

Abstract
Objective
To examine whether indices of Parkinson disease (PD) pathology and other brain pathologies are associated with the progression of parkinsonism in older adults.
Methods
We used data from decedents who had undergone annual clinical testing prior to death and structured brain autopsy. Parkinsonism was based on assessment with a modified Unified Parkinson's Disease Rating Scale and a clinical diagnosis of PD was based on medical history. We used a series of mixed-effects models controlling for age and sex to investigate the association of PD pathology (nigral neuronal loss and Lewy bodies) and indices of 8 other brain pathologies with the progression of parkinsonism prior to death.
Results
During an average of 8.5 years’ follow-up, more than half (771/1,430, 53.9%) developed parkinsonism proximate to death. On average, parkinsonism was progressive (estimate 0.130, SE 0.005, p < 0.001) in all older adults, but more rapid in adults with a clinical diagnosis of PD (n = 52; 3.6%) (estimate 0.066, SE 0.021, p < 0.001). Progression of parkinsonism was more rapid in adults with PD pathology (estimate 0.087, SE 0.013, p < 0.001). Alzheimer disease and several cerebrovascular pathologies were all independently associated with more rapid progression (all p values <0.05). The association between a higher person-specific weighted pathology score and more rapidly progressive parkinsonism did not differ between individuals with and without a clinical diagnosis of PD (estimate 0.003, SE 0.047, p = 0.957).
Conclusion
The rate of progressive parkinsonism in older adults with and without a clinical diagnosis of PD is related to the burden of mixed brain pathologies.
Following dementia and stroke, Parkinson disease (PD) is the third most familiar neurodegenerative disease, but it is uncommon in the general population, with a reported prevalence of 2% to 5% of persons by age 85.1,2 The pathology of PD, loss of nigral dopaminergic neurons and the formation of Lewy bodies in these same neurons, is known to accumulate over many years before a clinical diagnosis of PD.3,4 Hence, identifying clinical antecedents of PD prior to its clinical diagnosis, prodromal PD, offers the potential for early intervention and prevention.5 While PD is uncommon, accumulating evidence suggests that parkinsonism, a heterogeneous disorder, may affect more than half of older adults by age 85.6
Our prior studies have shown that indices of PD pathology including some degree of nigral degeneration and/or insoluble α-synuclein (Lewy bodies) may be present in up to 40% of older persons without PD and associated with parkinsonism.7 These data and other work8–10 show that the presence of PD pathology is much more common than the presence of clinical PD and that it is an underestimated cause of parkinsonism in older adults who do not meet clinical criteria for PD. However, the nosology of parkinsonism in older adults and its relationship to prodromal PD is still debated in part because there is a paucity of data about its pathologic substrate.11,12 In prior cross-sectional studies, we showed that PD and other brain pathologies are related to the severity of parkinsonism in older adults without a clinical diagnosis of PD proximate to death.13 However, it is unknown whether these pathologies are associated with the progression of parkinsonism over time and contribute to declining motor function in older adults. To address this question, we used data from more than 1,400 older adults who underwent annual assessment of parkinsonism and structured brain autopsy.
Methods
Participants
Participants came from 3 community-based longitudinal cohort studies of diverse participants, the Religious Orders Study (ROS), the Rush Memory and Aging Project (MAP), and the Minority Aging Research Study (MARS).14,15 Participants were enrolled without known dementia and agreed to annual clinical evaluations and autopsy. The ROS and MAP participants were predominantly Caucasian Americans and the MARS participants were all African Americans. Of note, all 3 studies share a large common core of testing batteries and uniform structured clinical evaluations facilitating combined analysis as done previously.16
At the time of these analyses, 4,084 individuals had been recruited to these studies (MAP, n = 1,939; ROS, n = 1,412; MARS, n = 731). We excluded 2,654 who were still living or did not undergo autopsy (MAP, n = 1,233; ROS, n = 712; MARS, n = 709). This left 1,430 participants who had undergone autopsy and completed baseline and follow-up assessment of parkinsonism for these analyses (MAP, n = 706; ROS, n = 702; MARS, n = 22). Their characteristics by study included the following: MAP: age at death 89.9 years (SD = 6.20); 485 (68.7%) women; years of education 14.5 (2.89); and last examination before death 1.6 years (1.82). ROS: age at death 87.9 years (SD = 6.97); 449 (64.0%) women; years of education 18.1 (3.48); last examination before death 1.2 years (1.39). MARS: age at death 82.8 years (SD = 9.54); 18 (81.8%) women; years of education 14.5 (2.89); last examination before death 1.6 years (1.95).
Assessment of parkinsonism and PD
Nurse clinicians assessed parkinsonian gait, rigidity, bradykinesia, and tremor annually using 26 items from a modified Unified Parkinson's Disease Rating Scale (UPDRS). These measures have high interrater reliability and short-term stability among nurses and compared with a movement disorders specialist.17
Global parkinsonian score
A continuous measure of parkinsonism is useful for examining the associations of postmortem indices with the progression of parkinsonism. A score for each of the 4 parkinsonian signs was based on the sum of the scores for each of its individual items assessed with the UPDRS. The scores for the 4 parkinsonian signs were averaged to provide a continuous global parkinsonian score as previously described.17
Clinical parkinsonism category
A categorical measure of parkinsonism is necessary to estimate the cases of incident parkinsonism. A previously validated parkinsonism category was constructed based on the number of the 4 parkinsonian signs present with UPDRS assessment. A parkinsonian sign was present if 2 or more of its items were scored as a mild abnormality. Clinical parkinsonism was present if at least 2 of the 4 parkinsonian signs were present.13
PD diagnosis
After categorizing whether an individual showed parkinsonism based on each annual UPDRS assessment, we also recorded whether they ever reported receiving a clinical diagnosis of PD for which they had received PD medications including levodopa or a dopamine agonist. Complementing self-reported history, our annual structured examination included inspection of all current medications to identify PD medications.7,18
Demographic covariates
Date of birth and sex were collected through a participant interview. Age in years was computed from self-reported date of birth and date of death.
