

ABSTRACT
Autophagy is a well-conserved process of self-digestion of intracellular components. T. cruzi is a protozoan parasite with a complex life-cycle that involves insect vectors and mammalian hosts. Like other eukaryotic organisms, T. cruzi possesses an autophagic pathway that is activated during metacyclogenesis, the process that generates the infective forms of parasites. In addition, it has been demonstrated that mammalian autophagy has a role during host cell invasion by T. cruzi, and that T. cruzi can modulate this process to its own benefit. This review describes the latest findings concerning the participation of autophagy in both the T. cruzi differentiation processes and during the interaction of parasites within the host cells. Data to date suggest parasite autophagy is important for parasite survival and differentiation, which offers interesting prospects for therapeutic strategies. Additionally, the interruption of mammalian autophagy reduces the parasite infectivity, interfering with the intracellular cycle of T. cruzi inside the host. However, the impact on other stages of development, such as the intracellular replication of parasites is still not clearly understood. Further studies in this matter are necessaries to define the integral effect of autophagy on T. cruzi infection with both in vitro and in vivo approaches.
KEYWORDS: T. cruzi, metacyclogenesis, Atg8.1, T. cruzi infection, host cell, LC3
Introduction
Successful parasites have evolved to live and develop in specific hosts during their biological cycle. They have accumulated adaptations in response to environmental changes such as the immune response of the host. The repetition of the parasite-host interaction cycle ensures the persistence of several species of parasites. In humans, this interaction causes health disorders known as vector-borne and parasitic diseases.
According to the WHO, human parasitic diseases cause more than 700,000 deaths each year (http://www.who.int/vector-control/en/). These infectious diseases are caused by parasites from the group of protozoa and helminths. Some species of arthropods are also parasites and produce the so-called ectoparasitic diseases. Many of these diseases are classified as Neglected Tropical Diseases (NTDs) because they are caused by pathogens present in tropical and sub-tropical areas, and are often overlooked by pharmaceutical companies and even the public health community.
Trypanosomatidae is a vast family of protozoan parasites characterized by the presence of a single flagellum and a compacted DNA called kinetoplast. All members are exclusively parasitic, found primarily in insects. A few genera have life-cycles involving a secondary host, which may be a vertebrate, invertebrate or plant. These include several species that cause major diseases in humans [1]. The three major human diseases caused by trypanosomatids are Leishmaniasis, caused by various species of the genus Leishmania; African trypanosomiasis (or “sleeping sickness”), caused by Trypanosoma brucei; and American trypanosomiasis or Chagas disease, caused by Trypanosoma cruzi. These digenetic parasites have complex biological cycles in response to the specific adaptations required to live within two very different types of hosts, the insect vector and the mammalian organism.
Autophagy is an intracellular catabolic pathway highly conserved in eukaryotic organisms. Experimental and genomic data showed that the autophagy machinery characterized in animals and fungi also appeared in protists and function for diverse lifestyle adaptations [2]. Pathogenic protists can either utilize their own autophagy mechanisms or manipulate host-cell autophagy in order to establish or maintain infection within a host. This work revises the involvement of T. cruzi and host cell autophagy during the T. cruzi life-cycle.
The T. cruzi life-cycle and the infection process
During its complex life cycle, T. cruzi is found in three different parasitic forms. Epimastigotes and amastigotes are the replicative stages found in the lumen of the intestine of triatomine insect vectors or in the cytosol of infected host cells, respectively. The infective forms of T. cruzi are metacyclic trypomastigotes and blood-stream trypomastigotes, derived from epimastigotes and amastigotes, respectively, both with the capacity to invade a large number of different cell types in a process independent of host cell actin polymerization [3]. The so-called extracellular amastigotes, originated from the premature rupture of infected cells or transformed from swimming trypomastigotes, are also infective forms that, in contrast to trypomastigotes, require functional intact microfilaments to infect non-phagocytic host cells [4,5].
