37.34. ESI-MS: m/z 514.3 (M + 1)+, 536.4 (M + 23)+, C28H27N5O5 (513.2).
4.1.5.17. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-((phenylth io)methyl)-1H-1,2,3-triazole-4-carboxamide (13q).
Yellow solid, yield: 32%. mp: 64-66°C. 1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 8.38 (d, J = 8.0 Hz, 1H), 7.42 (d, J = 7.3 Hz, 2H), 7.34 (q, J = 8.2, 7.5 Hz, 3H), 7.22 (d, J = 7.9 Hz, 2H), 7.15 (d, J = 6.4 Hz, 3H), 7.01 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 6.7 Hz, 2H), 5.99 (s, 2H), 4.64 (q, J = 7.4 Hz, 1H), 3.81 (s, 3H), 3.13 (s, 3H), 2.88 (d, J = 6.9Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.25, 159.37, 159.08, 142.64, 138.02, 135.93, 132.43, 131.29, 129.83, 129.32, 129.26, 128.59, 128.41, 126.89, 126.55, 115.20, 55.93, 52.57, 51.88, 37.89, 37.09. ESI-MS: m/z 502.3 (M + 1)+, 524.4 (M + 23)+, C27H27N5O3S (501.1).
4.1.5.18. N-((S)-1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-((phenylsu lfinyl)methyl)-1H-1,2,3-triazole-4-carboxamide (13r).
White solid, yield: 45%. mp: 77-79°C. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (t, J = 9.6 Hz, 1H), 8.34 (d, J = 25.1 Hz, 1H), 7.56 (d, J = 12.2 Hz, 5H), 7.37 – 7.10 (m, 5H), 7.03 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 6.8 Hz, 2H), 6.00 (d, J = 13.4 Hz, 1H), 5.77 (dd, J = 13.4, 3.6 Hz, 1H), 4.66 (q, J = 7.4 Hz, 1H), 3.82 (s, 3H), 3.14 (s, 3H), 2.90 (d, J = 6.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6 δ 171.31, 159.26, 159.10, 142.07, 142.01, 140.50, 140.42, 138.08, 135.95, 132.17, 129.74, 129.71, 129.32, 128.59, 128.28, 128.13, 126.89, 124.89, 124.84, 115.22, 68.55, 55.94, 51.97, 37.91, 37.03. ESI-MS: m/z 518.4 (M + 1)+, 540.4 (M + 23)+, C27H27N5O4S (517.1).
4.1.5.19. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1-((phenylsu lfonyl)methyl)-1H-1,2,3-triazole-4-carboxamide (13s).
Light greeen solid, yield: 56%. mp: 68-70°C. 1H NMR (400 MHz, DMSO-d6) δ 8.57 (d, J = 7.9 Hz, 1H), 8.50 (s, 1H), 7.85 – 7.59 (m, 5H), 7.34 – 7.12 (m, 5H), 7.09 – 6.99 (m, 2H), 6.91 – 6.80 (m, 2H), 6.40 (s, 2H), 4.65 (q, J = 7.2 Hz, 1H), 3.81 (s, 3H), 3.14 (s, 3H), 2.90 (d, J = 6.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.27, 159.11, 159.07, 142.60, 138.06, 136.27, 135.94, 135.47, 130.08, 129.31, 128.99, 128.60, 128.50, 126.90, 115.22, 67.46, 55.94, 52.05, 37.90, 37.02. ESI-MS: m/z 534.3 (M + 1)+, 556.3 (M + 23)+, C27H27N5O5S (533.1).
4.1.5.20. 1-(2-(hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-N-((S)-1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-1H-1,2,3-triazole-4-carboxamide (13t).
