

Abstract
Objective
To investigate the potential for a causal effect of age at puberty on multiple sclerosis (MS) susceptibility using a mendelian randomization (MR) approach.
Methods
We used 372 genetic variants strongly associated with age at menarche in a genome-wide association study (GWAS) involving 329,245 women. The genetic architecture of pubertal timing across both sexes is highly correlated (genetic correlation [rg] = 0.75, p = 1.2 × 10−79), allowing these variants to provide reliable insight into pubertal timing in males as well. The effect of pubertal timing on risk of MS was measured with summary statistics from a GWAS of 14,802 cases with MS and 26,703 controls from the International Multiple Sclerosis Genetics Consortium. Multivariable MR controlling for effects of body mass index (BMI) using genetic data from additional consortia investigated whether pubertal effects on MS were dependent on weight status.
Results
A 1-year increase in genetically predicted age at puberty decreased odds of MS by 8% (odds ratio [OR] 0.92, 95% confidence interval [CI] 0.86–0.99, p = 0.03). However, multivariable MR analysis showed that after accounting for effects on adult BMI, the association of age at puberty with MS susceptibility attenuated (OR 0.96, 95% CI 0.88–1.04, p = 0.36). Similar results were obtained when childhood BMI was incorporated. Sensitivity analyses provided no evidence of major bias from genetic pleiotropy.
Conclusions
We found support for an association between higher age at puberty and decreased risk of MS with a magnitude comparable to that reported in observational studies. This effect appears to be largely mediated by the strong association between age at puberty and obesity. A large causal effect of pubertal timing independent of BMI is unlikely.
Epidemiologic studies have reported an increased risk of multiple sclerosis (MS) with earlier age at puberty, particularly among women.1–4 However, others failed to replicate this finding.5–8 Pubertal timing has complex interactions with weight status, whereby higher childhood adiposity leads to earlier puberty, which in turn is associated with higher adult body mass index (BMI).9 Because evidence supports a role for increased BMI in MS pathogenesis,10,11 at least part of the observed link between pubertal timing and MS might be explained by BMI.
Establishing a causal contribution of pubertal timing in MS is crucial to understanding the effects of sex hormone exposure and sexual maturation on disease etiology. This in turn may lead to novel preventive or therapeutic strategies. It may also clarify how the decreasing age at puberty over the past decades12 might relate to observed increases in MS incidence.13
Some of the limitations faced by observational studies can be mitigated through instrumental variable methods, in which a variable is used as proxy for an exposure to explore the effect of that exposure on an outcome. In mendelian randomization (MR), genetic variants are used as instrumental variables to test for a causal association between a risk factor and an outcome.14 Because alleles linked with different traits are randomly assigned at conception, the MR design greatly limits confounding15 and avoids reverse causation.14 To assess the effect of age at puberty on MS susceptibility, we performed a 2-sample MR analysis using genetic associations from large genome-wide association studies (GWASs). Here, we first explore whether participants with genetic variants associated with later age at puberty also have a lower risk of MS. Second, we use recent developments in MR methodology to determine whether pubertal timing exerts direct effects on MS susceptibility independently of BMI.
Methods
Genetic variants associated with age at puberty
To select our genetic instruments, we used single-nucleotide polymorphisms (SNPs) associated with age at menarche from the largest GWAS meta-analysis to date by the Reproductive Genetics (ReproGen) consortium,16 combining 329,245 women of European ancestry (table). The effect of these variants was measured in years. The genetic architecture of pubertal timing across both sexes is highly correlated (genetic correlation [rg] = 0.75; p = 1.2 × 10−79), allowing these variants to provide insight into pubertal timing in males as well.16 Indeed, these variants have previously been used in an MR analysis of prostate cancer risk,16 and the strong overlap has provided a basis for a combined GWAS of men and women.17 Therefore, the genetic variants identified by the ReproGen consortium are hereafter referred to as being associated with puberty rather than menarche alone.