The T. cruzi entry process was classically divided into two specific steps (Figure 1, points 1 and 2): the adhesion process, related to the binding of T. cruzi antigens to host cell receptors which trigger cell signaling events [6,7]; and the internalization process that describes the mechanism that culminates in the formation of the T. cruzi parasitophorous vacuole (TcPV). Adhesion of trypomastigotes is driven by a repertoire of molecules present at the parasite surface or secreted during the infection process which bind to the specific receptors in the host cells [6,8,9]. Despite the advances made towards identification of the receptor molecule responsible for the adhesion of parasites, no single main host receptor candidate has been identified yet. In fact, the arsenal of parasite surface proteins and the multiple possible host receptors provide several opportunities for T. cruzi to recognize and contact cells and explains why this parasite is able to infect almost all types of mammalian cells [10,11]. The adhesion to cell surface triggers a host cell signaling cascade that culminates in the formation of the T. cruzi parasitophorous vacuole. Different processes have been described for the internalization of T. cruzi into non-phagocytic cells. Seminal studies showed two different models of trypomastigote invasion: by activating the lysosomal exocytosis or by inducing an endocytosis-like process. Both agree on the absence of plasma membrane protrusions (and microfilaments) but differ in the source of the membrane that initially envelops the parasite. The first model demonstrated that parasites elicited a Ca2+ signaling cascade in the host cells which activates lysosomal exocytosis [12]. The later fusion of lysosomes with the host plasma membrane allows the parasite entry and the formation of a parasitophorous vacuole with lysosomal characteristics [13]. This lysosomal exocytosis is produced with the peripheral pool of lysosomes and require microtubules and kinesin to the lysosomal transport toward the plasma membrane [14,15]. Autophagy has also been shown to participate in this process by the finding of autophagic proteins in the TcPV membrane [16] (see below). In contrast, the endocytosis-like process claims that at very early times after infection, the highest proportion of trypomastigotes enter host cells by invagination of the plasma membrane that generates an initial TcPV enriched in phosphoinositides derived from plasma membrane but not lysosomal markers [17]. This model reinforced previously published data that demonstrated the participation of endocytic GTPases such as Rab5 and Rab7 and dynamin in the T. cruzi infection process [18,19]. Later studies showed that T. cruzi exploited the plasma membrane wound repair mechanism [20] carried out by the exocytosis of lysosomes at the site of the injury followed by the compensatory endocytosis of injured membranes [21]. In summary, both events, exocytosis and endocytosis, previously classified as independent processes, now, under the light of these new data, have been shown to occur sequentially during parasite invasion. The different styles of T. cruzi entry were also associated with the different trypomastigote forms. Metacyclic trypomastigotes (MT) that express gp82, the surface molecule that mediates invasion, induce mTOR dephosphorylation and lysosome biogenesis and scattering, evidencing preferentially the lysosomal exocytosis process. In contrast, the entry of tissue culture trypomastigotes (TCT), the equivalent to blood-stream trypomastigotes, is predominantly related to endocytosis [22]. Other forms of T. cruzi entry into host cells, such as phagocytosis and macropinocytosis, have been also described [23].
Figure 1.
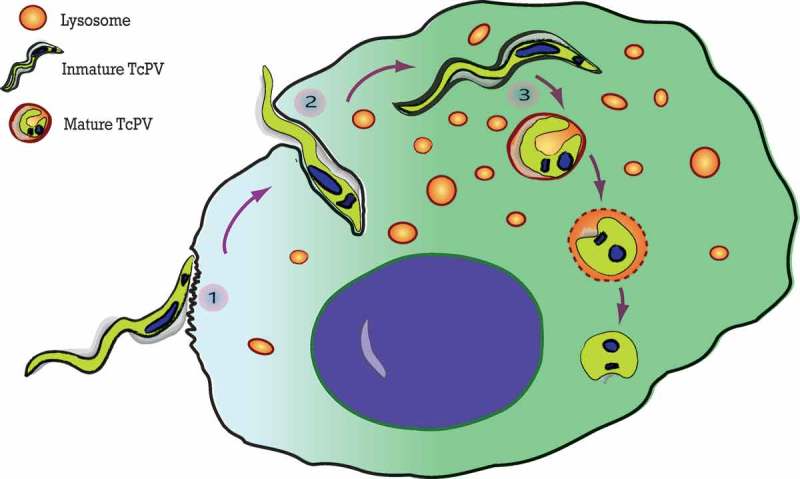
The three steps of the T. cruzi invasion. The scheme summarizes the process of T. cruzi entry into the host cell. (1) Adhesion of trypomastigotes to the host cell surface. (2) Internalization of trypomastigotes produced by invagination of the plasma membrane. (3) Vacuole maturation proceeds after the fusion with lysosomes which initiates the differentiation of T. cruzi from trypomastigotes to amastigotes.