White solid, yield: 50%. mp: 143-145°C. 1H NMR (400 MHz, DMSO-d6) δ 11.37 (s, 1H), 8.73 (s, 1H), 8.35 (d, J = 8.1 Hz, 1H), 7.89 – 7.76 (m, 1H), 7.18 (dt, J = 15.1, 8.1 Hz, 5H), 7.02 (d, J = 8.4 Hz, 2H), 6.95 – 6.80 (m, 2H), 6.42 (t, J = 6.6 Hz, 1H), 5.43 (dt, J = 8.6, 5.5 Hz, 1H), 5.28 (t, J = 5.2 Hz, 1H), 4.70 (q, J = 7.3 Hz, 1H), 4.23 (q, J = 4.0 Hz, 1H), 3.81 (s, 3H), 3.74 – 3.54 (m, 2H), 3.13 (s, 3H), 2.91 (d, J = 7.0 Hz, 2H), 2.83 – 2.59 (m, 2H), 1.81 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.25, 164.19, 159.52, 159.09, 150.89, 142.69, 137.98, 136.72, 135.92, 129.35, 129.25, 128.62, 126.90, 126.69, 115.22, 110.11, 84.73, 84.29, 61.09, 60.06, 55.93, 51.73, 37.90, 37.60, 37.29, 12.73. ESI-MS: m/z 604.5 (M + 1)+, 621.7 (M + 18)+, 626.5 (M + 23)+, C30H33N7O7 (603.2).
4.2. In vitro anti-HIV assay
4.2.1. Assessment of Inhibitory Activity on HIV-1 Replication in MT-4 Cells
Inhibitory activity of compounds against HIV-1 infection in MT-4 Cells was measured as the reduction in luciferase gene expression after multiple rounds of virus infection of the cells similar to that described previously[36]. Briefly, 200 TCID50 of virus (NL4-3) was used to infect MT-4 cells in the presence of various concentrations of compounds. Two days after infection, the culture medium was removed from each well and 100 μL of Bright Glo reagent (Promega, Luis Obispo, CA) was added to the cells for measurement of luminescence using a Victor 2 luminometer. The effective concentration (EC50) against HIV-1 strains was defined as the concentration that caused a 50% reduction of luciferase activity (Relative Light Units) compared to virus control wells.
4.2.2. Cytotoxicity Assay[37]
A CytoTox-Glo cytotoxicity assay (Promega) was used to determine the cytotoxicity of the synthesized compounds. Parallel to the antiviral assays, MT-4 cells were cultured in the presence of various concentrations of the compounds for 1 day. The percent of viable cells was determined by following the protocol provided by the manufacturer. The 50% cytotoxic concentration (CC50) was defined as the concentration that caused a 50% reduction in cell viability.
4.3. Binding to CA Proteins Analysis via Surface Plasmon Resonance (SPR)
All binding assays were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad Laboratories, Hercules, CA). The instrument temperature was set at 25°C for all kinetic analyses. ProteOn GLH sensor chips were preconditioned with two short pulses each (10 seconds) of 50 mM NaOH, 100 mM HCl, and 0.5% sodium dodecyl sulfide. Then the system was equilibrated with PBS-T buffer (20 mM sodium phosphate, 150 mM NaCl, and 0.005% polysorbate 20, pH 7.4). The surface of a GLH sensorchip was activated with a 1:100 dilution of a 1:1 mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (0.2 M) and sulfo-N-hydroxysuccinimide (0.05 M). Immediately after chip activation, the HIV-1 NL4-3 capsid protein constructs, purified as in Xu el al. [16], were prepared at a concentration of 100 μg/ml in 10 mM sodium acetate, pH 5.0 and injected across ligand flow channels for 5 min at a flow rate of 30 μl/min. Then, after unreacted protein had been washed out, excess active ester groups on the sensor surface were capped by a 5 minute injection of 1 M ethanolamine HCl (pH 8.0) at a flow rate of 5 μl/min. A reference surface was similarly created by immobilizing a non-specific protein (IgG b12 anti HIV-1 gp120; was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (IgG1 b12) from Dr. Dennis Burton and Carlos Barbas) and was used as a background to correct non-specific binding.