Table.
Details of datasets included in the MR analyses

For our main MR analysis, we included as candidate genetic instruments 377 genome-wide significant (p < 5 × 10−8) autosomal variants associated with pubertal timing. In a sensitivity analysis, we restricted the MR analysis to 311 SNPs with a concordant direction of effect in a GWAS of age at voice breaking in 54,871 men from 23andMe.16 To ensure that all selected SNPs are independent, we measured the degree of linkage disequilibrium between variants and excluded those with a value of r2 > 0.05 in the European subset of 1,000 Genomes18 using PLINK software version 1.9.19
Genetic variants associated with MS
Corresponding effects of the puberty-associated SNPs on MS susceptibility were derived from the discovery cohorts of the latest International Multiple Sclerosis Genetics Consortium (IMSGC) meta-analysis, which includes up to 41,505 participants (14,802 cases with MS and 26,703 controls).20 Further details on the eligibility criteria, MS case ascertainment, and demographic characteristics can be found in the original publication.20 For puberty-associated variants not directly ascertained in the IMSGC dataset, we identified proxy SNPs in high linkage disequilibrium (r2 > 0.8) using PLINK and samples of European descent from 1,000 Genomes18 or the UK10K consortium.21 The odds ratios (ORs) and p values of the summary statistics from the IMSGC were transformed into β coefficients and standard errors for subsequent analyses. If the OR was exactly 1 (which occurred for 2 variants), the β coefficient was closely approximated on the basis of the OR displayed by immediately neighboring SNPs.
For each genetic variant, alleles were aligned and matched so that their effects correspond to an increase in age at puberty. The orientation of proxy alleles was based on phasing information from 1,000 Genomes.18 We examined the datasets contributing to the genetic estimates for age at puberty and MS to identify overlapping sets of participants because this can introduce bias in 2-sample MR.22
Genetic variants associated with BMI
Pubertal timing and weight status share common regulatory mechanisms23 and show strong genetic correlation (rg = −0.35, p = 1.6 × 10−72).16 In general, BMI-increasing alleles lead to earlier puberty.16 To investigate the effect of pubertal timing on MS independently of weight status, we accounted for the effect of puberty-associated SNPs on both adult and childhood BMI. For adult BMI, we used a densely imputed GWAS of 87,048 European individuals from the European Network for Genetic and Genomic Epidemiology (ENGAGE) Consortium24 and adjusted for age, age2, and study-specific covariates. For childhood BMI, sex- and age-adjusted genetic data for 35,668 children of European descent were obtained from the Early Growth Genetics (EGG) consortium.25 Proxy identification and allele alignment were implemented as described for the MS susceptibility dataset.
MR analyses
For the main analysis, we applied inverse-variance weighted 2-sample MR26–29 to obtain effect estimates of genetically predicted pubertal timing on MS susceptibility. In brief, we weighted the effect of each SNP on MS susceptibility by its effect on age at puberty using the ratio method.28 We combined the individual effect estimates thus obtained into a summary measure reflecting the effect of pubertal timing on MS risk using an inverse-variance weighted random-effects model. We also measured the degree of heterogeneity across the individual effect estimates derived from each genetic variant using the Cochran Q test and I2 statistic. In a secondary analysis, we repeated this method using the restricted set of SNPs with directionally concordant effects on age at menarche and voice breaking.
Next, to account for potential effects of weight status, we performed a multivariable MR30,31 in which the genetic effects of the same puberty-associated variants on adult and childhood BMI were separately included in a weighted regression analysis. To further explore the effects of weight status, we stratified the age-at-puberty SNPs into those individually associated with BMI (p < 0.05 in the ENGAGE or EGG datasets) and those not associated with BMI. This p value threshold was used to obtain 2 SNP subsets that differ in the strength of their association with BMI, rather than as a measure of significance. The association with MS susceptibility was then tested separately within each of the SNP subsets.