In addition to the specific manner of TcPV formation, there is a third little described step during T. cruzi invasion that involves the TcPV maturation (Figure 1, point 3). Fusion of lysosomes with the early parasitophorous vacuole is crucial to the progress of infection. Previous works from the laboratory of Norma Andrews have demonstrated that when lysosomal fusion was inhibited, internalized parasites failed to be retained inside host cells and escaped to the extracellular environment [24,25]. The abolition of this phenomenon, known as reversal infection, by allowing the lysosomal fusion to TcPV, is a key process for the retention of T. cruzi inside the host cell [26]. In agreement with this, further studies from our group demonstrated that the SNARE VAMP7 and its partner Vti1b, both components of the molecular machinery that promotes lysosomal fusion, are required during T. cruzi vacuole development and, as a consequence, for the parasite infection [27]. We have also shown that migration of VAMP7 positive vesicles to the TcPV depends on KIF5, a motor protein belonging to the superfamily of kinesins. It was suggested that the highly motile trypomastigotes contained in vacuoles fail to associate with microtubules when lysosomal fusion is impaired, allowing the parasites to continue to move around the cytosol and eventually leave the cell. In the same way, the loss of parasite motility associated with the lysosomal fusion to the TcPV could be attributed to the beginning of the differentiation process from trypomastigotes to non-motile amastigotes induced in part by the acidic pH gained by the vacuole during this process. The striking morphological transition undergone by the parasite throughout the course of the infection process, from the typical elongated form at early times to the ovoid form at later times [27] strengthens this idea. Maturation of the vacuole is finally a key process for both the retention of T. cruzi inside the host cell and for the progression of the T. cruzi intracellular cycle by allowing the differentiation from trypomastigotes to amastigotes.
The autophagic pathway
Autophagy is a catabolic pathway conserved in most eukaryotes from yeast to mammalian cells [28]. This process involves the cellular self-digestion through the lysosomal machinery. Macroautophagy, the most widely studied type of autophagy, is important in many physiological situations such as cell development, cell growth, and cell differentiation. As a constitutive process, autophagy functions at basal levels in the turnover of long-lived proteins and old organelles for maintaining cellular homeostasis. Nutritional stress conditions are the main physiologic stimulus of autophagy. Under these situations, cells entrap cytoplasmic materials and degrade them to provide aminoacids that function as energy source for cell survival [29]. Besides this bulk degradation process, autophagy functions selectively to remove misfolded or aggregated proteins, damaged organelles, such as mitochondria, endoplasmic reticulum and peroxisomes, as well as to eliminate intracellular pathogens, being part of the innate immune response [30]. The autophagic pathway involves specific compartments inside the cell. Basically, the cytoplasmic cargo is captured through the intermediary of a double membrane-bound vesicle, referred to as autophagosome, which fuses with the lysosome to form an autolysosome. In mammalian cells, autophagosomes derived from an isolation membrane, also known as phagophore, which is likely derived from a lipid bilayer contributed by the endoplasmic reticulum and/or the trans-Golgi and late endosomes [31–33]. The outer mitochondrial membrane and the plasma membrane can also participate as membrane donor [34]; therefore, the exact origin of the phagophore in mammalian cells is still controversial. The isolation membrane grows around the cargo and finally closes to form the early autophagosome, easily recognized as a double-membrane vesicle by electron microscopy [35]. Autophagosomes can then fuse with endocytic compartments to form amphisomes, which, in turn, fuse with lysosomes, forming autolysosomes where the proteins are degraded to amino acids that are recycled via lysosomal permeases and other transporters [36].
