

Abstract
Circulating tumor DNA (ctDNA) can be detected in the blood and body fluids of patients using ultra-sensitive technologies which have the potential to improve cancer diagnosis, risk stratification, non-invasive tumor profiling, and tracking of treatment response and disease recurrence. As we begin to apply “liquid biopsy” strategies in children with cancer, it is important to tailor our efforts to the unique genomic features of these tumors and address the technical and logistical challenges of integrating biomarker testing. This article reviews the literature demonstrating the feasibility of applying liquid biopsy to pediatric solid malignancies and suggests new directions for future studies.
Keywords: Circulating tumor DNA, pediatric cancer, next generation sequencing, digital PCR, liquid biopsy
Introduction
As the field of precision oncology grows, the analysis of circulating tumor DNA (ctDNA) holds incredible potential for advancing cancer treatment. After cell-free DNA was first identified in the peripheral blood in 1948, ctDNA was described in the late 1970s and has since been quantified and characterized across a range of cancers.1–6 Potential applications of ctDNA are far-reaching, including cancer screening and diagnosis, non-invasive tumor profiling and identification of targetable somatic variants, pretreatment risk-stratification, tracking of treatment response, detection of minimal residual disease, and surveillance for relapse.7–9 For children with cancer, these applications could reduce exposure to anesthesia and radiation by decreasing reliance on surgical biopsies and serial imaging for diagnosis, staging, and disease monitoring. Furthermore, the opportunity to analyze serial liquid biopsy samples could vastly increase our understanding of tumor evolution and genomic patterns of treatment resistance in children with relapsed cancer.
Analysis of ctDNA in pediatric malignancies presents unique methodologic and clinical challenges, requiring tailored approaches in the liquid biopsy field. Much of the early success of ctDNA evaluation in adult malignancies focused on the identification of highly-recurrent hotspot mutations in oncogenes such as EGFR or KRAS, including polymerase chain reaction (PCR)-based assays that have gained regulatory approval in the United States and Europe.4–6,10–20 Such recurrent single nucleotide variants (SNVs) are rare in pediatric malignancies. Instead, pediatric tumors are more commonly characterized by recurrent copy-number changes and translocations.21–27 Detecting these structural variants requires customized DNA profiling approaches that differ from assays optimized for detecting somatic SNVs. Clinical implementation and validation of these custom assays is also challenging due to the rarity and diversity of pediatric cancers.28
Beyond the studies examining ctDNA in solid tumors, there is a growing body of literature evaluating ctDNA in adult patients with hematologic malignancies.29–36 While ctDNA has been evaluated in the peripheral blood of children with leukemia, these assays lack the sensitivity of modern minimal residual disease assays (MRD).37,38 In CNS tumors, detection of ctDNA in the peripheral blood is possible, but sensitivity appears to be a challenge in these patients.4,39–41 Instead, recent efforts have focused on detection of tumor DNA in the CSF.41,42 Given the paucity of data from studies of ctDNA in children with hematologic and CNS malignancies, our review will focus on current technologies being applied to interrogate ctDNA in children with non-CNS solid tumors. The goals of this paper are to familiarize pediatric oncologists with the principles and techniques used for ctDNA work and to highlight unique aspects of applying liquid biopsy approaches to pediatric cancer care and research.
Detection, quantification, and characterization of ctDNA in children with solid tumors
Cell-free DNA is composed of short fragments (~135-170 base pairs) of double-stranded DNA found in the non-cellular fraction of the blood. Cell-free DNA originates from multiple cellular sources in the body, including normal cells, injured tissues, fetal cells, and cancer cells and has a half-life in the circulation of less than 2 hours.43–52 In patients with cancer, ctDNA accounts for a portion of the cell-free DNA present in the circulation (Fig. 1). Circulating tumor DNA can be detected and quantified by measuring the presence of somatic events, including SNVs, insertion/deletions (indels), copy-number changes, translocations, and methylation patterns that differentiate tumor DNA from cell-free DNA originating from normal cells.53–55 One recent article demonstrated that methylation patterns in cell-free DNA can be used to detect the presence of ctDNA and correlate ctDNA methylation with specific cancer histologies.56 However, this strategy has yet to be applied to pediatric solid tumors. The two most common methods for detecting somatic variants in any DNA sample are PCR and next-generation sequencing (NGS)(Fig. 1). Both approaches can be applied to the detection of ctDNA but differ in how well they can be optimized for classes of somatic variants and clinical scenarios (Fig. 2). There are numerous technology-specific and context-specific considerations that must be evaluated when choosing an approach for the detection of ctDNA. We have summarized what we consider to be the most important and commonly encountered considerations in Table 1.57–63
Figure 1. Overview of cell-free DNA extraction and processing.
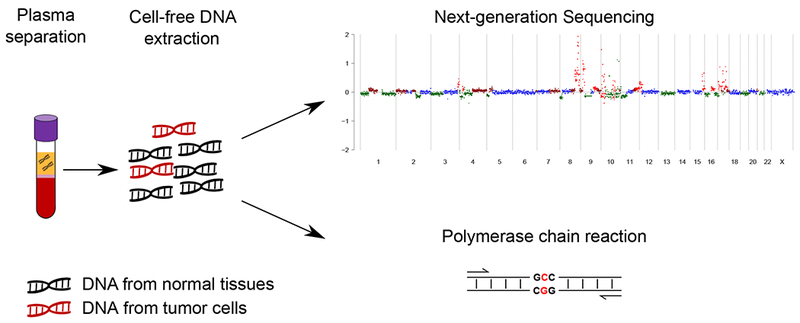
Cartoon depicts the presence of cell-free DNA in the plasma layer of a blood sample collected in an EDTA tube (left). The extracted cell-free DNA is a mixture of DNA originating from normal tissues (black strands of DNA) and DNA originating from tumor cells (red strands of ctDNA), the latter being often a small fraction of the total DNA in the sample (middle). Identifying and quantifying ctDNA from a cell-free DNA sample requires detection of somatic variants that are present only in the tumor. This is typically done with either next-generation sequencing or polymerase chain reaction assays (right).
Figure 2. PCR and next-generation sequencing approaches to detecting somatic variants in cell-free DNA.