To prepare a compound for direct binding analysis, compound stock solutions, along with 100% DMSO, and totaling 30μl was made to a final volume of 1 ml by addition of sample preparation buffer (PBS, pH 7.4). Preparation of analyte in this manner ensured that the concentration of DMSO was matched with that of running buffer with 3% DMSO. Serial dilutions were then prepared in the running buffer (PBS, 3% DMSO, 0.005% polysorbate 20, pH 7.4) and injected at a flow rate of 100 μl/min, for a 1 minute association phase, followed by up to a 5 minutes dissociation phase using the “one shot kinetics” capability of the Proteon instrument[38]. Data were analyzed using the ProteOn Manager Software version 3.0 (Bio-Rad). The responses from the reference flow cell were subtracted to account for the nonspecific binding and injection artifacts. The equilibrium dissociation constant (KD) for the interactions, and derived from a minimum of three experiments, were calculated in ProteOn Manager Version 3.1.0.6 (Bio-Rad, Hercules, CA), using the equilibrium analysis function.
4.4. Molecular Dynamics Simulation
4.4.1. Initial Structure Preparation
The initial X-ray structure of hexameric HIV-1 CA was downloaded from the protein databank (PDB code 5HGL with a resolution of 3.1 Å) [21]. Only one monomer of the hexameric structure was used for the entire MD study. This structure, which is used in the binding assay, has four mutated amino acids (M185A, E45C, A14C and W184A). 5HGL structure misses amino acids Ala88, Gly89, Pro90, Ile91, Ala92, Pro93, Gly94, Gln95, Lys182, Asn183, Ala184, and Ala185. Accordingly, we used HIV-1 CA X-ray structure 3GV2 to extract the missed amino acids with their corresponding tertiary structure and add them to 5HGL structure after their alignment using discovery studio software [39]. Then, the whole system was energy minimized to remove any strains due to the added amino acid residues, which is then used for further study.
13m structure was sketched by discovery studio in the S-configuration, and no ionizable groups were detected. Then, its conformers were generated by OMEGA module of OPENEYE Scientific Software Inc. using default parameters [40, 41]. 13p conformers were docked into the active site of the modeled 5HGL structure using OPENEYE Scientific Software Inc. module OEDOCKING 3.0.1 with chemgauss4 scoring function [42-45]. The docking procedure resulted in only one docked conformer to the active site of the X-ray structure. We used this structure, i.e the docked 13m to the modified 5HGL, in the subsequent molecular dynamics simulation.
4.4.2. Molecular Dynamics Simulation Production
Atomic point charges for 13m were derived from AM1-BCC charge model with ANTECHAMBER module of AMBER14 [46]. Coordinate and topology parameters were prepared using ff14sb force field for HIV-1 CA monomer and GAFF force field for 13m. The whole system was solvated in explicit water TIP3PBOX octahedral solvent box model with 9Å cut, and the system was neutralized by Na+ ions. Water was minimized for 10000 cycles using steepest descent and then conjugate gradient algorithms. The whole system was then minimized for 5000 cycles using steepest descent followed by conjugate gradient algorithms. Then, water was equilibrated for 20 ps at constant volume and periodic boundaries with weak strength restraints on the whole system through the equilibrium stage, a force constant of 10 Å as position restraint. The whole system was then equilibrated for 40 ps using constant pressure periodic boundaries with no restraints. The minimized and equilibrated structure was used in the molecular dynamics simulation for 1 μs at constant temperature (300 K) and constant pressure (1 atm). Non-bonded forces were calculated at a Cutoff distance of 10 Å. H Mass Repartition was used to shift the mass of all hydrogen atoms of solute to 3.024 Da [47]. This allowed us to use an integration time step of 4 fs during MD [47]. SHAKE bond length constraint involving hydrogen atoms was turned on.
4.4.3. Clustering
All frames were imaged and then water molecules and Na+ ions were stripped off. All frames were aligned against the first frame of the MD production using protein residues only. They were then clustered by DBSCAN algorithm [48] implemented in CPPTRAJ of AMBER14 on 13m using minimum points of 3 and epsilon of 2.5 with no frame orientation (no fit), which clustered all frames according to 13m to explore the binding site of 5HGL.