Specific molecules coordinate this complex process. To date, different autophagy-related genes (Atg) have been identified by genetic screening in yeast [28]. Many of them have been conserved in mammalian cells. These genes can be grouped according to their functions at key stages of the autophagy pathway: initiation, elongation, maturation, and fusion with the lysosomes. In mammalian systems, the phagophore is derived from membranes enriched in phosphatidyl inositol triphosphate (PI3P), the product of the Beclin1-Vps34 complex [37]. Phagophore elongation requires the sequential activation of two protein conjugation reactions. The mammalian Atg5-Atg12-Atg16L complex is recruited to the isolation membrane, favoring the extension of the precursor membrane [38]. LC3, the mammalian homolog of Atg8, is then conjugated with phosphatidylethanolamine (PE) to form LC3-II, which inserts into the autophagosome membrane and contributes to vesicle elongation [39]. After autophagic induction LC3-II is visualized by a punctuate pattern corresponding to autophagic vesicles. Unlike the Atg5-Atg12.Atg16L complex, LC3-II remains on completed autophagosomes and also on autolysosomes [40]. The final steps of autophagy involve docking and fusion of autophagosomes to lysosomes to generate autolysosomes which produce cargo degradation and recycling. Specific Rab GTPases and SNAREs proteins function as master regulators of these fusion events. Impairment of this process by the use of mutant proteins or chemicals prevents the normal maturation of the pathway leading to the accumulation of autophagosomes. Bafilomycin A1 is a broadly used autophagic flux inhibitor that prevents the fusion of autophagosomes with endosomes/lysosomes and the autophagic degradation [41].
Two key signaling processes converge to correlate canonical autophagy with cell nutrient or stress conditions, the mTOR and the PI3K pathways. The mTOR signaling cascade transduces the response from growth factors, via class I PI3K, Akt/PKB, and so forth, to negatively regulate autophagy [42]. Rapamycin is one of the classical inducers of autophagy due to inhibition of the mTOR signaling pathway [43]. As mentioned above, Beclin1-Vps34 complex is necessary to produce the PI3P required in the phagophore formation and elongation. This class III PI3K complex is inhibited by wortmannin, which blocks the autophagosome initiation process [44].
The process of autophagy can be monitored intracellularly by utilizing LC3 fused to a fluorescent protein such as GFP-LC3 or mCherry-LC3. Fluorescent LC3 is incorporated into the autophagosomes and could be observed as small dots inside the cell by light microscopy. The number of puncta in a cell at any specific time is a function of both the formation and the clearance of autophagosomes [45].
Selective autophagy refers to the different mechanisms of capture and degradation of specific cargos by autophagy. These non-canonical classes of autophagy are named according to the type of component that has been incorporated to the route, such as mitophagy, pexophagy and xenophagy [46], among others. Xenophagy is the process by which cells can degrade an intracellular microorganism present inside a phagosome [47,48] or free in the cytosol [49]. Due to this property, xenophagy has been classified as a component of the innate immune responses against intracellular pathogens. Xenophagy generally functions as a second barrier when phagocytosis or other defense mechanisms are exceeded. However, some pathogens have the capacity to evade autophagic responses or to subvert the autophagic pathway and to live and replicate inside an autophagosomal compartment (see below). Recent research has related LC3 protein to pathways other than autophagy. Since 2007, the concept of LAP (LC3-associated phagocytosis) has been introduced. Nowadays LAP is understood as a type of non-canonical autophagy. LAP uses some autophagic components but it differs mainly in that the LC3 positive vesicle is a simple membrane compartment [50,51].
Autophagy during the T. cruzi life-cycle
As eukaryotic organisms, protists possess a rudimentary autophagic pathway that functions at specific stages of their life-cycles mainly during the differentiation processes [2]. Autophagy is also activated when pathogenic protists are treated with anti-parasitic drugs, as a mechanism to overcome the stress caused by the toxic compounds before cell death [52–55]. In addition, a growing body of evidence shows that intracellular protists have the ability to manipulate host-cell autophagy in order to establish or maintain the infection within a host. Below we summarize the findings made on the T. cruzi autophagy pathway and its participation in the differentiation processes, and then we describe the recent advances concerning the interaction of T. cruzi with host cell autophagy.