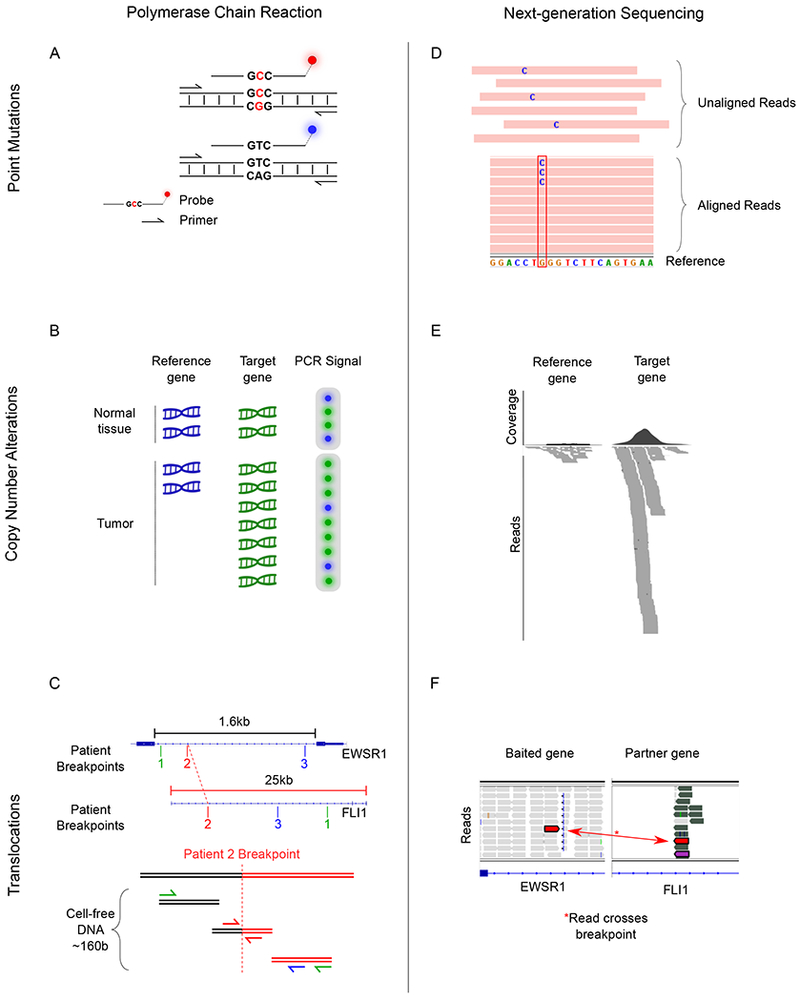
A) Image depicts the ability to detect DNA with single-nucleotide variants (top) from wild-type DNA (bottom) by utilizing sequence-specific fluorescent PCR probes. In this approach, primers are specific to the sequence flanking the region of interest while each fluorescent probe has a sequence complementary to either the mutated strand (red) or the wild-type strand (blue). DNA is depicted by two horizontal lines connected by small vertical lines with the three central base pairs indicating the sequence of interest. Red letters indicated mutated single-nucleotide variant. B) dPCR can be used to identify copy-number alterations by comparing the number of PCR reactions from the target gene to the number of PCR reactions from a reference gene. Top, DNA originating from normal tissue contains two copies of the reference gene (blue DNA) and two copies of the target gene (green DNA) and results in two droplets containing reference gene PCR product (blue circle) and two droplets containing the target gene product (green circles). Bottom, DNA originating from tumor has an amplification of the target gene (green DNA) resulting in more droplets with PCR product from the target gene (green circles) compared to the number of droplets with reference gene (blue circle). C) The EWSR1/FLI1 translocation breakpoints occur at intronic regions of each gene and are patient specific. Top, intronic regions of EWSR1 and FLI1 with patient-specific breakpoints indicated for hypothetical patients 1 through 3. Bottom, rearranged DNA from patient 2 is shown with the breakpoint indicated by a vertical dotted red line. Short fragments of cell-free DNA from patient 2 align to the same genomic region as the translocation. Only patient-specific primers designed for patient 2 (red) can successfully result in a PCR product. PCR primers are depicted as arrows and the color is specific to primers designed to amplify the matched patient-specific translocation D) Top, next-generation sequencing reads are generated from cell-free DNA. Bottom, reads are then aligned to the reference genome. Mutated DNA has a sequence mismatch at the site of the somatic single-nucleotide variant indicated by the letter “C” in blue. E) Average coverage and aligned sequencing reads for a reference gene (left) and the target gene (right). Amplification of the target gene results in many more sequencing reads compared to the reference gene. F) Using a hybrid-capture sequencing panel designed to enrich sequencing reads for the intronic region of EWSR1, DNA translocations are identified as reads that map on one side to the EWSR1 intron and on the other side map to the FLI1 intron. Two red rectangles represent the two sides of a single sequencing read.
TABLE 1. Features of PCR and NGS.
Listed are common features and limitations of PCR and NGS that should be considered when choosing a strategy for ctDNA assay development.
| PCR | Next Generation Sequencing | ||||
|---|---|---|---|---|---|
| Gene Panel1 | Translocation Panel1 | Shallow Sequencing (WIGS) | Deep Sequencing (WES/WGS) | ||
| Detectable variants | SNV/indel, translocation, CNA | SNV/indel, translocation, CNA | Translocation | CNA (Mb scale) | SNV/indel, CNA, Translocation (WGS) |
| Mutation-specific assay | Yes | No2 | No3 | No | No |
| Patient-specific assay | Yes (Translocation and non-recurrent variants) | No | No | No | No |
| Patient germline required | Variant dependent4 | Variant dependent4 | No | No | Yes |
| ctDNA quantification | ctDNA estimation complicated by CNA | Measuring multiple variants offsets effects of CNA | ctDNA estimation complicated by CNA | Multiple CNA improve ctDNA estimation | Measuring multiple variants offsets effects of CNA |
| Time to result | Day(s) | Week(s) | Week(s) | Week(s) | Week(s) |
| Relative cost | $ | $$($) | $$ | $$ | $$$$ |
| Sensitivity | Up to 0.005% of tumor fraction | Sensitivity increases with coverage5 | Sensitivity increases with coverage | ~3% (Adalsteinson et al.) | Sensitivity increases with coverage5 |
| Detects novel variants | No | Novel variants in targeted gene(s) | Unknown translocation partners | Yes | Yes |
Gene and translocation panels can be combined because they use the same hybrid capture technology.
Assays are not mutation specific but selected genes are typically restricted to those known to be mutated in the cohort of interest.
Assays are not breakpoint specific but are enriched for intronic regions typically involved in the translocations of interest.
For variants that can be observed in the germline (e.g. TP53), either germline control must be used or prior knowledge of germline status is required to interpret results.
There is a risk of confusing sequencing artifact for low-allelic SNV/indels events. Error suppression strategies need to be incorporated when targeting these events by NGS.
SNV, single nucleotide variation; indel, small insertion or deletion; CNA, copy-number alterations; Mb, megabase.
PCR-based assays
Many approaches to ctDNA analysis rely on PCR, a technology used to detect or quantify a segment of DNA by sequence-specific amplification (Table 1). In the late 1990s, it was shown that ctDNA microsatellites could be detected by PCR in patients with head and neck cancer.64 Since then, many studies have focused on using PCR to detect highly recurrent hotspot mutations in genes such as TP53, EGFR or BRAF (Fig. 2A). PCR can also be adapted to the detection of structural variants which are more common in pediatric malignancies, such as copy-number alterations (i.e. MYCN amplifications) and translocations (i.e. EWSR1/FLI1) (Figs. 2B and 2C). These assays rely on the development of mutation-specific primers and/or probes which are often employed in the setting of digital PCR (dPCR) and can attain a sensitivity to detect variants that are present in as little as 0.005% of a DNA sample.4,19,20,65–68 dPCR partitions DNA into a large number of PCR reactions, allowing for quantification, improved sensitivity, and some degree of multiplexing.69
Digital PCR-based assays can serve as noninvasive surrogates to biopsies for identification of potentially targetable oncogenic hotspot mutations, tracking treatment responses, and monitoring for the development of specific treatment resistant mutations.10,34,65–67,70–74 However, the use of PCR to detect ctDNA requires prior knowledge of the disease-specific or patient-specific mutations, including the exact location of the patient-specific DNA breakpoints in the case of oncogenic translocations.75,76 Furthermore, while ctDNA quantification by dPCR is highly sensitive, it only measures the mono-allelic fraction of the targeted genetic region. This mono-allelic fraction may not be an accurate estimate for the total abundance of ctDNA when the targeted region is affected by copy-number alterations, especially when the magnitude of those events is heterogeneous in the tumor (Table 1).