4.5. Determination of the action link of compound 13m
4.5.1. Cells
Human embryonic kidney 293T cells (a gift from Dr. Irwin Chaiken, Drexel University, Philadelphia, PA) were cultured in Dulbecco's Modified Eagle’s Medium (DMEM), 10% Fetal Bovine Serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2mM L-glutamine. Human astroglioma U87 cells stably expressing CD4/CCR5 (obtained from Professor Hongkui Deng, Peking University, and Prof. Dan Littman, New York University, USA, through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH)[35, 49, 50] were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine, 300μg/ml G418 (Thermo Scientific, Waltham, MA) and 1 μg/ml Puromycin (Thermo Scientific). All cells were incubated, unless otherwise stated, at 37°C in a humidified in a 5% CO2 air environment chamber.
4.5.2. Production of pseudotyped viruses
A dual transfection of two plasmids (3:4 ratio of the viral backbone vector to the viral envelope vector) in HEK293T cells (0.8×106 cells/well) produced single-round infectious HIV-1B41 pseudotyped luciferase-reporter viruses1. The viral backbone vector is an Env-deficient HIV-1 pNL4-3-LucR+E- plasmid that carries the luciferase-reporter gene [51]. The viral envelope vector is a plasmid expressing the HIV-1 gp160B41 envelope [52, 53]. A calcium phosphate transfection (ProFection Mammalian Transfection System, Promega, Madison, WI) was used to co-transfect these two plasmids. Following a 5-hour incubation, the media containing the transfection reagents and the DNA was removed, the cells were washed with DMEM, and fresh culture media was added. 72 hours post-transfection, the pseudovirus containing supernatants were clarified, filtered, and stored at −80°C.
4.5.3. Single-round infection assay
The details of the single-round HIV-1 infection assay for detecting viral infectivity have been published previously [51, 54, 55]. Briefly, U87.CD4.CCR5 (1.2 × 104 cells/well) target cells were seeded in 96-well luminometer-compatible tissue culture plates (Greiner bio-one). After a 24-hour incubation at 37°C, the compound or a DMSO vehicle control (Sigma) was mixed with pseudotyped virus and the mixture was added to the target cells. After a 48-hour 37°C incubation, the media was removed from each well and the cells were lysed by adding 50 μl/well of luciferase lysis buffer (Promega). Following one freeze-thaw cycle, 50ul/well of luciferase assay substrate (Promega) was added and a GloMax 96 microplate luminometer (Promega) was used to measure the luciferase activity of each well. Luciferase activity of virus produced with compound were normalized to the luciferase activity of virus produced from DMSO vehicle control treated cells. The compound-induced effects are manifested as a decrease in normalized luciferase activity which indicates a decrease in infectivity in the target cells when the compound was present.
4.5.4. Viral late-stage infection assay
Single-round infectious envelope-pseudotyped luciferase-reporter viruses were produced from 293T cells[35] in the presence of the compound (from 100 μM to 0.01 μM with the same DMSO concentration) or DMSO vehicle control (Sigma) and incubated for 72 hours at 37°C. The resulting pseudovirus-containing supernatants were clarified, filtered, and underwent one freeze-thaw cycle before being diluted ten-fold and used to infect U87.CD4.CCR5 target cells. Target cells with pseudotyped viruses were incubated for 48 hours at 37°C. Following the 48-hour incubation, the media was removed from each well and the cells were lysed by adding 50 μl/well of luciferase lysis buffer (Promega). Following one freeze-thaw cycle, 50ul/well of luciferase assay substrate (Promega) was added and a GloMax 96 microplate luminometer (Promega) was used to measure the luciferase activity of each well. Luciferase activity of virus produced with compound were normalized to the luciferase activity of virus produced from DMSO vehicle control treated cells. The compound-induced effects are manifested as a decrease in normalized luciferase activity which indicates a decrease in infectivity in the target cells when the compound was present.
Supplementary Material
Highlights.
20 new compounds designed and synthesized using a parallel “click” chemistry approach
Most potent new compounds are equipotent to PF-74 in assay utilized.