Autophagy is induced during the T. cruzi differentiation processes
The first evidence for the existence of an autophagic-like process in T. cruzi was provided, as in mammalian cells, by morphological studies showing the presence of double-membrane vesicles and multivesicular structures in parasites treated with trypanocidal drugs [56,57]. Later genome database searches supplemented with more advanced analyses revealed the presence of autophagy-associated components in T. cruzi [58–60]. These bioinformatics analyses showed that only half of the genes found in yeasts were present in trypanosomatids. T. cruzi possesses all components of the LC3/Atg8 conjugation system, but lacks the Atg5-Atg12 protein complex, indicative of a different manner to initiate and elongate the autophagosome. Two TcAtg8 homologs have been found in T. cruzi: TcAtg8.1 and TcAtg8.2. Only the first one was functionally linked to autophagy [60]. During starvation, TcAtg8.1 is efficiently cleaved by one of the two TcAtg4 homologs (autophagins) and then, after conjugation to PE, is inserted in the membrane of the autophagosome-like vesicles [60]. Further studies from our lab established the participation of autophagy during the differentiation of T. cruzi from epimastigotes to metacyclic trypomastigotes [61]. This process, called metacyclogenesis, takes place in the lumen of the intestine of the triatomine vector. The reduced nutrient availability produced by the high multiplication of epimastigotes is the main stimulus to induce it. By performing an in vitro method of differentiation, we showed an increment in the number of Atg8.1 positive vesicles during starvation. The same result was observed in the presence of other autophagy inducers such as rapamycin, the classical inhibitor of TOR kinase, and spermidine, a polyamine that induces autophagy in many eukaryotic organisms by regulation of gene expression and protein activity [62]. We also showed that classical autophagy inhibitors such as wortmannin impaired the autophagosome formation in T. cruzi [61]. In agreement with this, Schoijet et al. [63] showed that TcVps34 kinase (the parasitic counterpart of the mammalian Beclin-1-Vps34 kinase) is regulated by TcVps15 and that both form a complex that participates in starvation induced autophagy in T. cruzi. As mentioned above, bafilomycin is a H+ pump inhibitor that impairs the normal autophagic flux, producing an accumulation of autophagic structures [41]. In contrast to mammalian cells, in T. cruzi bafilomycin inhibits the first steps of autophagy, evidenced by the low number of Atg8.1 positive vesicles generated under this treatment. A previous work showed that formation of autophagosome in the related parasite T. brucei requires the normal acidification of acidocalcisomes, acidic parasitic compartments required for osmoregulation [64]. Acidification impairment of acidocalcisomes by bafilomycin treatment completely abrogates autophagic response, indicating that these compartments could be participating in the first steps of autophagy in trypanosomes in a not yet clearly understood mechanism.
Autophagy is also required during the differentiation of trypomastigotes to amastigotes, a process that starts in the acidic environment of the parasitophorous vacuole and finish in the host cell cytosol. Our previous data showed that starvation in PBS and rapamycin were able to induce this process similarly to low pH. Moreover, we observed Atg8-decorated vesicles in amastigotes present in the cytosol of infected cells, indicating that autophagy can be induced in the parasite inside the host [16]. Although more studies will be necessary in the future to elucidate the specific mechanisms that regulate autophagy in trypanosomatids, these data show that this process is an interesting target to be modulated to impair T. cruzi survival in the host.