Within the context of MR studies, pleiotropy denotes a situation in which genetic variants affect the outcome (MS risk) through pathways other than the exposure of interest (age at puberty). Whereas the association of genetic variants with pathways along the same causal chain as the effect of interest (vertical pleiotropy) does not breach the assumptions of MR, variants associated with multiple distinct pathways (horizontal pleiotropy) can induce bias. We took several steps to investigate the possibility of horizontal pleiotropy and to reduce its likelihood. First, we excluded from all analyses variants within human leukocyte antigen (HLA) genes given the strength of their association with MS and the complexity of linkage disequilibrium at this locus. Second, we inspected funnel plots of the precision of the association between pubertal timing and MS risk given by each SNP against the estimate of this association.32 Visual asymmetry on this plot indicates the presence of directional pleiotropy, that is, horizontal pleiotropy leading to bias in a given direction. Third, we applied multivariable MR-Egger regression to account for the possibility of unmeasured horizontal pleiotropy after accounting for the effects of BMI (adult and childhood).33 The intercept of this regression allows detection of directional pleiotropy, while the regression coefficient provides a causal estimate largely robust to horizontal pleiotropy, insofar as the magnitude of a potential pleiotropic effect is independent of the effects of the genetic variants on pubertal timing or BMI (described as the InSIDE assumption33,34).
To exclude the possibility of reverse causation, we tested whether genetically increased risk of MS influences pubertal timing. Here, we selected non-HLA variants associated with MS susceptibility in the latest IMSGC meta-analysis20 and applied an inverse-variance weighted MR with age at puberty as the outcome using the same methods as above.
All statistical analyses were performed in R (version 3.4.1). The α level for statistical significance was set to 0.05.
Standard protocol approvals, registrations, and patient consents
All data sources used in this study (IMSGC, ReproGen, ENGAGE, and EGG consortia) received approval from institutional review boards and obtained informed consent from all participants.16,20,24,25
Data availability
Summary-level data for the genetic associations with age at puberty, adult BMI, and childhood BMI are publicly available and can be obtained through the links provided in the table. In addition, the dataset used to generate the results in the current study is available from the corresponding author on request.
Results
Selection of genetic instruments
A flow diagram of the SNP selection process is presented in figure 1. None of the 377 candidate genetic instruments were in linkage disequilibrium. We excluded a SNP near HLA-DQB1 (rs3021057) because of its known pleiotropic effects, leaving 376 SNPs for the subsequent steps. For the genetic association with MS risk, a total of 345 autosomal non-HLA variants associated with pubertal timing were directly ascertained in the IMSGC meta-analysis. For another 27 variants, proxies were identified with a median value of r2 of 1.00 (range 0.83–1.00). Altogether, these 372 SNPs explained 7.1% of the variance in pubertal timing in an independent cohort16 and were carried forward for the main analysis. Of those, 311 SNPs showed directionally concordant effects on age at menarche and voice breaking and were selected for a secondary analysis (21 proxy SNPs, median r2 = 0.98, range 0.83–1.00). Participant overlap between the age at puberty and MS risk genetic datasets was negligible, representing at most 0.13% of the sample size.
Figure 1. Flowchart for selection of genetic variants associated with age at puberty.

BMI = body mass index; IMSGC = International Multiple Sclerosis Genetics Consortium; LD = linkage disequilibrium; MS = multiple sclerosis; 1KG = 1,000 Genomes; ReproGen = Reproductive Genetics; SNP = single-nucleotide polymorphism.
For the genetic association with BMI, we undertook a similar process. For adult BMI, all 372 SNPs associated with age at puberty included in the main analyses were extracted from the ENGAGE consortium dataset, either directly (n = 345) or through a proxy (n = 27, median r2 = 1.00, range 0.83–1.00). For childhood BMI, only 343 SNPs were present in the EGG consortium dataset, with 149 identical SNPs and 194 proxies (median r2 = 0.98, range 0.59–1.00).