Postmortem indices
Brain removal, tissue sectioning and preservation, and a uniform gross and microscopic examination with quantification of postmortem indices followed a standard protocol.19 Postmortem interval was 9.1 hours (SD = 8.55) after death. In prior work, we developed summary pathology measures combining samples from different brain regions as measures from diverse regions were highly correlated and composite measures improve their metric properties as previously described.20
PD pathology
Nigral neuronal loss was assessed in sections of the substantia nigra using a semiquantitative scale (0–3).21 Lewy body disease pathology was assessed in 6 brain regions using a monoclonal phosphorylated antibody to α-synuclein as previously described and was treated as present or absent in these analyses.21 PD pathology is defined as moderate to severe nigral neuronal loss and the presence of Lewy bodies. As we have done in prior studies, we stratified individuals into 4 groups based on the presence of moderate-severe nigral neuronal loss and/or any Lewy body pathology: (1) nigral neuronal loss alone, (2) Lewy body pathology alone, and (3) a pathologic diagnosis of PD pathology if both nigral neuronal loss and Lewy body pathology were present. These 3 groups were compared to a reference group in which neither nigral neuronal loss nor Lewy body pathology was observed.
Alzheimer pathology
Bielschowsky silver stain was used to visualize neuritic plaques, diffuse plaques, and neurofibrillary tangles in 5 brain regions, as previously described.22 Standardized scores for plaque and tangle counts in each were then averaged across the 5 regions. We averaged these 3 summary scores to yield a global measure of Alzheimer disease (AD) pathology used in these analyses.23
TDP-43
Immunohistochemistry with TAR5P-1D3 antibody was used to assess TAR DNA-binding protein 43 (TDP-43) in 5 brain regions. Three stages of TDP-43 distribution were recognized (stage 1, localized to amygdala; stage 2, extension to hippocampus and/or entorhinal cortex; stage 3, extension to the neocortex) and scores ranged from 0 to 3 as described previously.24
Hippocampal sclerosis
Hippocampal sclerosis was evaluated unilaterally in a coronal section and graded as absent or present or absent based on severe neuronal loss and gliosis in CA1 and/or subiculum.25
Macroscopic cerebral infarcts
We reviewed 1-cm slabs and recorded the age, volume (in mm3), side, and location of all cerebral infarcts visible to the eye as previously reported.26 Only chronic infarcts were included in these analyses. Macroinfarcts were treated as present or absent for these analyses.
Microscopic cerebral infarcts
Nine brain regions were dissected, processed, and embedded for review by a neuropathologist to identify microscopic infarcts as previously described.27 Microinfarcts were treated as present or absent for these analyses.
Cerebral atherosclerosis
This was assessed on gross examination of the anterior, middle, and posterior cerebral arteries and their proximal branches at the circle of Willis using a semiquantitative scale (0–3) of all visualized arteries.
Cerebral arteriolosclerosis
Arteriolosclerosis describes degenerative and fibrohyalinotic thickening of arterioles with narrowing of the vascular lumen. We evaluated the vessels of the anterior basal ganglia with a semiquantitative grading system (0–3) as previously described.19
Cerebral amyloid angiopathy
Immunohistochemistry was used to score (0–4) β-amyloid deposition in meningeal and parenchymal vessels in 5 neocortical regions. The maximum score between the meningeal and parenchymal amyloid angiopathy scores was used as the amyloid angiopathy pathology score for that region. Scores were then averaged across the 5 regions. We used this overall average score to develop a 4-level rating scale, based on the data distribution and pathology severity at the different levels as determined by the neuropathologist.28
Analyses
We used a series of linear mixed-effect models to examine the association of indices of PD and other brain pathologies with the progression of parkinsonism prior to death. The distribution of global parkinsonism was positively skewed and was square-root transformed before these analyses. The outcome for these models was repeated annual measures of the global parkinsonism score. Each model included a term, time, the number of years prior to death, whose coefficient estimates the mean annual rate of change in parkinsonism (i.e., slope). Each predictor added to the model included a term for its estimated association with global parkinsonism proximate to death and a second term for its interaction with time, the annual rate of change in global parkinsonism. All the models included terms for age and sex and their interaction with time. First, we calculated the progression of parkinsonism in all individuals and included a term for the diagnosis of PD and its interaction with time to determine whether the rate of progression differed in older adults with and without PD.
Next, to examine the association of PD pathology (nigral neuronal loss and Lewy body pathology) with progressive parkinsonism, we compared the rate of progression for 3 groups with one or both elements of PD pathology to a reference group, individuals without evidence of either element of PD pathology.
In further analyses, we retained terms for age, sex, and PD pathology (nigral neuronal loss and Lewy body pathology) while adding additional terms for each of 8 other postmortem indices to determine whether they were independently associated with the progression of parkinsonism.
There are about 250 combinations of mixed pathologies in this sample of more than 1,400 autopsied older persons analyzed in this study.29 This highlights the need for an approach to illustrate the additive effect of multiple pathologies on progressive parkinsonism. Risk scores have been used to aggregate the additive risks of multiple genes or clinical risk factors.30,31 Therefore, we constructed a person-specific weighted pathology score to illustrate the aggregated effects of varied combinations of mixed pathologies on the progression of parkinsonism. The weighted pathology score calculated for each person was based on the estimated coefficients (β values) obtained in table 4, model 1, for each pathology independently associated with the progression of parkinsonism.