New compounds retain binding specificity to HIV-1 CA
Most potent compound inhibits HIV-1 in both early and late stages of replication.
Acknowledgements
Financial support from the National Natural Science Foundation of China (NSFC Nos. 81273354, 81573347), Key Project of NSFC for International Cooperation (No. 81420108027), Young Scholars Program of Shandong University (YSPSDU, No. 2016WLJH32), the Fundamental Research Funds of Shandong University (No. 2017JC006), Key research and development project of Shandong Province (No. 2017CXGC1401), Major Project of Science and Technology of Shandong Province (No. 2015ZDJS04001) and NIH/NIAID grants 1 R56 AI118415-01A1 and 1 R01 GM125396-01A1 (Cocklin, PI) are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Zhan P, Pannecouque C, De Clercq E, Liu X, Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends, J Med Chem, 59 (2016) 2849–2878. [DOI] [PubMed] [Google Scholar]
- [2].Iyidogan P, Anderson KS, Current perspectives on HIV-1 antiretroviral drug resistance, Viruses, 6 (2014) 4095–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Le VS, Mouland AJ, Valienteecheverria F, Roles of HIV-1 capsid in viral replication and immune evasion, Virus Res, 193 (2014) 116–129. [DOI] [PubMed] [Google Scholar]
- [4].Perrier M, Bertine M, Le Hingrat Q, Joly V, Visseaux B, Collin G, Landman R, Yazdanpanah Y, Descamps D, Charpentier C, Prevalence of gag mutations associated with in vitro resistance to capsid inhibitor GS-CA1 in HIV-1 antiretroviral-naive patients, J Antimicrob. Chemother 72 (2017) 2954–2955. [DOI] [PubMed] [Google Scholar]
- [5].Ambrose Z, Aiken C, HIV-1 uncoating: connection to nuclear entry and regulation by host proteins, Virology, 454-455 (2014) 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hulme AE, Perez O, Hope TJ, Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription, Proc. Natl. Acad. Sci. U S A 108 (2011) 9975–9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI, Assembly properties of the human immunodeficiency virus type 1 CA protein, J. Virol 78 (2004) 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].López CS, Eccles JD, Still A, Sloan RE, Barklis RL, Tsagli SM, Barklis E, Determinants of the HIV-1 core assebly pathway, Virology, 417 (2011) 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thenin-Houssier S, Valente ST, HIV-1 capsid inhibitors as antiretroviral agents, Curr HIV Res. 14(2016) 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Campbell EM, Hope TJ, HIV-1 capsid: the multifaceted key player in HIV-1 infection, Nat. Rev. Microbiol 13 (2015) 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL, HIV capsid is a tractable target for small molecule therapeutic intervention, PLoS Pathog. 6 (2010) e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang J, Liu X, De Clercq E, Capsid (CA) protein as a novel drug target: recent progress in the research of HIV-1 CA inhibitors, Mini Rev. Med. Chem 9 (2009) 510–518. [DOI] [PubMed] [Google Scholar]
- [13].Xu JP, Francis AC, Meuser ME, Mankowski M, Ptak RG, Rashad AA, Melikyan GB, Cocklin S, Exploring modifications of an HIV-1 capsid inhibitor: design, synthesis, and mechanism of action, J. Drug Des. Res 5 (2018) 1070. [PMC free article] [PubMed] [Google Scholar]
- [14].Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB 3rd, Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid, J. Virol 86 (2012) 8472–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kortagere S, Xu JP, Mankowski MK, Ptak RG, Cocklin S, Structure-activity relationships of a novel capsid targeted inhibitor of HIV-1 replication, J. Chem. Inf. Model 54 (2014) 3080–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu JP, Branson JD, Lawrence R, Cocklin S, Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer, Bioorg. Med. Chem. Lett 26 (2016) 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Curreli F, Zhang H, Zhang X, Pyatkin I, Victor Z, Altieri A, Debnath AK, Virtual screening based identification of novel small-molecule inhibitors targeted to the HIV-1 capsid, Bioorg. Med. Chem 19 (2011) 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier JF, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C, Sundquist WI, Mason SW, Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes, Antimicrob. Agents Chemother 57 (2013) 4622–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP, Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein, J. Mol. Biol 373 (2007) 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF, Antiviral inhibition of the HIV-1 capsid protein, J. Mol. Biol 327 (2003) 1013–1020. [DOI] [PubMed] [Google Scholar]