MR estimates
Inverse-variance weighted MR analysis revealed that a 1-year increase in age at puberty was associated with an 8% lower risk of MS (OR 0.92, 95% confidence interval [CI] 0.86–0.99, p = 0.032) (figure 2). A secondary analysis using the restricted set of 311 SNPs with directionally concordant effects on age at menarche and voice breaking resulted in similar findings (OR 0.92, 95% CI 0.86–0.99, p = 0.040). The Cochran Q test and I2 statistic revealed moderate heterogeneity among the individual SNP effect estimates in the main analysis (Q = 665.9, p = 1.1 × 10−16; I2 = 44% [37%–51%]), which decreased only slightly with the restricted set of SNPs (Q = 506.8, p = 1.1 × 10−11; I2 = 39% [30%–47%]).
Figure 2. MR estimates of the association between age at puberty and risk of MS.

aGenetic association with age at voice breaking derived from a genome-wide association study of 54,871 men from 23andMe.16 BMI = body mass index; CI = confidence interval; IVW = inverse-variance weighted; MR = mendelian randomization; MS = multiple sclerosis; SNP = single-nucleotide polymorphism.
In a multivariable MR analysis, the effect of age at puberty on MS after accounting for the contribution of adult BMI attenuated (OR 0.96, 95% CI 0.89–1.04, p = 0.36). Repeating the multivariable analysis after adjusting for the effects of childhood BMI yielded similar results (OR 0.97, 95% CI 0.89–1.05, p = 0.40). A univariable inverse-variance weighted analysis with a set of 115 variants associated with age at puberty and nominally associated with either adult or childhood BMI resulted in a strong association between age at puberty and MS susceptibility (OR 0.81, 95% CI 0.70–0.93, p = 0.002). Conversely, the 257 age-at-puberty SNPs that were not associated with BMI demonstrated a lack of association (OR 0.99, 95% CI 0.91–1.08, p = 0.829).
The funnel plot was symmetric and therefore not suggestive of directional pleiotropy (figure 3). Similarly, the multivariable MR-Egger regression intercept was estimated to be centered around zero (intercept −0.002, 95% CI −0.009 to 0.005, p = 0.622), and the estimate from the regression slope was consistent with the absence of an independent effect of pubertal timing on MS risk after adjustment for BMI (OR 1.01, 95% CI 0.82–1.24, p = 0.929).
Figure 3. Funnel plot for the effect of age at puberty on risk of MS.
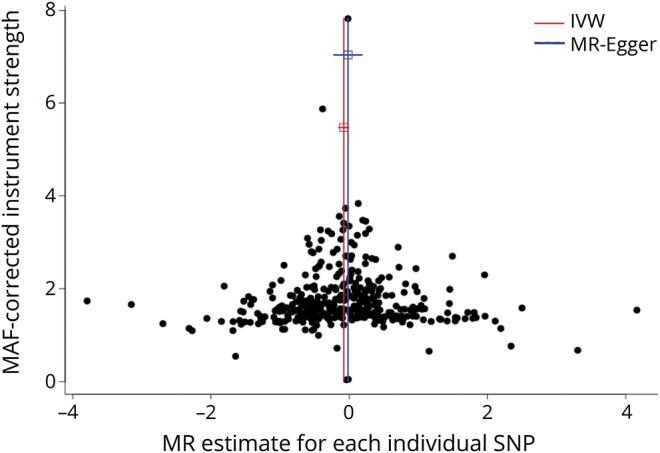
For each single-nucleotide polymorphism (SNP), the resulting mendelian randomization (MR) estimate is plotted against the minor allele frequency (MAF)–corrected association with pubertal timing. Red vertical line represents the summary measure of the effect of a 1-year increase in age at puberty on risk of multiple sclerosis (MS) on the log-odds ratio scale. Symmetry noted in this plot provides evidence against the presence of directional horizontal pleiotropy. IVW = inverse-variance weighed.