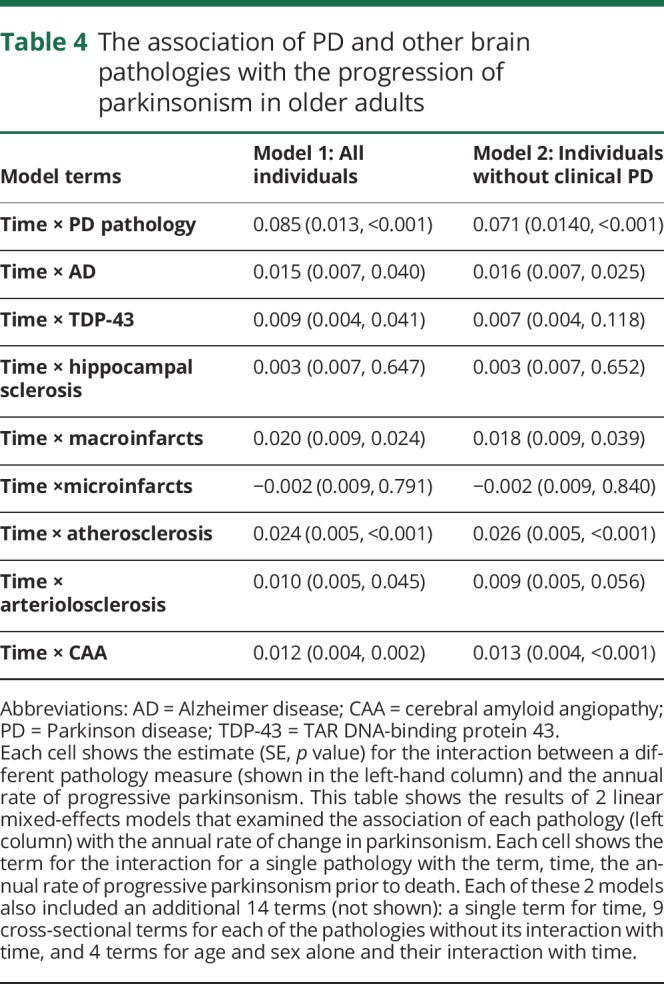
Table 4.
The association of PD and other brain pathologies with the progression of parkinsonism in older adults
Standard protocol approvals, registrations, and patient consents
The study was approved by the institutional review board of Rush University Medical Center. Written informed consent was obtained from all study participants as was an Anatomical Gift Act for organ donation.
Data availability
All data included in these analyses are available via the Rush Alzheimer's Disease Center Research Resource Sharing Hub, which can be found at radc.rush.edu. It has descriptions of the studies and available data. Any qualified investigator can create an account and submit requests for deidentified data.
Results
Description of the analytic group
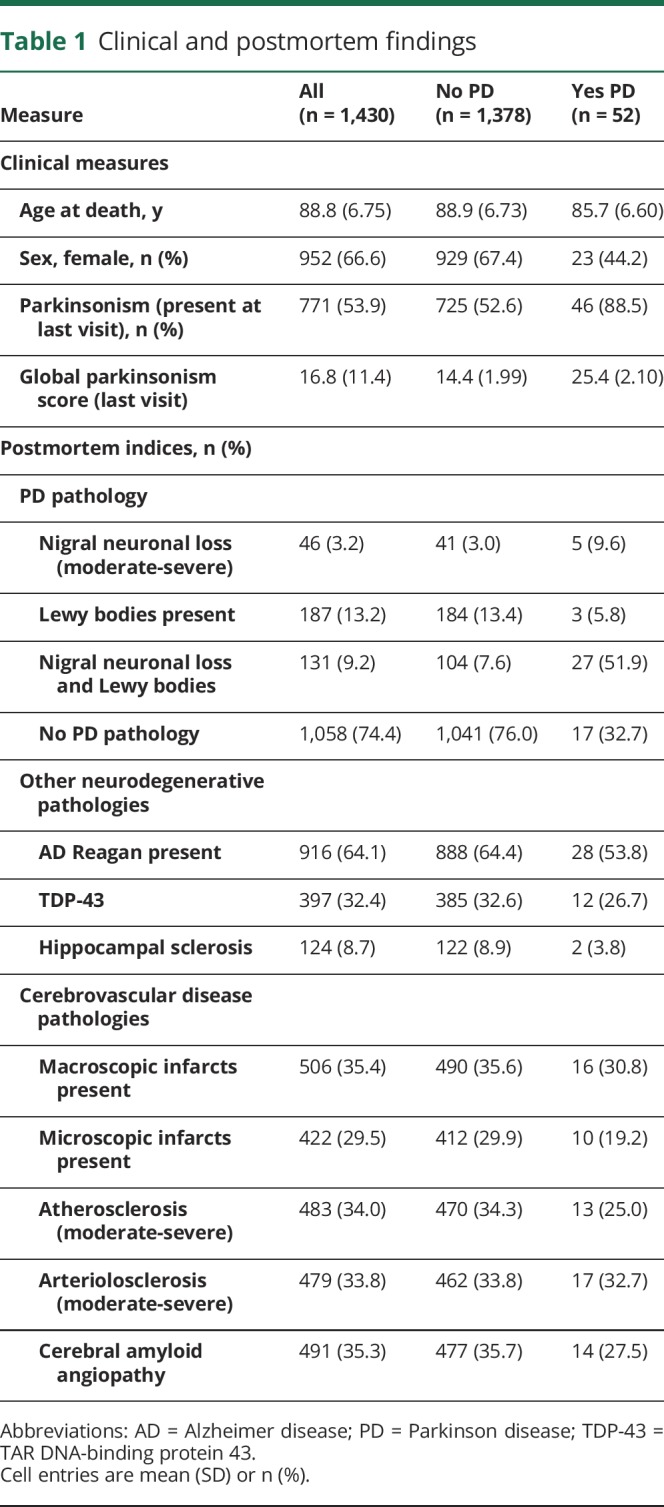
There were 1,430 adults included in these analyses, and their clinical characteristics proximate to death and autopsy findings are summarized in table 1. During more than 8.5 years of follow-up (mean 8.7 years [SD = 4.82]), 771 individuals without parkinsonism at baseline (53.9%) developed parkinsonism. On average, the last clinical assessment of parkinsonism was 1.4 years (SD = 1.72) before death.