Next-generation sequencing
Next-generation sequencing is a powerful tool for ctDNA analysis. Unlike PCR, NGS technology allows for evaluation of somatic events without previous knowledge of the abnormality of interest. These assays can be developed to sequence individual genes, selected regions of the genome, the exome, or the entire genome (Table 1). For example, recurrent mutations in driver oncogenes, such as oncogenic EGFR mutations in patients with lung cancer, can be detected and monitored by NGS while also having the capacity to identify previously unknown or uncommon variants in this gene (Fig. 2D).10 NGS gene panels of multiple known oncogenes (i.e. BRAF, EGFR, KRAS, and PIK3CA) have been used in patients with advanced cancer to identify targetable mutations for therapy selection without the need to perform numerous tests for individual variants.77
Next-generation sequencing can also be utilized to detect chromosomal copy-number alterations in ctDNA, a key feature of many pediatric malignancies (Fig. 2E).21–23,78 Leary et al. developed an algorithm to infer presence of ctDNA and its concentration from an analysis of chromosomal copy-number variants by whole-genome sequencing (WGS) performed with an average sequencing coverage of only 8 reads per base pair (8x).79 Economical shallow WGS, with a coverage of 0.1× and a turnaround time of 2 days, was first used to identify ctDNA in prostate cancer by detecting copy-number changes.80 It has also been used in neuroblastoma, allowing a reliable noninvasive copy-number profiling.81 Adalsteinsson et al. showed that ctDNA can be quantified by measuring genome-wide segmental copy-number changes from ultra-low passage WGS (ULP-WGS) using the ichorCNA algorithm.60 We recently applied this approach to the most common non-CNS pediatric solid tumors and we demonstrated that the majority of pediatric malignant tumors shed ctDNA at detectable levels.82,83 One advantage of this approach is that quantification of ctDNA does not require prior tumor profiling or the need for a germline sample. Therefore, ctDNA analyses can be performed in settings where access to such samples may be limited or incomplete, as is often the case in multi-institutional prospective clinical trials of rare cancers.
High-throughput NGS can also be used for translocation detection in pediatric cancer. Translocations typically occur in introns that frequently span greater than 1,000 to 100,000 bases, with unique DNA break-points seen in each patient. Given that ctDNA fragments are less than 200 base pairs in length, identifying translocation breakpoints in the cell-free DNA by PCR requires the development of patient-specific PCR assays and previous knowledge of the translocation break-point (Fig. 2C). Recently, NGS hybrid capture assays have been adapted to detect these fusions by enriching sequencing libraries for regions of the genome commonly involved in oncogenic translocations, such as those described in Ewing sarcoma and alveolar rhabdomyosarcoma (Fig. 2F).82,84 Such an approach has allowed for detection of oncogenic translocations directly from cell-free DNA specimens without the need to profile tumor biopsy material. This technology may be readily adapted to other fusion-positive tumors not previously evaluated.
Conceptually, ultra-deep WGS of matched cell-free DNA and germline samples would allow investigators to detect and quantify ctDNA by identifying any type of somatic variant without the need for previous knowledge of the tumor genome. However, this approach is currently cost prohibitive. Therefore, NGS strategies must be selected to balance three main profiling considerations: 1) the proportion of the genome targeted for sequencing; 2) the depth of sequencing desired; and 3) the cost of sequencing each sample (Fig. 3). For example, low-passage whole-genome sequencing can identify ctDNA by detecting chromosomal copy-number events.62,79,81–83,85 This approach efficiently identifies copy-number events in samples with a high fraction of tumor DNA (≥ 3%) at a relatively low cost but is unable to identify specific base-pair substitutions, focal copy-number events, or translocations. Conversely, very deep sequencing (i.e. 10,000 – 100,000× coverage) of a small panel of genomic regions can identify SNVs, indels, and focal copy-number alterations. With the appropriate error-suppression techniques,86 this approach can detect variants with allelic fractions of < 0.1%, but is unable to detect chromosomal copy-number events, translocations, or SNVs occurring outside the targeted regions.
Figure 3. Relationship between sequencing depth and DNA coverage for next-generation sequencing.
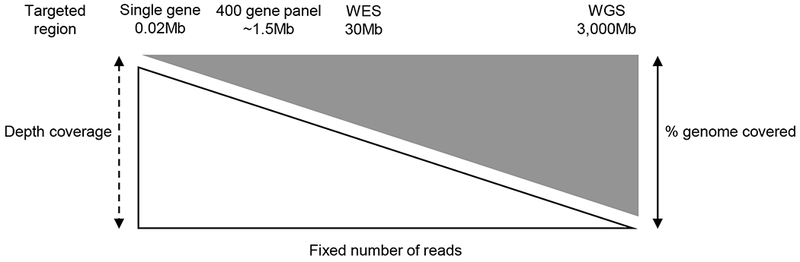
The graph assumes that a fixed number of sequencing reads are generated for each sequencing strategy. As the targeted region increases (left to right), the depth of coverage decreases. These changes in depth can be overcome by increasing the number of sequencing reads generated, but that also results in a significant increase in cost.
Clinical exploration of ctDNA in pediatric solid tumors
The available data indicate that circulating tumor DNA is detectable in a range of pediatric solid tumors at diagnosis, during treatment, and at the time of relapse (Table 2). Criteria for determining how ctDNA assays should adopted into clinical practice were recently outlined in an American Society of Clinical Oncology and College of American Pathologists joint review.8 They concluded that ctDNA assays must demonstrate 1) analytical validity, meaning the assay is able to detect a targeted variant with accuracy, reproducibility, and reliability, 2) clinical validity, meaning that the assay can divide a clinical group in to one or more cohorts with significantly different outcomes, and 3) clinical utility, that the knowledge gained from the assay significantly improves clinical care. At the time of this review, there are no ctDNA assays that have gained regulatory approval for clinical use in pediatric oncology. However, numerous opportunities exist for incorporating ctDNA assays into the care of patients with childhood cancers. Results from ongoing and future studies are needed to validate these assays and justify their use in routine clinical care.
TABLE 2:
Clinical evaluation of ctDNA in children with cancer.
| First Author | Year of publication | Disease(s) evaluated | Number of patients | Key finding |
|---|---|---|---|---|
| Combaret87 | 2002 | Neuroblastoma | 102 patients 72 controls |
MYCN amplification is detectable in ctDNA with high sensitivity and specificity using rtPCR. |
| Gotoh89 | 2005 | Neuroblastoma | 87 patients | MYCN amplification is detectable in ctDNA with high sensitivity and specificity using rtPCR. |
| Yagyu90 | 2008 | Neuroblastoma | 86 patients | Methylated-DCR2 gene is detected using rtPCR in ctDNA is associated with inferior outcomes. |
| Combaret88 | 2009 | Neuroblastoma | 267 patients | MYCN amplification is detectable in ctDNA with high sensitivity and specificity using rtPCR, especially in patients with Stage 3 or 4 disease. |
| Combaret104 | 2011 | Neuroblastoma | 142 patients 16 controls |
17q gain is variably detectable in ctDNA using rtPCR. |
| Yagyu105 | 2011 | Neuroblastoma | 24 patients | 11q loss is detectable in ctDNA with high sensitivity and specificity via rapid analysis of microsatellites using polymorphic markers. |
| Charlton96 | 2014 | Wilms tumor | 120 patients | ctDNA can be identified through methylome analysis and may be useful for disease monitoring. |
| Combaret91 | 2015 | Neuroblastoma | 114 patients | ALK mutations are found in ctDNA using ddPCR with high sensitivity and specificity. |
| Chicard78 | 2016 | Neuroblastoma | 70 patients | ctDNA copy-number profiling using NGS is feasible and shows high concordance with the tumor genomic profile. |
| Ferreira94 | 2016 | DSRCT | 1 patient | Patient-specific ddPCR probes can be created from tumor-specific breakpoint sequencing. |
| Hayashi93 | 2016 | Ewing sarcoma | 3 patients | Patient-specific ddPCR probes can be used to detect tumor-specific breakpoint DNA fragments with high sensitivity. |
| Krumbholz76 | 2016 | Ewing sarcoma | 20 patients | ddPCR detects patient-specific EWSR1 fusion sequences with high sensitivity and correlates with tumor volume and disease status. |
| Chicard63 | 2017 | Neuroblastoma | 19 patients | Using WES and deep target sequencing can identify tumor heterogeneity and evolution of treatment resistant clones. |
| Lodrini72 | 2017 | Neuroblastoma | 10 patients | ddPCR can be used to accurately discriminate ALK and MYCN copy-number changes. |