Finally, testing whether MS susceptibility influences age at puberty with an inverse-variance weighted MR, we found that genetically increased MS susceptibility was not associated with age at puberty (OR 0.99 per log-odds increase in MS, 95% CI 0.98–1.01, p = 0.328).
Discussion
Using an MR design in 14,802 cases with MS and 26,703 controls, this study found evidence that genetically predicted later puberty is associated with a protective effect on MS risk. Specifically, each 1-year increase in age at puberty conferred an 8% decrease in odds of MS. After adjustment for genetically predicted BMI in both adulthood and childhood, this association was attenuated, and its CIs included the null. In addition, stratified MR analyses of the puberty-related variants based on their association with BMI further support this evidence because puberty-related variants not associated with BMI had no influence on MS risk, while puberty-related variants associated with BMI did influence MS risk. This suggests that effects specific to pubertal timing such as longer duration of exposure to sex hormones do not have large effects on MS susceptibility. While small effects independent of BMI cannot be excluded, the relatively narrow CIs do not support a clinically relevant influence.
The association between pubertal timing and weight status is complex and plausibly bidirectional. Increased adiposity in childhood has been linked to earlier pubertal maturation,35 although this relationship may be nonlinear in boys.36 Furthermore, several studies report evidence for an association between earlier age at puberty and later obesity.9 Therefore, we sought to control for both genetically predicted adult and childhood BMI, and we observed a similar magnitude of attenuation in the association between pubertal timing and risk of MS. However, there is a strong association between childhood and adult BMI,25,37 which limits the exploration of age-specific effects. Nonetheless, postpubertal rather than childhood obesity is most clearly related to MS susceptibility,38,39 making the association between pubertal timing and adult obesity the most likely mediator of the effect of age at puberty on risk of MS. Because it appears that BMI and pubertal timing are in the same causal biological pathway, the association of the selected genetic variants with both exposures represents an example of vertical pleiotropy due to shared biological underpinnings and thus does not bias the MR estimates.
Previous observational studies reporting the relevance of pubertal timing in MS have yielded conflicting results. An early study observed a lower age at menarche in 118 female cases with MS compared to an equivalent number of controls (12.3 vs 12.7 years, p = 0.01).2 A larger case-control study reproduced this finding in 4,472 female cases with MS and 658 controls (12.4 vs 12.6 years, p = 1.7 × 10−4).3 Consistent with our findings, the authors found a 10% decrease in MS risk per 1-year increase in age at puberty.3 No association was seen in male cases, although the sample size was 4-fold smaller. Recently, a study using the Danish National Birth Cohort reported 77,330 women, 226 of whom eventually developed MS. In this cohort, a 1-year increase in age at puberty translated in a 13% reduction in MS risk.1 In contrast, other case-control studies5,7,8,40 did not find an association between age at puberty and MS susceptibility, while another even reported a higher age at menarche among female cases.6 However, most of these discordant studies were limited by small numbers and low power.
In addition to these variable results, the contribution of weight status to the association between pubertal timing and MS had been little studied. In the study based on the Danish National Birth Cohort, the authors controlled for the effect of BMI in their analysis.1 While they observed little attenuation in the association between age at puberty and MS in females, BMI was available only at a mean age of 30 years. Because the association with MS seems most robust for adolescent obesity,38,39 residual mediating effects by BMI cannot be excluded. In contrast, the MR method in this study accounted for the lifetime effects of genetic variants on BMI.