Table 1.
Clinical and postmortem findings
Approximately 25% had one or both elements of PD pathology as illustrated in figure 1. The distributions of the other 8 pathologies measured are summarized in table 1.
Figure 1. PD pathology in older adults with and without clinical parkinsonism.

The number of individuals with clinical parkinsonism (red) and without clinical parkinsonism (blue) at the last visit proximate to death for each of the 3 Parkinson disease (PD) pathology groups (LB [Lewy bodies present], NNL [nigral neuronal loss], PD pathology [presence of both LB and NNL]) and a fourth reference group of individuals without either of the elements of PD pathology. The majority of older adults who developed parkinsonism proximate to death (red) did not show evidence of either of the elements of PD pathology.
For descriptive purposes, we dichotomized PD pathology and the other 8 pathologies measured to count how many pathologies were observed in each individual examined (table 1). The average individual had 3 of the 9 pathologies (median 3.0, interquartile range 2.0). One or more pathologies were observed in almost all persons (94.1%); 0: n = 84 (5.9%); 1: n = 227 (15.9%); 2: n = 269 (18.8%); 3: n = 301 (21.1%); 4: n = 271 (19.0%); 5: n = 165 (11.5%); 6–9: n = 113 (7.9%).
The average individual with and without a clinical diagnosis of PD who developed parkinsonism had 3 of 9 pathologies (median 3.00, interquartile range 2.0). Figure 2 illustrates the proportions of individuals with each of the different brain pathologies observed in persons developing parkinsonism. More than 80% of individuals developing parkinsonism with and without elements of PD pathology showed mixed brain pathologies.
Figure 2. Proportions of individuals with each of 9 brain pathologies in older adults with parkinsonism before death.

The majority of older individuals with clinical evidence of parkinsonism prior to death, with and without a clinical diagnosis of PD, showed mixed brain pathologies. AD = Alzheimer disease; CAA = cerebral amyloid angiopathy; CVDA = atherosclerosis; HS = hippocampal sclerosis; LIPO = arteriolosclerosis; MACRO = macroinfarcts; MICRO = microinfarcts; PD = Parkinson disease; TDP = TAR DNA-binding protein.
Progressive parkinsonism in older adults
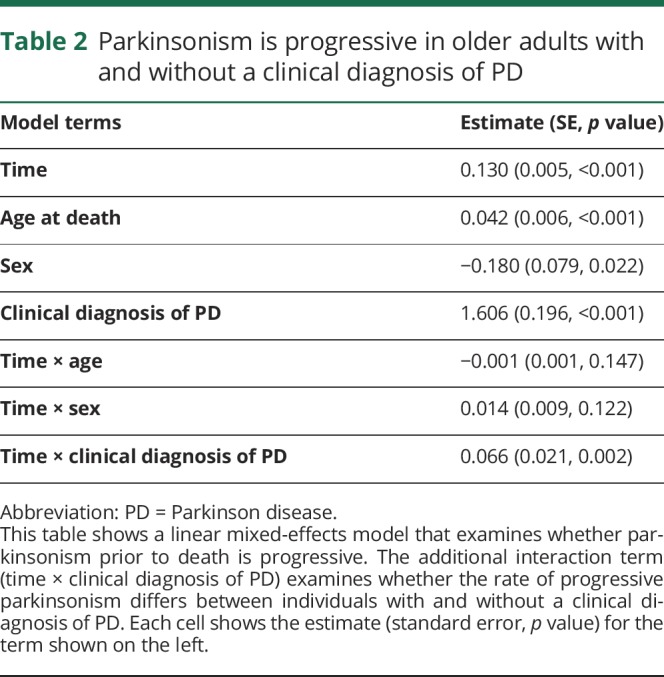
We examined the rate of progression of parkinsonism using a linear mixed-effects model that included terms for time, age, sex, and the clinical diagnosis of PD as well as their interaction with time. On average, parkinsonism was progressive in persons with and without a clinical diagnosis of PD (table 2, Time). The progression of parkinsonism was more rapid (table 2, time × PD), and the estimated severity of parkinsonism at time of death was worse in individuals with a clinical diagnosis of PD (table 2, PD).
Table 2.