| Shukla84 | 2017 | Ewing sarcoma, DSRCT | Ewing sarcoma (11), DSRCT (6) | EWSR1 fusions are detectable using ddPCR and NGS approaches, and NGS identifies TP53 and STAG2 in patients with Ewing sarcoma. |
| Barris106 | 2018 | Osteosarcoma | 10 patients | Targeted NGS can be used to identify somatic aberrations in osteosarcoma tissue and ctDNA at diagnosis and during treatment, with the capacity to detect new alterations in serial ctDNA samples. |
| Klega82 | 2018 | Osteosarcoma, neuroblastoma, Ewing sarcoma, alveolar rhabdomyosarcoma, and WT | Ewing sarcoma (11), osteosarcoma (10), neuroblastoma (10), WT (8), alveolar rhabdomyosarcoma (7) | Changes in ctDNA levels correlate with treatment response and disease-specific genomic biomarkers are identifiable in ctDNA. |
| Shulman83 | 2018 | Ewing sarcoma, osteosarcoma | Ewing sarcoma (94), osteosarcoma (72) | Hybrid capture (Ewing sarcoma) and ultra-low pass WGS (osteosarcoma) identify ctDNA in ~50% of banked samples from diagnosis and detection/quantification of ctDNA is associated with inferior outcome. NGS identifies genomic features (TP53 mutation and STAG2 loss in Ewing sarcoma and 8q gain in osteosarcoma). |
DSRCT, Desmoplastic small round cell tumor; WT, Wilms tumor; rtPCR, real-time PCR; dPCR, droplet digital PCR
Diagnosis and prognostication
Circulating tumor DNA may provide a means of early cancer detection or of obtaining a diagnosis in cases where viable diagnostic tumor material cannot be obtained. To date, there have been no systematic evaluations of ctDNA for the purpose of early detection or diagnosis, however a number of studies have demonstrated the feasibility of using ctDNA for diagnostic purposes. We recently demonstrated that custom hybrid-capture NGS assays as well as an ULP-WGS NGS assay can be utilized in Ewing sarcoma, rhabdomyosarcoma, neuroblastoma, osteosarcoma and Wilms tumor to identify ctDNA without prior sequencing of tumor biopsy material.82,84 For now, it is unlikely that such technology would replace conventional biopsies given the frequent need to assess histologic and pathologic characteristics of the tumor tissue for making a definitive diagnosis and risk group classification. However, in instances where tissue cannot be obtained, analysis of ctDNA using a validated assay may ultimately provide a diagnostic alternative, particularly in diseases with pathognomonic translocations.
A number of studies have examined ctDNA for the purpose of detecting prognostic genomic features. For example, studies have demonstrated the feasibility of detecting MYCN amplification in the peripheral blood of patients with neuroblastoma using real-time PCR with a sensitivity of 75-100% and specificity of 100%.87–89 Another study utilized methylation of the DCR2 gene in serum using real-time PCR to identify patients with an inferior event-free survival in patients with and without MYCN amplification.90 Proof of concept studies found a correlation between plasma dPCR and tumor genomic analysis for detection of MYCN amplification, ALK amplification, and ALK hotspot mutations in neuroblastoma. 72,82,91,92 NGS assays have also been utilized to detect variants associated with a worse outcome, including TP53 mutations and STAG2 loss in Ewing sarcoma, 8q gain in osteosarcoma, MYCN amplification in neuroblastoma, 1q gain in Wilms tumor, and PAX3 gene rearrangements in alveolar rhabdomyosarcoma.82,83
While the prior studies identified known prognostic genetic features in ctDNA, we demonstrated that detection of ctDNA itself provided prognostic information at diagnosis in patients with Ewing sarcoma and osteosarcoma.83 Using a hybrid-capture NGS assay, ctDNA could be detected in over half of patients with Ewing sarcoma from a cooperative group biobank of plasma samples. In these patients, the detection of ctDNA was associated with an inferior EFS and OS. Similarly, using an ULP-WGS approach capitalizing on the complex CNVs in osteosarcoma, ctDNA could be detected in over half of all baseline plasma samples. For patients with newly diagnosed localized osteosarcoma, increasing ctDNA levels were associated with incrementally inferior EFS and OS. These assays are now undergoing clinical validation in a prospective, multicenter biomarker study.
Measurement of residual disease and disease surveillance
Multiple studies have evaluated ctDNA levels throughout treatment for the purposes of assessing response to therapy and post-treatment disease surveillance. In this setting, one would expect that a very high sensitivity may be required to detect the earliest signs of relapse, although the sensitivity needed for these assays to demonstrate clinical utility is unknown. In Ewing sarcoma, ctDNA levels by patient-specific dPCR were shown to drop early in treatment, and rise with disease recurrence, sometimes prior to radiologic detection.76,82,93 A case report described a similar approach utilized in a patient with desmoplastic small round cell tumor.94 In one study of patients with MYCN-amplified neuroblastoma, MYCN was detected by real-time PCR in plasma. The authors demonstrated that MYCN levels remained elevated in patients with sub-total resections, but returned to baseline levels in patients with complete resections.95 Wilms tumor has a genome with few recurrent mutations, but frequently has a characteristic aberrant methylation at 11p15. As proof of concept, differentially methylated regions have been used to identify ctDNA in patients with Wilms’ tumor.96
Highly sensitive measurements of treatment response, such as MRD testing in hematologic malignancies, are lacking in solid tumors. Instead, much reliance is placed on radiologic evaluations often carrying increased radiation exposure and cancer risk, or sedation in young children, possibly affecting neuro-developmental outcomes.97–99 In some cases, serial radiographic measurements of tumor burden have failed to be significantly predictive of outcome.100,101 If validated and utilized appropriately, ctDNA assays may provide a sensitive biomarker of treatment response for solid tumors and decrease reliance on serial imaging and sedation in young children.
Exploration of tumor biology and identification of targets for therapy
Increasingly, ctDNA is recognized as an avenue to explore tumor biology that may exceed what is possible with tumor tissue alone. Particularly in solid tumors, biopsies may under-represent tumor heterogeneity and evaluation of ctDNA may provider further insight into spatially diverse aspects of the tumor genome. Furthermore, serial surgical biopsies of tumors are not currently feasible, limiting our ability to assess tumor evolution in solid tumor malignancies. However, serial liquid biopsy sampling is readily implemented and may provide insights into tumor heterogeneity and its evolution in response to conventional and novel therapies. For example, sequencing of matched pre-treatment tumor samples, relapsed biopsy material and plasma samples from patients with relapsed breast cancer following chemotherapy allowed for longitudinal comparison of somatic copy-number alterations. While the majority of copy-number alterations were present in primary, metastatic, and ctDNA samples, copy-number events that were specific for metastatic samples, such as NOTCH2, AKT2 and AKT3, were also observed in the plasma samples obtained at the time of relapse.102
In pediatrics, tumor genomic heterogeneity and tumor evolution over time can also be characterized from the peripheral blood using ctDNA. Chicard et al. provides two illustrative examples of this approach in the context of neuroblastoma.78,103 In the first study of 70 patients, somatic copy-number alterations found in ctDNA were highly correlated with those in matched tumor samples. Interestingly, additional alterations were found in the ctDNA of some patients that were not present in the match tumor biopsy, indicating the possibility of heterogeneity among metastatic tumors.78 In particular, there were two alterations involving IGF1R and two involving TERT, genes thought to confer increased metastatic potential, that were identified only in the ctDNA. In the second study, a combination of whole-exome sequencing and targeted sequencing demonstrated the utility of ctDNA to understand tumor heterogeneity and tumor evolution of neuroblastoma.63 The authors found that sub-clonal events present at diagnosis frequently evolved into clonal mutations at relapse. These studies provide the most compelling evidence thus far that ctDNA may provide an avenue for exploration of tumor biology in pediatric solid tumors.