While this study does not support substantial direct effects of pubertal timing on MS independently of weight status, it did not ascertain its potential role on disease course after onset, as some studies have suggested.41,42 Furthermore, puberty attainment itself, rather its timing, may still play direct roles in MS initiation because this transition period relates to important hormonal and immune changes, as well as brain maturation.43 For instance, sexual maturity was shown to enhance CNS autoimmunity in female experimental autoimmune encephalomyelitis, a murine model for MS.4
A major strength of this study is the MR design, which reduces confounding15 and contributes to establishing causality. The 2-sample approach also allowed us to maximize statistical power by leveraging genetic data from large cohorts, totaling >450,000 individuals and including 14,802 cases with MS. This study also has some limitations. First, the use of summary-level statistics from the IMSGC precluded separate analysis of male and female cases in the total sample. Second, the possibility of pleiotropy can be addressed only indirectly, and while combining genetic variants allowed us to achieve adequate power, some may relate to MS risk through pathways other than pubertal timing or BMI. However, we undertook several steps to ensure that this had not biased our results. We excluded from all analyses the pleiotropic SNP rs3021057 near HLA-DQB1. In addition, the presence of moderate heterogeneity between the MR estimates from the inverse-variance weighted analysis is likely due to the SNPs showing a variable relationship with BMI, in addition to their association with age at puberty. While heterogeneity can be indicative of horizontal pleiotropy,44 the association of the SNPs with those 2 phenotypes is expected to result in the detection of heterogeneity on the Cochran Q test and I2 statistic.45 Conversely, the funnel plot and multivariable MR-Egger intercept did not reveal evidence of directional, or unbalanced, pleiotropy. Moreover, the multivariable MR-Egger regression was consistent with the finding that age at puberty is related to MS risk through BMI-related pathways. This method is largely robust to pleiotropic effects as long as the InSIDE assumption is satisfied.33,34 Consequently, it seems unlikely that genetic pleiotropy can account for our results. Third, MR studies can be confounded by population stratification if SNPs are associated with subpopulations of different ancestry and carrying distinct MS risks. To avoid this, we included only individuals of European ancestry and used GWASs that included genomic control. Lastly, the effect estimates reported in this study assume the presence of a linear relationship between age at puberty and MS risk. While nonlinear associations have been reported for associations between age at puberty and cardiovascular disease46 and diabetes mellitus,47 a population-based study did not support this being the case for MS,1 at least in female cases.
This study provides evidence that higher genetically predicted age at puberty is protective against the development of MS. The magnitude of this association appears to be dependent on obesity, suggesting that pathways specific to pubertal timing are less likely to be direct determinants of MS risk. Prevention strategies should therefore be aimed at decreasing rates of obesity.
Acknowledgment
The authors thank the IMSGC, ReproGen Consortium, ENGAGE Consortium, and EGG Consortium for access to their summary statistics data.
Glossary
- BMI
body mass index
- CI
confidence interval
- EGG
Early Growth Genetics
- ENGAGE
European Network for Genetic and Genomic Epidemiology
- GWAS
genome-wide association study
- HLA
human leukocyte antigen
- IMSGC
International Multiple Sclerosis Genetics Consortium
- MR
mendelian randomization
- MS
multiple sclerosis
- OR
odds ratio
- ReproGen
Reproductive Genetics
- SNP
single-nucleotide polymorphism
Appendix. Authors


Footnotes
Editorial, page 735
Study funding
The Richards laboratory is supported by the Canadian Institutes of Health Research, the Canadian Foundation for Innovation, and the Fonds de Recherche Santé Québec (FRSQ). Dr. Richards is supported by a FRSQ Clinical Research Scholarship and received research support from the National MS Society and the MS Society of Canada. TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, National Institute for Health Research–funded BioResource, Clinical Research Facility, and Biomedical Research Centre based at Guy's and St. Thomas' NHS Foundation Trust in partnership with King's College London.
Disclosure
A. Harroud and J. Morris report no disclosures relevant to the manuscript. V. Forgetta received personal compensation as a consultant for Precision Analytics. R. Mitchell and G. Davey Smith report no disclosures relevant to the manuscript. S. Sawcer received research support from Merck Sharp & Dohme Corp. B. Richards received research support from Eli Lilly and Company, Merck Sharp & Dohme Corp, and GlaxoSmithKline. Go to Neurology.org/N for full disclosures.