Parkinsonism is progressive in older adults with and without a clinical diagnosis of PD
PD pathology and progressive parkinsonism
To examine the association of PD pathology with the progression of parkinsonism, we used a linear mixed-effects model in which we compared each of the 3 groups with one or more elements of PD pathology to a reference group of individuals without evidence of PD pathology. The rate of change in parkinsonism was similar in the individuals with either nigral neuronal loss or Lewy body pathology alone as compared to the reference group (table 3, model 1). However, individuals with PD pathology (both nigral neuronal loss and Lewy body pathology) had more rapid progression of parkinsonism and more severe parkinsonism at the time of death as compared to the reference group (table 3, model 1). PD pathology remained associated with the rate of change in parkinsonism when the analysis was restricted to individuals without a clinical diagnosis of PD (table 3, model 2).
Table 3.
PD pathology groups and progression of parkinsonism in older adults

Other brain pathologies and progressive parkinsonism
Since most adults who developed parkinsonism did not have any elements of PD pathology and the majority of individuals had evidence of multiple brain pathologies, we repeated a linear mixed-effects model, which controlled for age and sex, to determine whether other brain pathologies independently contribute to the progression of parkinsonism. In a single model controlling for PD pathology with terms for 8 other brain pathologies, AD, TDP-43, macroinfarcts, atherosclerosis, arteriolosclerosis, and cerebral amyloid angiopathy (CAA) were independently associated with the rate of progression of parkinsonism, but microinfarcts and hippocampal sclerosis were not (table 4, model 1). Demographics accounted for 4.5% and pathologies 18.5% of the variance of the rate of progression of parkinsonism. Similar results were obtained when we excluded individuals with a clinical diagnosis of PD (table 4, model 2). Age and sex accounted for 2.6% and pathologies 17.1% of the variance.
Burden of diverse brain pathologies and progressive parkinsonism
Almost 80% of individuals examined in this study showed evidence of multiple brain pathologies. To illustrate the cumulative effect of the burden of these pathologies on the rate of progression of parkinsonism, we constructed a weighted pathology score based on all the pathologies that were independently associated with progressive parkinsonism (table 4).
The average weighted pathology score was 0.51 (SD = 0.263). The weighted pathology score was higher in persons with a clinical history of PD (PD 0.68 [SD = 0.274] vs no PD 0.51 [SD = 0.261]; t test: t1,128 = −404, p ≤ 0.001).
We used a mixed-effect model with terms for age, sex, and average weighted pathology score and their interaction with the annual rate of change in parkinsonism (time). A higher weighted pathology score was associated with a more rapid progression of parkinsonism (estimate 0.172, SE 0.015, p < 0.001). Adding a term for education and its interaction with time did not change the association of the weighted pathology score and the rate of progressive parkinsonism (estimate 0.172, SE 0.015, p < 0.001). The trajectories for the progression of parkinsonism for 5 average individuals with increasing weighted pathology scores are shown in figure 3. For example, the progression of parkinsonism was about 4× faster in an individual with a very high weighted pathology score, 90th percentile weighted pathology score (gray line) compared to an individual without any pathology (black line) (gray line slope: 0.19/y vs black line slope: 0.05/y).
Figure 3. A higher burden of brain pathologies is related to more rapid progression of parkinsonism.
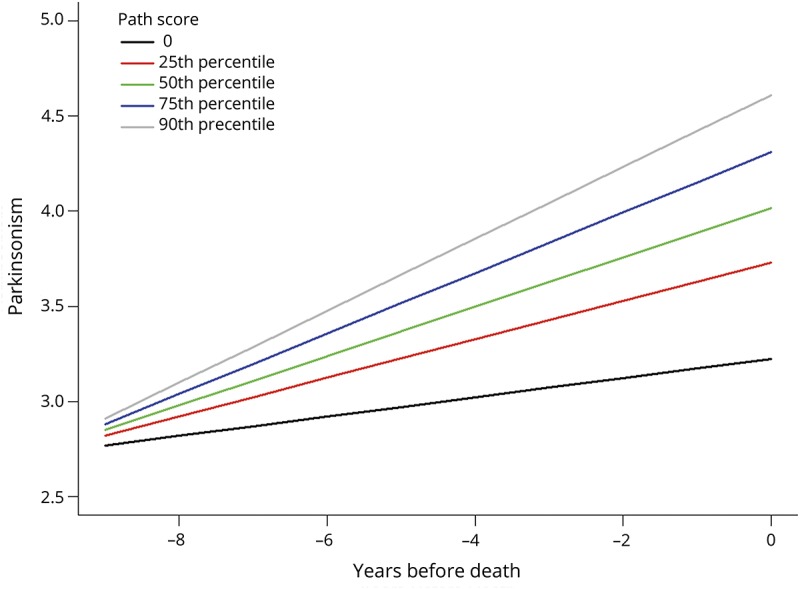
Model-derived trajectories of progressive parkinsonism for 5 average participants, an 89-year-old woman, with increasing weighted pathology scores derived from the associations between different postmortem indices and the rate of progressive parkinsonism. Since a higher global parkinsonism score (y-axis) represents more severe parkinsonism, the slope for more rapid progression is positive and larger. Slope values increase with a higher burden of pathology as illustrated by showing increasing weighted pathology scores and slope values: black line (no pathology, rate: 0.051/y); red line (25th percentile, weighted pathology score 0.316; rate: 0.101/y); green line (50th percentile weighted pathology score 0.483; rate: 0.130/y); blue line (75th percentile, weighted pathology score 0.678; rate: 0.159/y); and gray line (90th percentile weighted pathology score 0.868; rate: 0.189/y).
Since the weighted pathology score was higher in adults with a clinical history of PD, we added interaction terms to the previous mixed-effect model and found that the association of the weighted pathology score with the progression of parkinsonism did not vary between individuals with and without a clinical diagnosis of PD (time × weighted pathology score × PD diagnosis; estimate −0.011, SE 0.074, p = 0.878). This demonstrates that the association of mixed pathologies with progressive parkinsonism is on a continuum from low to high pathology burden, rather than any qualitative distinction by the presence or absence of a clinical diagnosis of PD.