Challenges in clinical implementation
With an array of technologies now available for detection and characterization of ctDNA in pediatric solid tumors, it is important to consider the relevance of the information that can be derived from each assay as well as the logistical limitations of each approach. For example, NGS may be able to identify pathognomonic translocations in the plasma of patients with an undiagnosed tumor, however, clinical sequencing cannot currently be performed quickly enough for diagnostic purposes. In the case of prognostication, dPCR assays may have a greater sensitivity than NGS assays in the context of a minimal residual disease testing, but these assays often first require profiling of a tumor biopsy followed by the development and validation of patient-specific assays. While there may be sufficient time to develop such patient-specific assays for use in surveillance, after completion of therapy, this workflow is unlikely to be feasible for pre-treatment or early response-based risk-stratification. Therefore, we believe that a complement of approaches are needed to meet the clinical needs that could be addressed by ctDNA and we provide one example of how such assays could be applied longitudinally to patient care (Fig. 4).
Figure 4. Potential workflow for clinical application of different ctDNA assays.
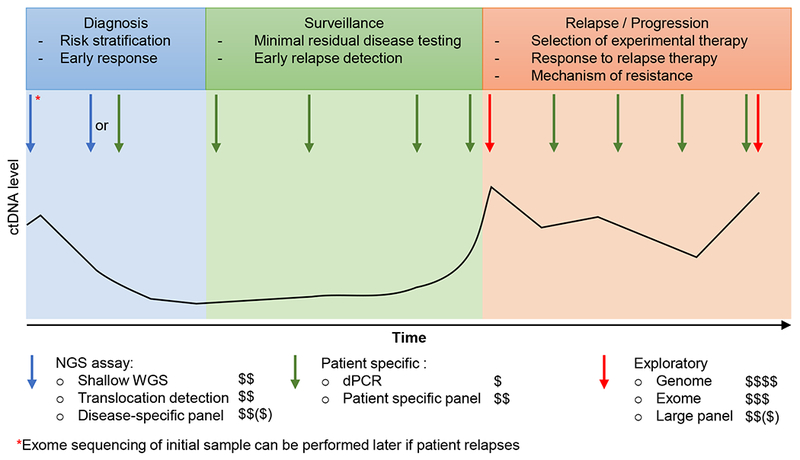
This graph depicts one potential strategy to combine complementary technologies to detect, quantify, and profile ctDNA throughout the course of a patient’s care. At diagnosis and early in therapy, focused NGS assays can be used to detect and quantify ctDNA without requiring existing genomic data from the tumor (blue arrows). Highly-sensitive patient-specific assays can be developed for use later in therapy to detect minimal residual disease and for surveillance (green arrows). Broad genomic profiling can performed on ctDNA at relapse or progression (red arrows) and compared to broad profiling of the initial diagnostic sample (red asterisk) to identify patterns of tumor evolution, treatment resistance, and identify new targetable variants for clinical trial enrollment. Dollar signs indicate relative cost of each approach. The black line indicates ctDNA levels in the patient throughout the course of treatment.
Future directions
The feasibility of detecting, quantifying, and profiling ctDNA in patients with pediatric solid tumors has now been established. A variety of technical approaches can be readily adapted to the development of ctDNA assays. Choosing the optimal approach depends on a thorough understanding of the strengths and limitations of each technology as well as the goals of each clinical scenario. This review provides a framework for making such decisions. Translating these new tools into clinically meaningful and validated assays will require a focused and coordinated effort within the pediatric oncology community. Validation of clinically relevant biomarkers for pediatric cancer can be challenging due to the relative rarity of these diseases. Each biomarker must be studied in the context of a well-defined patient cohort treated with a unified approach as part of a prospective analysis. Such studies in pediatric solid tumors are generally performed through multi-institutional cooperative efforts. Furthermore, studies sufficiently large to provide statistical power to validate a prognostic biomarker are typically conducted over several years. Given the rarity of these opportunities, it is critical that the appropriate samples be collected on multi-institutional trials whenever possible. The collection of blood is a minimal-risk procedure and as we explore which timepoints during therapy provide the most useful information, we recommend collecting blood samples prior to therapy, frequently during planned treatment, and serially while patients remain at risk for relapse. Other considerations, such as methods to collect and store samples for central processing have largely been standardized, making these studies immediately feasible.
As a scientific research tool, ctDNA provides a new avenue for understanding aspects of tumor biology that were largely inaccessible until now. The paucity of clinically annotated, matched diagnostic and relapsed tumor biopsy samples has resulted in few opportunities to study patterns of tumor evolution and treatment resistance in pediatric solid tumors. Rigorous efforts to collect and annotate matched diagnostic and relapsed blood samples from patients on banking studies and prospective trials will yield a new source of tumor DNA. Similarly, ctDNA technologies may help us explore the prevalence of tumor heterogeneity, a challenge that was previously restricted by a lack of access to geographically distinct biopsy samples collected simultaneously from a single tumor and from multiple metastatic tumors.
Beyond these immediate applications, ctDNA assays may ultimately have the potential to change the way patients are diagnosed by generating genomic information which can be integrated with clinical and pathologic data even when tumor biopsy material is significantly limited. As novel therapies become available for use in pediatric cancers, ctDNA may facilitate the identification of targetable mutations that direct the selection of specific agents and may also allow the detection of resistance mutations for patients undergoing targeted therapy. The early detection of ctDNA may inform the use of maintenance therapies designed to keep minimal residual disease in check and could play a role in surveillance strategies for patients with cancer predisposition syndromes.
Through the inherent collaborative nature of the pediatric oncology community and the tradition of enrolling our patients in prospective clinical trials, we have a unique opportunity to rapidly validate and implement ctDNA studies into the care of our patients. With plasma samples now being appropriately collected for ctDNA studies in the Children’s Oncology Group banking study (Project: EveryChild) and into the majority of recent prospective trials, we are well poised to validate the clinical and scientific potential of liquid biopsy studies in pediatric oncology.
Acknowledgments
Funding: We thank the Nuovo Soldati Fundation for the financial support (Research grant, S.Abbou).
Abbreviation table:
- ctDNA
Circulating tumor DNA
- PCR
Polymerase chain reaction
- SNVs
Single nucleotide variants
- MRD
minimal residual disease assays
- Indels
insertion/deletions
- NGS
next-generation sequencing
- dPCR
digital PCR
- WGS
whole-genome sequencing
- ULP-WGS
ultra-low passage WGS
Footnotes
Conflict of Interest statement: The authors declare that there is no conflict of interest regarding the publication of this article.