References
- 1.Nielsen NM, Harpsoe M, Simonsen J, et al. Age at menarche and risk of multiple sclerosis: a prospective cohort study based on the Danish National Birth Cohort. Am J Epidemiol 2017;185:712–719. [DOI] [PubMed] [Google Scholar]
- 2.Operskalski EA, Visscher BR, Malmgren RM, Detels R. A case-control study of multiple sclerosis. Neurology 1989;39:825–829. [DOI] [PubMed] [Google Scholar]
- 3.Ramagopalan SV, Valdar W, Criscuoli M, et al. Age of puberty and the risk of multiple sclerosis: a population based study. Eur J Neurol 2009;16:342–347. [DOI] [PubMed] [Google Scholar]
- 4.Ahn JJ, O'Mahony J, Moshkova M, et al. Puberty in females enhances the risk of an outcome of multiple sclerosis in children and the development of central nervous system autoimmunity in mice. Mult Scler 2015;21:735–748. [DOI] [PubMed] [Google Scholar]
- 5.Antonovsky A, Leibowitz U, Smith HA, et al. Epidemiologic study of multiple sclerosis in Israel, I: an overall review of methods and findings. Arch Neurol 1965;13:183–193. [DOI] [PubMed] [Google Scholar]
- 6.Berr C, Puel J, Clanet M, Ruidavets JB, Mas JL, Alperovitch A. Risk factors in multiple sclerosis: a population-based case-control study in Hautes-Pyrenees, France. Acta Neurol Scand 1989;80:46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gustavsen MW, Page CM, Moen SM, et al. Environmental exposures and the risk of multiple sclerosis investigated in a Norwegian case-control study. BMC Neurol 2014;14:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurtzke JF, Hyllested K, Arbuckle JD, et al. Multiple sclerosis in the Faroe Islands, 7: results of a case control questionnaire with multiple controls. Acta Neurol Scand 1997;96:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prentice P, Viner RM. Pubertal timing and adult obesity and cardiometabolic risk in women and men: a systematic review and meta-analysis. Int J Obes 2013;37:1036–1043. [DOI] [PubMed] [Google Scholar]
- 10.Mokry LE, Ross S, Timpson NJ, Sawcer S, Davey Smith G, Richards JB. Obesity and multiple sclerosis: a mendelian randomization study. PLoS Med 2016;13:e1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gianfrancesco MA, Barcellos LF. Obesity and multiple sclerosis susceptibility: a review. J Neurol Neuromedicine 2016;1:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aksglaede L, Olsen LW, Sorensen TI, Juul A. Forty years trends in timing of pubertal growth spurt in 157,000 Danish school children. PLoS One 2008;3:e2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koch-Henriksen N, Sorensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol 2010;9:520–532. [DOI] [PubMed] [Google Scholar]
- 14.Smith GD, Ebrahim S. Mendelian randomization: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32:1–22. [DOI] [PubMed] [Google Scholar]
- 15.Smith GD, Lawlor DA, Harbord R, Timpson N, Day I, Ebrahim S. Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med 2007;4:e352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Day FR, Thompson DJ, Helgason H, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet 2017;49:834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Day FR, Bulik-Sullivan B, Hinds DA, et al. Shared genetic aetiology of puberty timing between sexes and with health-related outcomes. Nat Commun 2015;6:8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.1000 Genomes Project Consortium; Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patsopoulos N, Baranzini SE, Santaniello A, et al. The Multiple Sclerosis Genomic Map: role of peripheral immune cells and resident microglia in susceptibility. bioRxiv 2018. Available at: 10.1101/143933. Accessed July 13, 2017. [DOI] [Google Scholar]