As most individuals were from MAP and ROS, we excluded individuals from MARS and conducted a sensitivity analysis to ensure that our primary findings were observed in both MAP and ROS separately. We repeated our primary analyses separately in MAP and ROS. In both studies, mixed pathologies were associated with progressive parkinsonism (data available from Dryad, table 1, doi.org/10.5061/dryad.vn42tj2), and a greater weighted pathology score was associated with a more rapid rate of progression (data available from Dryad, table 2).
Discussion
The nature and nosology of subclinical PD remains controversial. This is largely because the association of PD pathology with progressive parkinsonism in older persons without PD is not well characterized. Using prospectively documented annual parkinsonism scores in more than 1,400 community-dwelling older adults for an average of 8 years, this study found that parkinsonism is progressive in older adults with and without a clinical diagnosis of PD, but was more rapidly progressive in adults with PD. PD pathology measured in these same individuals was associated with a more rapid rate of progression in the years prior to death in both adults with and without a clinical diagnosis of PD. However, more than 70% who developed parkinsonism did not show evidence of PD pathology. Individuals who developed parkinsonism but did not have PD pathology showed diverse brain pathologies including AD, TDP-43, macroinfarcts, atherosclerosis, arteriolosclerosis, and CAA, which were independently associated with more rapidly progressive parkinsonism. Furthermore, almost 80% of adults included in this study showed evidence of multiple brain pathologies, and the rate of progressive parkinsonism was related to the burden of mixed brain pathologies in older adults with and without a clinical diagnosis of PD. These data suggest that the clinical phenotype of parkinsonism in older adults is not specific for a particular brain pathology and is most commonly associated with mixed brain pathologies.
In this prospective study, we evaluated the clinical heterogeneity of progressive parkinsonism associated with PD pathology by assessing parkinsonism in all individuals who were recruited and collecting postmortem indices from autopsies without exclusion of individuals based on clinical or postmortem findings. This study design allowed us to compare both the rate of progression of parkinsonism and its pathologic basis for adults with and without a clinical diagnosis of PD. In the current analysis, parkinsonism was progressive in all older adults but its rate was more rapid in adults with a clinical diagnosis of PD. PD pathology was associated with more severe parkinsonism proximate to death as well as a more rapid rate of progression many years prior to death in both adults with and without a clinical diagnosis of PD.
In this study 3.6% of individuals had a clinical diagnosis of PD, similar to population estimates for PD.1 By contrast, 7.6% of individuals without clinical PD had evidence of PD pathology, although the presence of nigral neuronal loss or Lewy bodies alone was not related to the rate of progressive parkinsonism. Approximately 25% of all individuals without a clinical diagnosis of PD showed accumulation of either or both elements of PD pathology. Further work is needed to develop more accurate clinical risk profiles to identify those with progressive parkinsonism due to PD pathology, potentially including nonmotor functions along with neuroimaging, genetic, and biochemical biomarkers. Such efforts may increase the specificity for predicting distinct pathologies.32
These data showing that PD pathology is associated with progressive parkinsonism in older adults is an important finding. Yet, the majority of individuals in this study who developed parkinsonism did not have PD pathology (figure 1). This highlights the importance of our findings that other brain pathologies including AD, TDP-43, macroinfarcts, atherosclerosis, arteriolosclerosis, and CAA are independently associated with progressive parkinsonism when controlling for PD pathology.
Most older adults in this study showed evidence of multiple brain pathologies, and there are hundreds of possible combinations of mixed pathologies in this large sample.29 Risk scores have been used to aggregate the additive risks associated with multiple genes or clinical risk factors.30,31 Therefore, we constructed a person-specific weighted pathology score to illustrate the aggregated risks of the varied combinations of mixed pathologies on the progression of parkinsonism (figure 3). Of note, while on average the burden of pathology was greater in adults with clinical PD, the association of a higher burden of pathologies with more rapid progressive parkinsonism did not vary with a clinical diagnosis of PD. These data suggest that the association of mixed pathologies with progression of parkinsonism is on a continuum from low to high pathology burden, rather than any qualitative distinction by the presence or absence of a clinical diagnosis of PD. Thus, for a similar level of mixed pathologies, individuals with and without PD may have similar rates of progressive parkinsonism.
These findings have important translational consequences as they suggest that the clinical phenotype of parkinsonism in older adults is not specific for a single specific brain pathology but is most commonly associated with mixed brain pathologies.33 Thus, parkinsonism in older adults is not simply an early manifestation of PD pathology but rather a phenotype associated with diverse pathologies. Moreover, since most individuals developing clinical parkinsonism did not have PD pathology, but rather several other brain pathologies, it suggests that currently available clinical constructs for the assessment of parkinsonism lack the requisite specificity to identify older adults at risk of specific brain pathologies. Moreover, as we have shown in prior publications, given the numerous combinations of mixed pathologies that may manifest in a population of older adults, it is likely to be difficult to develop cost-efficient therapies for mixed pathologies.29,32 An alternative and parallel approach focusing on identifying molecular or other resilience factors unrelated to pathologies that may mitigate the untoward effects of mixed pathologies on parkinsonism in older adults circumvents the difficulties of focusing on targeted treatments of mixed pathologies.34,35
Nonetheless, the current findings may have important clinical implications. For example, microvascular pathologies cannot be assessed with conventional brain imaging but nonetheless may offer an important new direction for preventing or decreasing the burden of parkinsonism in older adults. The prominence of microvascular pathologies including arteriolosclerosis, atherosclerosis, and CAA, which were as common as macroinfarcts, lends support to the notion that small vessel disease may make important contributions to late-life motor and cognitive impairment.19
Concepts of chronic age-related neurologic diseases are evolving. For example, we now recognize that the pathology of AD is common in persons without clinically diagnosed dementia. Recognition of the heterogeneity of cognition associated with pathologic AD and its reconceptualization as a clinical continuum required the prospective cognitive assessment of large numbers of older adults undergoing autopsy without case selection based on postmortem findings. Similar to AD, PD pathology accumulates over many years before a clinical diagnosis of PD. Leveraging the successful approach used for pathologic AD, this prospective study examined all brains collected without exclusion of individuals based on clinical or postmortem findings to avoid truncating the clinical heterogeneity of parkinsonism. This study design allowed us to compare both the progression of parkinsonism and its pathologic basis in adults with and without a clinical diagnosis of PD.