References
- 1.Mandel P, METAIS P. Comptes rendus des seances de la Societe de biologie et de ses filiales. 1948;142(3-4):241–243. [PubMed] [Google Scholar]
- 2.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Research. 1977;37(3):646–650. [PubMed] [Google Scholar]
- 3.Stroun M, Anker P, Lyautey J, Lederrey C, Maurice PA. Isolation and characterization of DNA from the plasma of cancer patients. European Journal of Cancer and Clinical Oncology. 1987;23(6):707–712. doi: 10.1016/0277-5379(87)90266-5 [DOI] [PubMed] [Google Scholar]
- 4.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Science translational medicine. 2014;6(224):224ra24. doi: 10.1126/scitranslmed.3007094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dawson S-J, Tsui DWY, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. The New England journal of medicine. 2013;368(13):1199–1209. doi: 10.1056/NEJMoa1213261 [DOI] [PubMed] [Google Scholar]
- 6.Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nature medicine. 2014;20(5):548–554. doi: 10.1038/nm.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nature Reviews Cancer. 2017;17(4):223–238. doi: 10.1038/nrc.2017.7 [DOI] [PubMed] [Google Scholar]
- 8.Merker JD, Oxnard GR, Compton C, et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. Journal of Clinical Oncology. 2018:JCO.2017.76.867. doi: 10.1200/JCO.2017.76.8671 [DOI] [PubMed] [Google Scholar]
- 9.Corcoran RB, Chabner BA. Application of Cell-free DNA Analysis to Cancer Treatment. New England Journal of Medicine. 2018;379(18):1754–1765. doi: 10.1056/NEJMra1706174 [DOI] [PubMed] [Google Scholar]
- 10.Remon J, Caramella C, Jovelet C, et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Annals of oncology : official journal of the European Society for Medical Oncology. 2017;28(4):784–790. doi: 10.1093/annonc/mdx017 [DOI] [PubMed] [Google Scholar]
- 11.Lecomte T, Berger A, Zinzindohoué F, et al. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. International Journal of Cancer. 2002;100(5):542–548. doi: 10.1002/ijc.10526 [DOI] [PubMed] [Google Scholar]
- 12.Kimura H, Kasahara K, Kawaishi M, et al. Detection of Epidermal Growth Factor Receptor Mutations in Serum as a Predictor of the Response to Gefitinib in Patients with Non ^ Small-Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(13):3915–3921. doi: 10.1158/1078-0432.CCR-05-2324 [DOI] [PubMed] [Google Scholar]
- 13.Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(45):16368–16373. doi: 10.1073/pnas.0507904102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian X, Liu JJ, Sun Y, et al. Circulating cell-free DNA has a high degree of specificity to detect exon 19 deletions and the single-point substitution mutation L858R in non-small cell lung cancer. Oncotarget. 2016;7(20). doi: 10.18632/oncotarget.8684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forshew T, Murtaza M, Parkinson C, et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Science translational medicine. 2012;4(136):136ra68–136ra68. doi: 10.1126/scitranslmed.3003726 [DOI] [PubMed] [Google Scholar]
- 16.Jenkins S, Yang JCH, Ramalingam SS, et al. Plasma ctDNA Analysis for Detection of the EGFR T790M Mutation in Patients with Advanced Non-Small Cell Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2017;12(7):1061–1070. doi: 10.1016/j.jtho.2017.04.003 [DOI] [PubMed] [Google Scholar]
- 17.Schmiegel W, Scott RJ, Dooley S, et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: concordance of results from circulating tumor DNA and tissue-based RAS testing. Molecular oncology. 2017;11(2):208–219. doi: 10.1002/1878-0261.12023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vidal J, Muinelo L, Dalmases A, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Annals of Oncology. 2017;28(6):1325–1332. doi: 10.1093/annonc/mdx125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in egfrmutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clinical Cancer Research. 2014;20(6):1698–1705. doi: 10.1158/1078-0432.CCR-13-2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA oncology. 2016;2(8):1014–1022. doi: 10.1001/jamaoncol.2016.0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harttrampf AC, Lacroix L, Deloger M, et al. Molecular Screening for Cancer Treatment Optimization (MOSCATO-01) in pediatric patients: A single-institutional prospective molecular stratification trial. Clinical Cancer Research. 2017;23(20):6101–6112. doi: 10.1158/1078-0432.CCR-17-0381 [DOI] [PubMed] [Google Scholar]
- 22.Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321–327. doi: 10.1038/nature25480 [DOI] [PubMed] [Google Scholar]
- 23.Harris MH, DuBois SG, Glade Bender JL, et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA oncology. 2016;02176(5):608–615. doi: 10.1001/jamaoncol.2015.5689 [DOI] [PubMed] [Google Scholar]
- 24.Huether R, Dong L, Chen X, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nature Communications. 2014;5:1–7. doi: 10.1038/ncomms4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555(7696):371–376. doi: 10.1038/nature25795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siegel DA, King J, Tai E, Buchanan N, Ajani UA, Li J. Cancer incidence rates and trends among children and adolescents in the United States, 2001–2009. Pediatrics. 2014;134(4):e945–55. doi: 10.1542/peds.2013-3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hocking J, Mithraprabhu S, Kalff A, et al. Liquid biopsies for liquid tumors: emerging potential of circulating free nucleic acid evaluation for the management of hematologic malignancies. Cancer Biology & Medicine. 2016;13(2):215–225. doi: 10.20892/j.issn.2095-3941.2016.0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frickhofen N, Müller E, Sandherr M, et al. Rearranged Ig heavy chain DNA is detectable in cell-free blood samples of patients with B-cell neoplasia. Blood. 1997;90(12):4953–4960. [PubMed] [Google Scholar]
- 31.Hohaus S, Giachelia M, Massini G, et al. Cell-free circulating DNA in Hodgkin’s and non-Hodgkin’s lymphomas. Annals of Oncology. 2009;20(8):1408–1413. doi: 10.1093/annonc/mdp006 [DOI] [PubMed] [Google Scholar]
- 32.Kurtz DM, Green MR, Bratman SV, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. The American Society of Hematology. 2015;125(24):3679–3688. doi: 10.1182/blood-2015-03-635169.D.M.K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armand P, Oki Y, Neuberg DS, et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. British Journal of Haematology. 2013;163(1):123–126. doi: 10.1111/bjh.12439 [DOI] [PubMed] [Google Scholar]
- 34.Camus V, Sarafan-Vasseur N, Bohers E, et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B-cell lymphoma. Leukemia & Lymphoma. 2016;57(9):2171–2179. doi: 10.3109/10428194.2016.1139703 [DOI] [PubMed] [Google Scholar]
- 35.Kwok M, Wu SP, Mo C, Summers T, Roschewski M. Circulating Tumor DNA to Monitor Therapy for Aggressive B-Cell Lymphomas. Current Treatment Options in Oncology. 2016;17(9). doi: 10.1007/s11864-016-0425-1 [DOI] [PubMed] [Google Scholar]
- 36.Spina V, Bruscaggin A, Cuccaro A, et al. Circulating tumor DNA reveals genetics, clonal evolution and residual disease in classical Hodgkin lymphoma. Blood. 2018;131(22):blood-2017-11-812073. doi: 10.1182/blood-2017-11-812073 [DOI] [PubMed] [Google Scholar]
- 37.Schwarz AK, Stanulla M, Cario G, et al. Quantification of free total plasma DNA and minimal residual disease detection in the plasma of children with acute lymphoblastic leukemia. Annals of Hematology. 2009;88(9):897–905. doi: 10.1007/s00277-009-0698-6 [DOI] [PubMed] [Google Scholar]
- 38.Cheng SH, Lau KM, Li CHCK, et al. Minimal Residual Disease-Based Risk Stratification in Chinese Childhood Acute Lymphoblastic Leukemia by Flow Cytometry and Plasma DNA Quantitative Polymerase Chain Reaction. PLoS ONE. 2013;8(7):e69467. doi: 10.1371/journal.pone.0069467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balaña C, Ramirez JL, Taron M, et al. O6-methyl-guanine-DNA methyltransferase Methylation in Serum and Tumor DNA Predicts Response to 1,3-Bis(2-Chloroethyl)-1-nitrosourea but not to Temozolamide Plus Cisplatin in Glioblastoma Multiforme. Clinical Cancer Research. 2003;9(April):1461–1468. [PubMed] [Google Scholar]