Host autophagy during T. cruzi infection
Invasion of trypomastigotes
As mentioned above, the interaction between host autophagy and intracellular pathogens has different fates according to the type of microorganism and host cell. Many microorganisms can be engulfed by autophagic compartments and eliminated in an autolysosome by xenophagy. Another group of pathogens has the ability to subvert the autophagic pathway to its own benefit. Some pathogens inhibit the autophagic flux to replicate in an autophagic niche which does not fuse with lysosomes [65]. Others have evolved to live and replicate inside a compartment with autolysosomal characteristics [66–69]. In the case of T. cruzi, our data showed that this parasite exploits autophagy to support invasionof the host cell. Previous studies from our laboratory demonstrated that parasite entry by the lysosomal route is favored under conditions that increase autophagy [16,70]. Induction of autophagy before infection by starvation or other means significantly increased the percentage of infected cells by enhancing the number of lysosomal compartments (lysosomes and autolysosomes) available for T. cruzi infection, resulting in an increased colonization of the host cell. We also showed that the T. cruzi parasitophorous vacuole is decorated with LC3 protein (Figure 2) and that the autophagic inhibitors wortmannin, 3-methyladenine or vinblastine suppress this recruitment and also significantly reduce the intracellular infection. Interestingly, infection was diminished in the absence of specific autophagy genes Beclin1 or Atg5, which are required for initiation of autophagy, indicating that autophagic-derived compartments are required for the increased entry of T. cruzi into the host cell. The pro-pathogen effects of autophagy on T. cruzi infection were observed in different types of host cells and of T. cruzi strains, demonstrating that this interaction is a widespread phenomenon [71]. These data were obtained with the TCT forms of T. cruzi. Quite different features were described by the group of Nobuko Yoshida, working with metacyclic trypomastigotes. They showed that, in contrast to TCT, pre-starvation of cells by 2 h or rapamycin treatment reduced MT invasion whereas other conditions that promote lysosomal exocytosis increased it [72]. Further data from the same group demonstrated that conditions that promote lysosome biogenesis and scattering such us 1 h starvation or treatment with sucrose, increase cell susceptibility to MT and resistance to TCT [22]. Due to cellular distribution of lysosomes have different outcomes related to the treatment used (1 h or 2 h starvation or rapamycin), these data indicate that MT invasion is mainly related to lysosomal exocytosis, whereas TCT entry is predominantly an endocytosis–like process.
Figure 2.
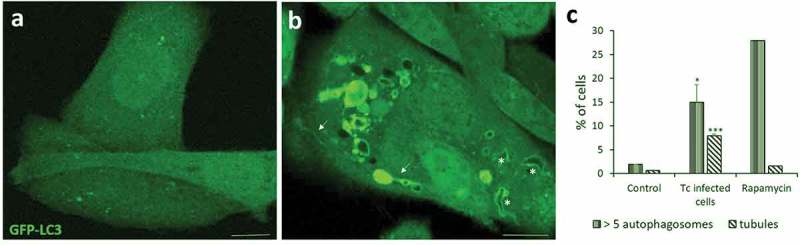
Autophagy is induced during T. cruzi infection. CHO cells overexpressing GFP-LC3 were infected with tissue culture trypomastigotes (TCT) of T. cruzi Y strain (MOI = 50) during 3 h. After fixation, cells were washed, mounted in mowiol and analyzed by confocal microscopy. (a) Control (non-infected) cells depicting the homogeneous distribution of GFP-LC3 in the soluble form. (b) Infected cell displaying numerous autophagic vacuoles and tubules (arrows) decorated with membrane-associated GFP-LC3. Note the recruitment of this protein in the membrane of the T. cruzi parasitophorous vacuole (asterisks) that were recognized by the typical form of trypomastigotes. (c) Percentage of cells containing more than 5 LC3-positive vesicles/cell and at least one tubule/cell at the indicated conditions. Rapamycin (50 ng/μl) were incubated for 2 h in control conditions. Scale bar: 5 μm. Data shown represent the mean ± SE from 3 independent experiments. *p < 0.05, ***p < 0.001 (Student´s t-test).
Concerning the mechanism of T. cruzi invasion, it is important to note that, as mentioned above, a growing body of evidence showed that trypomastigotes exploit the lysosome-dependent membrane repair mechanism to enter the host cell [21]. In this sense, autophagy has been shown to play a role in the fixing of damaged pathogen containing vacuoles such as Salmonella [73]. The rupture of the cell surface produced by T. cruzi during invasion may trigger the autophagic response as a mechanism to restore the plasma membrane integrity. In agreement with this hypothesis, our results using live imaging by confocal microscopy showed that GFP-LC3 positive vesicles move towards the plasma membrane and contact the sites where trypomastigotes bind to the membrane [16,71].