- 21.UK10K Consortium; Walter K, Min JL. The UK10K project identifies rare variants in health and disease. Nature 2015;526:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample mendelian randomization. Genet Epidemiol 2016;40:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day FR, Perry JR, Ong KK. Genetic regulation of puberty timing in humans. Neuroendocrinology 2015;102:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horikoshi M, Mgi R, van de Bunt M, et al. Discovery and fine-mapping of glycaemic and obesity-related trait loci using high-density imputation. PLoS Genet 2015;11:e1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Felix JF, Bradfield JP, Monnereau C, et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum Mol Genet 2016;25:389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37:658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dastani Z, Hivert MF, Timpson N, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet 2012;8:e1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 2008;27:1133–1163. [DOI] [PubMed] [Google Scholar]
- 29.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 2014;23:R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgess S, Dudbridge F, Thompson SG. Re: Multivariable mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 2015;181:290–291. [DOI] [PubMed] [Google Scholar]
- 31.Burgess S, Thompson DJ, Rees JMB, Day FR, Perry JR, Ong KK. Dissecting causal pathways using mendelian randomization with summarized genetic data: application to age at menarche and risk of breast cancer. Genetics 2017;207:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rees JMB, Wood AM, Burgess S. Extending the MR-Egger method for multivariable mendelian randomization to correct for both measured and unmeasured pleiotropy. Stat Med 2017;36:4705–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmgren A, Niklasson A, Nierop AF, et al. Pubertal height gain is inversely related to peak BMI in childhood. Pediatr Res 2017;81:448–454. [DOI] [PubMed] [Google Scholar]
- 36.Lee JM, Wasserman R, Kaciroti N, et al. Timing of puberty in overweight versus obese boys. Pediatrics 2016;137:e20150164. [DOI] [PubMed] [Google Scholar]
- 37.Silventoinen K, Pietilainen KH, Tynelius P, Sorensen TI, Kaprio J, Rasmussen F. Genetic and environmental factors in relative weight from birth to age 18: the Swedish Young Male Twins Study. Int J Obes 2007;31:615–621. [DOI] [PubMed] [Google Scholar]
- 38.Hedstrom AK, Olsson T, Alfredsson L. Body mass index during adolescence, rather than childhood, is critical in determining MS risk. Mult Scler 2016;22:878–883. [DOI] [PubMed] [Google Scholar]
- 39.Munger KL, Chitnis T, Ascherio A. Body size and risk of MS in two cohorts of US women. Neurology 2009;73:1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chitnis T, Graves J, Weinstock-Guttman B, et al. Distinct effects of obesity and puberty on risk and age at onset of pediatric MS. Ann Clin Transl Neurol 2016;3:897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lulu S, Graves J, Waubant E. Menarche increases relapse risk in pediatric multiple sclerosis. Mult Scler 2016;22:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D'Hooghe MB, Haentjens P, Nagels G, D'Hooghe T, De Keyser J. Menarche, oral contraceptives, pregnancy and progression of disability in relapsing onset and progressive onset multiple sclerosis. J Neurol 2012;259:855–861. [DOI] [PubMed] [Google Scholar]
- 43.Blakemore SJ, Burnett S, Dahl RE. The role of puberty in the developing adolescent brain. Hum Brain Mapp 2010;31:926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in mendelian randomization studies. Hum Mol Genet 2018;27:R195–R208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanderson E, Smith GD, Windmeijer F, Bowden J. An examination of multivariable mendelian randomization in the single sample and two-sample summary data settings. bioRxiv 2018. Available at: 10.1101/306209. Accessed April 27, 2018. [DOI] [Google Scholar]
- 46.Lakshman R, Forouhi NG, Sharp SJ, et al. Early age at menarche associated with cardiovascular disease and mortality. J Clin Endocrinol Metab 2009;94:4953–4960. [DOI] [PubMed] [Google Scholar]
- 47.Elks CE, Ong KK, Scott RA, et al. Age at menarche and type 2 diabetes risk: the EPIC-InterAct study. Diabetes Care 2013;36:3526–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Summary-level data for the genetic associations with age at puberty, adult BMI, and childhood BMI are publicly available and can be obtained through the links provided in the table. In addition, the dataset used to generate the results in the current study is available from the corresponding author on request.