- [21].Jacques DA, Mcewan WA, Hilditch L, Price AJ, Towers GJ, James LC, HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis, Nature, 536 (2016) 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou J, Price AJ, Halambage UD, James LC, Aiken C, HIV-1 resistance to the capsid-targeting inhibitor PF74 results in altered dependence on host factors required for virus nuclear entry, J. Virol 89 (2015) 9068–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saito A, Ferhadian D, Sowd GA, Serrao E, Shi J, Halambage UD, Teng S, Soto J, Siddiqui MA, Engelman AN, Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74, J. Virol 90 (2016) 5808–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, Ivanov DN, Yeager M, Structural basis of HIV-1 capsid recognition by PF74 and CPSF6, Proc. Natl. Acad. Sci. U S A 111 (2014) 18625–18630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, Sarafianos SG, Structural virology. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability, Science, 349 (2015) 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, James LC, Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly, PLoS. Pathog 10 (2014) e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bender JA, Gentles RG, Pendri A, Inhibitors of human immunodefiency virus replication, WO2016/172424A1, (2016). [Google Scholar]
- [28].Bondy SS, Chou CH, Link JO, Tse WC, Therapeutic compounds, WO2015/130964 A1, (2015). [Google Scholar]
- [29].Cavalli S, Tipton AR, Overhand M, Kros A, The chemical modification of liposome surfaces via a copper-mediated [3 + 2] azide-alkyne cycloaddition monitored by a colorimetric assay, Chem. Commun 30 (2006) 3193–3195. [DOI] [PubMed] [Google Scholar]
- [30].Said HF, Frisch B, Schuber F, Targeted liposomes: convenient coupling of ligands to preformed vesicles using "click chemistry", Bioconjug. Chem 17 (2006) 849–854. [DOI] [PubMed] [Google Scholar]
- [31].Spanedda MV, Salomé C, Hilbold B, Berner E, Heurtault B, Fournel S, Frisch B, Bourelbonnet L, Smart tools and orthogonal click-like reactions onto small unilamellar vesicles: Additional molecular data, Date Brief. 5 (2015) 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Artyushin OI, Sharova EV, Vinogradova NM, Genkina GK, Moiseeva AA, Klemenkova ZS, Orshanskaya IR, Shtro AA, Kadyrova RA, Zarubaev VV, Synthesis of camphecene derivatives using click chemistry methodology and study of their antiviral activity, Bioorg. Med. Chem. Lett 27 (2017) 2181–2184. [DOI] [PubMed] [Google Scholar]
- [33].Dang Z, Zhu L, Lai W, Bogerd H, Lee KH, Huang L, Chen CH, Aloperine and its derivatives as a new class of HIV-1 entry inhibitors, ACS Med. Chem. Lett 7 (2016) 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Meuser ME, Murphy MB, Rashad AA, Cocklin S, Kinetic characterization of novel HIV-1 entry inhibitors: discovery of a relationship between off-rate and potency, Molecules. 23 (2018) pii: E1940. doi: 10.3390/molecules23081940.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zentner I, Sierra LJ, Fraser AK, Maciunas L, Mankowski MK, Vinnik A, Fedichev P, Ptak RG, Martin-Garcia J, Cocklin S, Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein, ChemMedChem. 8 (2013) 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dang Z, Lai W, Qian K, Ho P, Lee KH, Chen CH, Huang L, Betulinic acid derivatives as human immunodeficiency virus type 2 (HIV-2) inhibitors, J. Med. Chem 52 (2009) 7887–7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu N, Wei L, Huang L, Yu F, Zheng W, Qin B, Zhu DQ, Morris-Natschke SL, Jiang S, Chen CH, Lee KH, Xie L, Novel HIV-1 non-nucleoside reverse transcriptase inhibitor agents: Optimization of diarylanilines with high potency against wild-type and rilpivirine-resistant E138K mutant virus, J. Med. Chem 59 (2016) 3689–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG, Exploring "one-shot" kinetics and small molecule analysis using the ProteOn XPR36 array biosensor, Anal. Biochem 358 (2006) 281–288. [DOI] [PubMed] [Google Scholar]
- [39].Ester MK, Kriegel HP, Sander J, Xu X, In proceedings of 2nd international conference on knowledge discovery and data mining, Simoudis E, Han J, Fayyad U, Eds., AAAI Press: Menlo Park, CA: (1996) 226–231. [Google Scholar]
- [40].S.F. OMEGA 3.0.0.1: OpenEye Scientific Software, NM., http://www.eyesopen.com., Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT.