A recent work showed that autophagosome formation was induced by T. cruzi infection. By quantifying the number of LC3 puncta in cells infected with T. cruzi, the authors demonstrated a gradual increment of these vesicles, which reach a maximum level at 9 h post infection [74]. Unpublished data from our laboratory show that the percentage of cells with more than five LC3-positive vesicles/cell was significantly increased in cells infected with TCT of T. cruzi Y strain, compared to control (Figure 2). Interestingly, we also observed a significantly higher number of LC3-positive tubules in these cells, which has not been observed either in the uninfected control cells or in rapamycin treated cells (Figure 2). Some of these tubules are even associated with the TcPV. Previous works have associated this type of dynamic tubules with efficient pathogen proliferation [75,76]. Induction of these tubules in cells infected with T. cruzi could favor the arrival or elimination of specific molecules into the TcPV, creating a favorable niche for the parasite. More experiments will need to confirm this hypothesis.
Onisuka and colleagues [74] also showed that T. cruzi infection inhibited the normal maturation of autophagosomes to autolysosomes (autophagic flux). Our investigation concerning this point was not conclusive. By using the GFP-RFP-LC3 tandem, we did not observe a different pattern of green and red colocalization, compared to control. Western blot experiments also yielded the same ambiguous results (data not shown). Unexpectedly, colocalization between DQ-BSA, one of the best markers of hydrolytic compartments, with the T. cruzi vacuole at different times after infection was lower compared to the marker of acidic compartments lysosensor (data not shown). On the other hand, many authors, including our laboratory, found the classical late endosomal/lysosomal markers, such as VAMP7, Lamp 1 and 2 and Cathepsin D, in the TcPV by indirect immunofluorescence [13,16,27]. These evidences suggest that although lysosomal fusion to TcPV is produced during vacuole maturation, the final mature TcPV might not be as hydrolytic as supposed. If this is the case, lysosomal enzymes might not be fully delivered to the vacuole or remain with low activity inside de TcPV because of the sub-optimal pH. Another possibility is that lysosomal enzymes were not activated inside the TcPV by a currently unknown mechanism. This hypothesis may explain, in addition to other mechanisms, the resistance of the parasites to lysosomal degradation. More studies will be necessary to confirm this assumption.
Intracellular differentiation and replication of amastigotes
As mentioned above, previous studies from our laboratory showed that conditions that increase autophagy favor the differentiation in vitro from trypomastigotes to amastigotes [16]. Atg8 positive vesicles were also visualized in amastigotes developed in the host cells [16]. Although more studies will need to confirm these findings, these evidences suggest the participation of autophagy in the T. cruzi intracellular differentiation. The following stage in the T. cruzi intracellular cycle, the proliferation of amastigotes, was not significantly impacted by the autophagic modulation in non-phagocytic host cells, compared to cells maintained in control conditions [16]. Since T. cruzi has its own autophagic pathway that can be modulated by the same inducers and inhibitors of host autophagy [61], other experimental approaches will be necessary to decipher the possible action of autophagy modulation on T. cruzi infected cells. In this context, autophagy-deficient host cell lines can be used to specifically assess the impact of host autophagy loss. In contrast, growth of amastigotes on professional phagocytes might be affected by the autophagic response of these cells as it is described in the following section.
Autophagy on in vivo models of infection
To date there is not published data about the outcome of T. cruzi infection in vivo under autophagic modulation. The pathogen/autophagy relationship on models of murine infections, with the exception of a few cases, remains little understood. The effect of autophagy as an innate immune component is easier to understand, particularly with the use of knockout mice. Studies on M. tuberculosis murine infections showed that Atg5 deficient mice relative to autophagy-proficient littermates resulted in increased bacillary burden and excessive pulmonary inflammation evidencing the role of autophagy in vivo by suppressing both M. tuberculosis growth and damaging inflammation [77]. Furthermore, mice deficient in either IFN-γ or IFN-γ receptors are highly susceptible to M. tuberculosis infection [78]. It is known that IFN-γ is a key cytokine in the regulation of the immune responses [79] that explains the high susceptibility of these mice to mycobacterial infection. This cytokine is also a strong inducer of autophagy in macrophages [80]. Taking into account that macrophage activation is a key process to control M. tuberculosis infection, the absence of an appropriate autophagy/xenophagy response from macrophages might also explain the higher bacterial burden observed in these mice.