- 40.Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro-Oncology. 2010;12(2):173–180. doi: 10.1093/neuonc/nop041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Connolly ID, Li Y, Gephart MH, Nagpal S. The “Liquid Biopsy”: the Role of Circulating DNA and RNA in Central Nervous System Tumors. Current Neurology and Neuroscience Reports. 2016;16(3):1–8. doi: 10.1007/s11910-016-0629-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez-Ricarte F, Mayor R, Martínez-Sáez E, et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2018;24(12):2812–2819. doi: 10.1158/1078-0432.CCR-17-3800 [DOI] [PubMed] [Google Scholar]
- 43.Panditharatna E, Kilburn LB, Aboian MS, et al. Clinically Relevant and Minimally Invasive Tumor Surveillance of Pediatric Diffuse Midline Gliomas Using Patient-Derived Liquid Biopsy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018. doi: 10.1158/1078-0432.CCR-18-1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer and Metastasis Reviews. 2016;35(3):347–376. doi: 10.1007/s10555-016-9629-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan EM, Schur PH, Carr RI, Kunkel HG. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. The Journal of Clinical Investigation. 1966;45(11):1732–1740. doi: 10.1172/JCI105479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vakrakou AG, Boiu S, Ziakas PD, Xingi E, Boleti H, Manoussakis MN. Systemic activation of NLRP3 inflammasome in patients with severe primary Sjögren’s syndrome fueled by inflammagenic DNA accumulations. Journal of Autoimmunity. March 2018. doi: 10.1016/j.jaut.2018.02.010 [DOI] [PubMed] [Google Scholar]
- 47.Davis GL, Davis JS. Detection of circulating DNA by counterimmunoelectrophoresis (CIE). Arthritis and Rheumatism. 1973;16(1):52–58. [DOI] [PubMed] [Google Scholar]
- 48.Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet (London, England). 1997;350(9076):485–487. doi: 10.1016/S0140-6736(97)02174-0 [DOI] [PubMed] [Google Scholar]
- 49.Hayward J, Chitty LS. Beyond screening for chromosomal abnormalities: Advances in non-invasive diagnosis of single gene disorders and fetal exome sequencing. Seminars in Fetal & Neonatal Medicine. January 2018. doi: 10.1016/j.siny.2017.12.002 [DOI] [PubMed] [Google Scholar]
- 50.Rodrigues Filho EM, Simon D, Ikuta N, et al. Elevated cell-free plasma DNA level as an independent predictor of mortality in patients with severe traumatic brain injury. Journal of Neurotrauma. 2014;31(19):1639–1646. doi: 10.1089/neu.2013.3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vajpeyee A, Wijatmiko T, Vajpeyee M, Taywade O. Cell free DNA: A Novel Predictor of Neurological Outcome after Intravenous Thrombolysis and/or Mechanical Thrombectomy in Acute Ischemic Stroke Patients. Neurointervention. 2018;13(1):13–19. doi: 10.5469/neuroint.2018.13.1.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vlaminck I De, Martin L, Kertesz M, et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proceedings of the National Academy of Sciences. 2015;112(43):13336–13341. doi: 10.1073/pnas.1517494112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nature medicine. 2008;14(9):985–990. doi: 10.1038/nm.1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mouliere F, Robert B, Peyrotte EA, et al. High Fragmentation Characterizes Tumour-Derived Circulating DNA. PLOS ONE. 2011;6(9):e23418. doi: 10.1371/journal.pone.0023418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Underhill HR, Kitzman JO, Hellwig S, et al. Fragment Length of Circulating Tumor DNA. PLOS Genetics. 2016;12(7):e1006162. doi: 10.1371/journal.pgen.1006162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563(7732):579–583. doi: 10.1038/s41586-018-0703-0 [DOI] [PubMed] [Google Scholar]
- 57.Goodall J, Mateo J, Yuan W, et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discovery. 2017;7(9):1006–1017. doi: 10.1158/2159-8290.CD-17-0261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quigley D, Alumkal JJ, Wyatt AW, et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discovery. 2017;7(9):999–1005. doi: 10.1158/2159-8290.CD-17-0146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manier S, Park J, Capelletti M, et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nature Communications. 2018;9(1):1691. doi: 10.1038/s41467-018-04001-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vandekerkhove G, Todenhöfer T, Annala M, et al. Circulating Tumor DNA Reveals Clinically Actionable Somatic Genome of Metastatic Bladder Cancer. Clinical Cancer Research. 2017;23(21):6487–6497. doi: 10.1158/1078-0432.CCR-17-1140 [DOI] [PubMed] [Google Scholar]
- 61.Favero F, Joshi T, Marquard AM, et al. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Annals of Oncology. 2015;26(1):64–70. doi: 10.1093/annonc/mdu479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nature communications. 2017;8(1):1324. doi: 10.1038/s41467-017-00965-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chicard M, Colmet Daage L, Clement N, et al. Whole exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clinical Cancer Research. 2017:clincanres.1586.2017. doi: 10.1158/1078-0432.CCR-17-1586 [DOI] [PubMed] [Google Scholar]
- 64.Nawroz H, Koch W, Anker P, Stroun M, Sidransky D. Microsatellite alterations in serum DNA of head and neck cancer patients. Nature Medicine. 1996;2(9):1035–1037. [DOI] [PubMed] [Google Scholar]
- 65.Garlan F, Blanchet B, Kramkimel N, et al. Circulating Tumor DNA Measurement by Picoliter Droplet-Based Digital PCR and Vemurafenib Plasma Concentrations in Patients with Advanced BRAF-Mutated Melanoma. Targeted Oncology. 2017;12(3):365–371. doi: 10.1007/s11523-017-0491-8 [DOI] [PubMed] [Google Scholar]
- 66.van Ginkel JH, Huibers MMH, van Es RJJ, de Bree R, Willems SM. Droplet digital PCR for detection and quantification of circulating tumor DNA in plasma of head and neck cancer patients. BMC Cancer. 2017;17(1):428. doi: 10.1186/s12885-017-3424-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanmamed MF, Fernandez-Landazuri S, Rodriguez C, et al. Quantitative Cell-Free Circulating BRAFV600E Mutation Analysis by Use of Droplet Digital PCR in the Follow-up of Patients with Melanoma Being Treated with BRAF Inhibitors. Clinical Chemistry. 2015;61(1):297–304. doi: 10.1373/clinchem.2014.230235 [DOI] [PubMed] [Google Scholar]
- 68.Douillard J-Y, Ostoros G, Cobo M, et al. Gefitinib Treatment in EGFR Mutated Caucasian NSCLC. Journal of Thoracic Oncology. 2014;9(9):1345–1353. doi: 10.1097/JTO.0000000000000263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pekin D, Skhiri Y, Baret JC, et al. Quantitative and sensitive detection of rare mutations using droplet-based microfluidics. Lab on a Chip. 2011;11(13):2156–2166. doi: 10.1039/c1lc20128j [DOI] [PubMed] [Google Scholar]
- 70.Jia S, Zhang R, Li Z, Li J. Clinical and biological significance of circulating tumor cells, circulating tumor DNA, and exosomes as biomarkers in colorectal cancer. Oncotarget. 2017;8(33):55632–55645. doi: 10.18632/oncotarget.17184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Do H, Cameron D, Molania R, et al. Digital PCR of Genomic Rearrangements for Monitoring Circulating Tumour DNA. Advances in Experimental Medicine and Biology. 2016;924:139–146. doi: 10.1007/978-3-319-42044-8_27 [DOI] [PubMed] [Google Scholar]
- 72.Lodrini M, Sprüssel A, Astrahantseff K, et al. Using droplet digital PCR to analyze MYCN and ALK copy number in plasma from patients with neuroblastoma. Oncotarget. 2017;8(49):85234–85251. doi: 10.18632/oncotarget.19076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yung TKF, Chan KCA, Mok TSK, Tong J, To K-F, Lo YMD. Single-Molecule Detection of Epidermal Growth Factor Receptor Mutations in Plasma by Microfluidics Digital PCR in Non–Small Cell Lung Cancer Patients. Clinical Cancer Research. 2009;15(6):2076–2084. doi: 10.1158/1078-0432.CCR-08-2622 [DOI] [PubMed] [Google Scholar]
- 74.Olsson E, Winter C, George A, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO molecular medicine. 2015;7(8):1034–1047. doi: 10.15252/emmm.201404913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Swisher EM, Wollan M, Mahtani SM, et al. Tumor-specific p53 sequences in blood and peritoneal fluid of women with epithelial ovarian cancer. American Journal of Obstetrics and Gynecology. 2005;193(3):662–667. doi: 10.1016/j.ajog.2005.01.054 [DOI] [PubMed] [Google Scholar]