- [41].Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT, Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and Cambridge structural database, J. Chem. Inf. Model 50 (2010) 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Druillennec S, Caneparo A, de Rocquigny H, Roques BP, Evidence of interactions between the nucleocapsid protein NCp7 and the reverse transcriptase of HIV-1, J. Biol. Chem 274(1999) 11283–11288. [DOI] [PubMed] [Google Scholar]
- [43].Kelley BP, Brown SP, Warren GL, Muchmore SW, POSIT: Flexible shape-guided docking for pose prediction, J. Chem. Inf. Model 55 (2015) 1771–1780. [DOI] [PubMed] [Google Scholar]
- [44].Mcgann M, FRED pose prediction and virtual screening accuracy, J. Chem. Inf. Model 51 (2011) 578–596. [DOI] [PubMed] [Google Scholar]
- [45].McGann M, FRED and HYBRID docking performance on standardized datasets, J. Comput. Aided Mol. Des 26 (2012) 897–906. [DOI] [PubMed] [Google Scholar]
- [46].Case DA, Betz RM, Botello-Smith W, Cerutti DS, Cheatham TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, York DM Kollman PA. AMBER 2016, University of California, San Francisco, 2016. [Google Scholar]
- [47].Hopkins CW, Grand SL, Walker RC, Roitberg AE, Long time step molecular dynamics through hydrogen mass repartitioning, J. Chem. Theory & Computation, 11 (2015) 1864–1874. [DOI] [PubMed] [Google Scholar]
- [48].Shao J, Tanner SW, Thompson N, Cheatham TE, Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms, J. Chem. Theory Comput 3 (2007) 2312–2334. [DOI] [PubMed] [Google Scholar]
- [49].Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D, Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1, J. Virol 72 (1998) 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bjorndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, Albert J, Scarlatti G, Littman DR, Fenyo EM, Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype, J. Virol 71 (1997) 7478–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Connor RI, Chen BK, Choe S, Landau NR, Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes, Virology. 206 (1995) 935–944. [DOI] [PubMed] [Google Scholar]
- [52].Hofmann W, Schubert D, LaBonte J, Munson L, Gibson S, Scammell J, Ferrigno P, Sodroski J, Species-specific, postentry barriers to primate immunodeficiency virus infection, J. Virol 73 (1999) 10020–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Marcon L, Choe H, Martin KA, Farzan M, Ponath PD, Wu L, Newman W, Gerard N, Gerard C, Sodroski J, Utilization of C-C chemokine receptor 5 by the envelope glycoproteins of a pathogenic simian immunodeficiency virus, SIVmac239, J. Virol 71 (1997) 2522–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cocklin S, Gopi H, Querido B, Nimmagadda M, Kuriakose S, Cicala C, Ajith S, Baxter S, Arthos J, Martin-Garcia J, Chaiken IM, Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor, J. Virol 81 (2007) 3645–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Billich A, Hammerschmid F, Peichl P, Wenger R, Zenke G, Quesniaux V, Rosenwirth B, Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions, J. Virol 69 (1995) 2451–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.