The clinical categories of parkinsonism examined in the current study might be conceptualized as a continuum of progressively more severe parkinsonian motor signs. For example, a clinical diagnosis of PD manifests more severe and rapidly progressive parkinsonian signs in individuals compared to those with and without parkinsonism but without a clinical diagnosis of PD. This clinical spectrum of increasing severity and rapidly progressive parkinsonism is analogous to cognitive phenotypes, wherein individuals with no cognitive impairment and mild cognitive impairment have slower rates of cognitive decline than adults with AD dementia.36
Parkinsonism in older adults is most commonly associated with mixed brain pathologies. Thus, identifying this behavioral phenotype does not have a one-to-one correspondence to a specific underlying pathologic process. This is similar to mixed brain pathologies underlying AD dementia such that the clinical diagnosis is not specific for AD pathology. Further work is needed to develop clinical risk profiles, potentially including clinical, genetic, imaging, or fluid biomarkers. Such efforts may increase the specificity for predicting distinct etiologies of parkinsonism, facilitating early treatments.32
There are several reasons to think that our study may have underestimated the contributions of common neuropathologies to parkinsonism in older adults, an underestimation that might explain why the brain pathologies explained only about 20% of the variation in the rate of change of parkinsonism. Postmortem indices were preferentially collected from traditional cognitive-related brain regions and did not include motor-related regions in the cerebrum or regions caudal to the substantia nigra including brainstem and spinal cord, which we have shown to make separate contributions to the severity of parkinsonism.18,37,38 In addition, the current study did not assess white matter integrity, which may make a substantial contribution to motor phenotypes in older adults.39 Finally, we used semiquantitative scales to measure pathologies rather than quantifying overall burden throughout the brain. Thus, our results are likely to underestimate the full extent to which pathologies underlie parkinsonism. Further studies are needed to replicate our findings and to determine the full range of neuropathologies as well as other molecular mechanisms unrelated to known pathologies underlying parkinsonism in old age.35
There are several limitations to the current analyses. First, similar to other community- or population-based cohort studies, the diagnosis of PD in these analyses was based on history and medical records and was not based on structured examination by a movement disorders specialist.1,40 Recent work suggests that early in its course, it may be difficult even for an expert to make an accurate diagnosis of PD.41,42 However, even if the clinical diagnosis of PD was suboptimal, since we obtained postmortem indices on all individuals, this would not have affected our main findings as the majority of adults developing clinical parkinsonism did not have evidence of PD pathology. This study has several strengths. One important strength was that by using the same validated instrument to measure parkinsonism from prospectively collected data, we were able to compare the trajectories of progressive parkinsonism in older community-dwelling older adults with and without a clinical diagnosis of PD. In addition, all decedents had a structured brain autopsy that included not only PD pathology but 8 other age-related brain pathologies, allowing us to assess the heterogeneity of pathologies underlying progressive parkinsonism in older adults.
Acknowledgment
The authors thank all the participants in the Rush Memory and Aging Project, the Religious Orders Study, and the Minority Aging Research Study, and the staff of the Rush Alzheimer's Disease Center (RADC). More information regarding obtaining RADC clinical and postmortem data for research use can be found at the RADC Research Resource Sharing Hub (radc.rush.edu).
Glossary
- AD
Alzheimer disease
- CAA
cerebral amyloid angiopathy
- MAP
Memory and Aging Project
- MARS
Minority Aging Research Study
- PD
Parkinson disease
- ROS
Religious Orders Study
- TDP-43
TAR DNA-binding protein 43
- UPDRS
Unified Parkinson's Disease Rating Scale
Appendix. Authors
Study funding
This work was supported by NIH (P30AG10161, R01AG15819, R01AG17917, RF1AG22018, R01AG44379, R01NS78009, R01AG47976, R01AG56352); the Illinois Department of Public Health; and the Robert C. Borwell Endowment Fund. The funding organizations had no role in the design or conduct of the study; collection, management, analysis, or interpretation of the data; or preparation, review, or approval of the manuscript.