- 76.Krumbholz M, Hellberg J, Steif B, et al. Genomic EWSR1 Fusion Sequence as Highly Sensitive and Dynamic Plasma Tumor Marker in Ewing Sarcoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(17):4356–4365. doi: 10.1158/1078-0432.CCR-15-3028 [DOI] [PubMed] [Google Scholar]
- 77.Janku F, Angenendt P, Tsimberidou AM, et al. Actionable mutations in plasma cell-free DNA in patients with advanced cancers referred for experimental targeted therapies. Oncotarget. 2015;6(14):12809–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chicard M, Boyault S, Daage LC, et al. Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(22):5564–5573. doi: 10.1158/1078-0432.CCR-16-0500 [DOI] [PubMed] [Google Scholar]
- 79.Leary RJ, Sausen M, Kinde I, et al. Detection of Chromosomal Alterations in the Circulation of Cancer Patients with Whole-Genome Sequencing. Science translational medicine. 2012;4(162):162ra154. doi: 10.1126/scitranslmed.3004742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heitzer E, Ulz P, Belic J, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Medicine. 2013;5(4):30. doi: 10.1186/gm434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Roy N, Van Der Linden M, Menten B, et al. Shallow whole genome sequencing on circulating cell-free DNA allows reliable noninvasive copy-number profiling in neuroblastoma patients. Clinical Cancer Research. 2017;23(20):6305–6315. doi: 10.1158/1078-0432.CCR-17-0675 [DOI] [PubMed] [Google Scholar]
- 82.Klega K, Imamovic-Tuco A, Ha G, et al. Detection of Somatic Structural Variants Enables Quantification and Characterization of Circulating Tumor DNA in Children With Solid Tumors. JCO precision oncology. 2018;2018(2):1–13. doi: 10.1200/PO.17.00285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shulman DS, Klega K, Imamovic-Tuco A, et al. Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group. British journal of cancer. 2018;(July):1–7. doi: 10.1038/s41416-018-0212-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shukla NN, Patel JA, Magnan H, et al. Plasma DNA-based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion-positive sarcomas. JCO precision oncology. 2017;2017(1):1–11. doi: 10.1200/PO.16.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ulz P, Auer M, Heitzer E. Detection of Circulating Tumor DNA in the Blood of Cancer Patients: An Important Tool in Cancer Chemoprevention. Methods in Molecular Biology (Clifton, NJ). 2016;1379:45–68. doi: 10.1007/978-1-4939-3191-0_5 [DOI] [PubMed] [Google Scholar]
- 86.Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nature biotechnology. 2016;34(5):547–555. doi: 10.1038/nbt.3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Combaret V, Audoynaud C, Iacono I, et al. Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer research. 2002;62(13):3646–3648. [PubMed] [Google Scholar]
- 88.Combaret V, Hogarty MD, London WB, et al. Influence of neuroblastoma stage on serum-based detection of MYCN amplification. Pediatric blood & cancer. 2009;53(3):329–331. doi: 10.1002/pbc.22009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gotoh T, Hosoi H, Iehara T, et al. Prediction of MYCN amplification in neuroblastoma using serum DNA and real-time quantitative polymerase chain reaction. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(22):5205–5210. doi: 10.1200/JCO.2005.02.014 [DOI] [PubMed] [Google Scholar]
- 90.Yagyu S, Gotoh T, Iehara T, et al. Circulating methylated-DCR2 gene in serum as an indicator of prognosis and therapeutic efficacy in patients with MYCN nonamplified neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(21):7011–7019. doi: 10.1158/1078-0432.CCR-08-1249 [DOI] [PubMed] [Google Scholar]
- 91.Combaret V, Iacono I, Bellini A, et al. Detection of tumor ALK status in neuroblastoma patients using peripheral blood. Cancer Medicine. 2015;4(4):540–550. doi: 10.1002/cam4.414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kurihara S, Ueda Y, Onitake Y, et al. Circulating free DNA as non-invasive diagnostic biomarker for childhood solid tumors. Journal of Pediatric Surgery. 2015;50(12):2094–2097. doi: 10.1016/j.jpedsurg.2015.08.033 [DOI] [PubMed] [Google Scholar]
- 93.Hayashi M, Chu D, Meyer CF, et al. Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer. 2016;122(19):3015–3023. doi: 10.1002/cncr.30144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferreira EN, Barros BDF, De Souza JE, et al. A genomic case study of desmoplastic small round cell tumor: comprehensive analysis reveals insights into potential therapeutic targets and development of a monitoring tool for a rare and aggressive disease. Human Genomics. 2016;10(1):1–13. doi: 10.1186/s40246-016-0092-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kojima M, Hiyama E, Fukuba I, et al. Detection of MYCN amplification using blood plasma: Noninvasive therapy evaluation and prediction of prognosis in neuroblastoma. Pediatric Surgery International. 2013;29(11):1139–1145. doi: 10.1007/s00383-013-3374-9 [DOI] [PubMed] [Google Scholar]
- 96.Charlton J, Williams RD, Weeks M, et al. Methylome analysis identifies a Wilms tumor epigenetic biomarker detectable in blood. Genome Biology. 2014;15(8):1–8. doi: 10.1186/s13059-014-0434-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davidson AJ, Disma N, De Graaff JC, et al. Neurodevelopmental outcome at 2 years of age after general anaesthesia and awake-regional anaesthesia in infancy (GAS): An international multicentre, randomised controlled trial. The Lancet. 2016;387(10015):239–250. doi: 10.1016/S0140-6736(15)00608-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun LS, Li G, Miller TLK, et al. Association between a single general anesthesia exposure before age 36 months and neurocognitive outcomes in later childhood. JAMA - Journal of the American Medical Association. 2016;315(21):2312–2320. doi: 10.1001/jama.2016.6967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pearce MS, Salotti JA, Little MP, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. The Lancet. 2012;380(9840):499–505. doi: 10.1016/S0140-6736(12)60815-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guenther LM, Rowe RG, Acharya PT, et al. Response Evaluation Criteria in Solid Tumors (RECIST) following neoadjuvant chemotherapy in osteosarcoma. Pediatric Blood & Cancer. 2017;(September):e26896. doi: 10.1002/pbc.26896 [DOI] [PubMed] [Google Scholar]
- 101.Harrison DJ, Parisi MT, Shulkin BL, et al. 18F 2Fluoro-2deoxy-D-glucose positron emission tomography (FDG-PET) response to predict event-free survival (EFS) in intermediate risk (IR) or high risk (HR) rhabdomyosarcoma (RMS): A report from the Soft Tissue Sarcoma Committee of the Children’s Oncolog. Journal of Clinical Oncology. 2016;34(15_suppl):10549. doi: 10.1200/JCO.2016.34.15_suppl.10549 [DOI] [Google Scholar]
- 102.Stover DG, Parsons HA, Ha G, et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. Journal of Clinical Oncology. 2018;36(6):543–553. doi: 10.1200/JCO.2017.76.0033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chicard M, Colmet-Daage L, Clement N, et al. Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(4):939–949. doi: 10.1158/1078-0432.CCR-17-1586 [DOI] [PubMed] [Google Scholar]
- 104.Combaret V, Bréjon S, Iacono I, et al. Determination of 17q gain in patients with neuroblastoma by analysis of circulating DNA. Pediatric blood & cancer. 2011;56(5):757–761. doi: 10.1002/pbc.22816 [DOI] [PubMed] [Google Scholar]
- 105.Yagyu S, Iehara T, Gotoh T, et al. Preoperative analysis of 11q loss using circulating tumor-released DNA in serum: a novel diagnostic tool for therapy stratification of neuroblastoma. Cancer letters. 2011;309(2):185–189. doi: 10.1016/j.canlet.2011.05.032 [DOI] [PubMed] [Google Scholar]
- 106.Barris DM, Weiner SB, Dubin RA, et al. Detection of circulating tumor DNA in patients with osteosarcoma. Oncotarget. 2018;9(16):12695–12704. doi: 10.18632/oncotarget.24268 [DOI] [PMC free article] [PubMed] [Google Scholar]