Disclosure
A. Buchman, L. Yu, R. Wilson, S. Leurgans, S. Nag, J. Shulman, and L. Barnes report no disclosures relevant to the manuscript. J. Schneider reports disclosures for this manuscript including scientific advisory board for Grifols, Lilly, and Genentech, and consultant for the Michael J. Fox Foundation and the National Hockey League. D. Bennett reports no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson's disease: a systematic review and meta-analysis. Mov Disord 2014;29:1583–1590. [DOI] [PubMed] [Google Scholar]
- 2.Hofman A, de Jong PT, van Duijn CM, Breteler MM. Epidemiology of neurological diseases in elderly people: what did we learn from the Rotterdam Study? Lancet Neurol 2006;5:545–550. [DOI] [PubMed] [Google Scholar]
- 3.Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 4.Burke RE, Dauer WT, Vonsattel JP. A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 2008;64:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Obeso JA, Stamelou M, Goetz CG, et al. Past, present, and future of Parkinson's disease: a special essay on the 200th anniversary of the shaking palsy. Mov Disord 2017;32:1264–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louis ED, Bennett DA. Mild parkinsonian signs: an overview of an emerging concept. Mov Disord 2007;22:1681–1688. [DOI] [PubMed] [Google Scholar]
- 7.Buchman AS, Shulman JM, Nag S, et al. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol 2012;71:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol 2009;68:816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adler CH, Connor DJ, Hentz JG, et al. Incidental Lewy body disease: clinical comparison to a control cohort. Mov Disord 2010;25:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jellinger KA. Lewy body-related alpha-synucleinopathy in the aged human brain. J Neural Transm 2004;111:1219–1235. [DOI] [PubMed] [Google Scholar]
- 11.Lerche S, Hobert M, Brockmann K, et al. Mild parkinsonian signs in the elderly—is there an association with PD? Cross-sectional findings in 992 individuals. PLoS One 2014;9:e92878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berg D, Postuma RB, Bloem B, et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson's disease. Mov Disord 2014;29:454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchman AS, Wilson RS, Shulman JM, Leurgans SE, Schneider JA, Bennett DA. Parkinsonism in older adults and its association with adverse health outcomes and neuropathology. J Gerontol A Biol Sci Med Sci 2016;71:549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis 2018;64:S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes LL, Shah RC, Aggarwal NT, Bennett DA, Schneider JA. The minority aging research study: ongoing efforts to obtain brain donation in African Americans without dementia. Curr Alzheimer Res 2012;9:734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu L, Lutz MW, Wilson RS, et al. APOE ε4-TOMM40 '523 haplotypes and the risk of Alzheimer's disease in older Caucasian and African Americans. PLoS One 2017;12:e0180356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett DA, Shannon KM, Beckett LA, Goetz CG, Wilson RS. Metric properties of nurses' ratings of parkinsonian signs with a modified Unified Parkinson's Disease Rating Scale. Neurology 1997;49:1580–1587. [DOI] [PubMed] [Google Scholar]
- 18.Buchman AS, Nag S, Leurgans SE, et al. Spinal Lewy body pathology in older adults without an antemortem diagnosis of Parkinson's disease. Brain Pathol 2018;28:560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchman AS, Leurgans SE, Nag S, Bennett DA, Schneider JA. Cerebrovascular disease pathology and parkinsonian signs in old age. Stroke 2011;42:3183–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett DA, Wilson RS, Schneider JA, et al. Apolipoprotein E ε4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology 2003;60:246–252. [DOI] [PubMed] [Google Scholar]
- 21.Schneider JA, Li JL, Li Y, Wilson RS, Kordower JH, Bennett DA. Substantia nigra tangles are related to gait impairment in older persons. Ann Neurol 2006;59:166–173. [DOI] [PubMed] [Google Scholar]
- 22.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 23.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 2004;61:378–384. [DOI] [PubMed] [Google Scholar]
- 24.Wilson RS, Yu L, Trojanowski JQ, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 2013;70:1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol 2015;77:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider JA, Wilson RS, Cochran EJ, et al. Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology 2003;60:1082–1088. [DOI] [PubMed] [Google Scholar]
- 27.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke 2011;42:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011;69:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018;83:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf PA, D'Agostino RB, Belanger AJ, Kannel WB. Probability of stroke: a risk profile from the Framingham Study. Stroke 1991;22:312–318. [DOI] [PubMed] [Google Scholar]
- 31.Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med 2017;14:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berg D, Lang AE, Postuma RB, et al. Changing the research criteria for the diagnosis of Parkinson's disease: obstacles and opportunities. Lancet Neurol 2013;12:514–524. [DOI] [PubMed] [Google Scholar]
- 33.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69:2197–2204. [DOI] [PubMed] [Google Scholar]
- 34.Mostafavi S, Gaiteri C, Sullivan SE, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat Neurosci 2018;21:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu L, Petyuk VA, Gaiteri C, et al. Targeted brain proteomics uncover multiple pathways to Alzheimer's dementia. Ann Neurol 2018;84:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu L, Boyle P, Wilson RS, et al. A random change point model for cognitive decline in Alzheimer's disease and mild cognitive impairment. Neuroepidemiology 2012;39:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buchman AS, Nag S, Shulman JM, et al. Locus coeruleus neuron density and parkinsonism in older adults without Parkinson's disease. Mov Disord 2012;27:1625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchman AS, Leurgans SE, Nag S, et al. Spinal arteriolosclerosis is common in older adults and associated with parkinsonism. Stroke 2017;48:2792–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchman AS, Dawe RJ, Yu L, et al. Brain pathology is related to total daily physical activity in older adults. Neurology 2018;90:e1911–e1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sonnen JA, Postupna N, Larson EB, et al. Pathologic correlates of dementia in individuals with Lewy body disease. Brain Pathol 2010;20:654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson disease: a systematic review and meta-analysis. Neurology 2016;86:566–576. [DOI] [PubMed] [Google Scholar]
- 42.Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 2014;83:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data included in these analyses are available via the Rush Alzheimer's Disease Center Research Resource Sharing Hub, which can be found at radc.rush.edu. It has descriptions of the studies and available data. Any qualified investigator can create an account and submit requests for deidentified data.