The main concerns with the in vivo models arise from the cases of pathogens for which in vitro studies have shown them to be favored by autophagy induction. No current evidence demonstrates that autophagy gene deletion in the host attenuates microbial disease in these cases. Therefore, the physiological significance of microbial utilization of autophagy for “promicrobial” effects remains to be established [81]. In line with these thoughts, our group observed a more aggressive infection of T. cruzi in an autophagy deficient mice model compared to the wild type counterpart (Casassa et al, unpublished). We also observed higher levels of infection in peritoneal cells obtained from autophagy deficient mice than cells from control animals. This means that contrary to what was observed on in vitro infections, deficiency of autophagy on in vivo models resulted in an increased infection and parasite burden. These findings highlight the role of autophagy as a component of immune responses against T. cruzi. Future studies on the participation of autophagy at different levels of the immunity against T. cruzi will allow understanding the mechanisms that prevail in the establishment of the infection, particularly in the case of the chronic stage which is characterized by T. cruzi persistence, tissue damage and inflammatory response.
Concluding remarks
As one of the main processes that regulate cell survival, differentiation or death, autophagy has a major participation in the T. cruzi life-cycle. Evidence shows that parasitic autophagy is required during the interconversion between epimastigotes, trypomastigotes and amastigotes. These changes confer to the parasite the capacity to adapt to the following host in their life-cycle making parasite autophagy an excellent target for trypanocidal drugs.
Host autophagy has also been described as a one of the main regulators of T. cruzi invasion with possible effects on the maturation of the parasitophorous vacuole. Therefore, modulation of this process could favor the infection control by the cell. Furthermore, the recruitment of LC3 to the T. cruzi parasitophorous vacuole constitutes an interesting finding that needs to be better characterized in the future, to understand which type of process, e.g. canonical autophagy; xenophagy or even LAP are involved.
The pivotal role of autophagy in pathogenicity and virulence demonstrated in T. cruzi suggests that autophagy machinery is a possible good target for anti-parasitic intervention. Further studies, especially using different classes of cells, phagocytic and non-phagocytic and alsoon in vivo models of infection by T. cruzi, will be mandatory to confirm the possible beneficial effect of autophagy modulation that might offer the perspective to interfere with the infective cycle of T. cruzi in the host.
Funding Statement
This work was supported by the Fondo para la Investigación Científica y Tecnológica [PICT 2013-2757]; Secretaria de Ciencia, Técnica y Posgrado, Universidad Nacional de Cuyo (AR) [Proyectos de Investigación 2016-2018]; Consejo Nacional de Investigaciones Científicas y Tecnológicas [CONICET-PIP 2014-2016].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Simpson AGB, Stevens JR, Lukeš J.. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006;22:168–174. [DOI] [PubMed] [Google Scholar]
- [2].Duszenko M, Ginger ML, Brennand A, et al. Autophagy in protists. Autophagy. 2011;7:127–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schenkman S, Robbins ES, Nussenzweig V. Attachment of Trypanosoma cruzi to mammalian cells requires parasite energy, and invasion can be independent of the target cell cytoskeleton. Infect Immun. 1991;59:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fernandes MC, Flannery AR, Andrews N, et al. Extracellular amastigotes of Trypanosoma cruzi are potent inducers of phagocytosis in mammalian cells. Cell Microbiol. 2013;15:977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bonfim-Melo A, Ferreira ÉR, Mortara RA. Rac1/WAVE2 and Cdc42/N-WASP participation in actin-dependent host cell invasion by extracellular amastigotes of Trypanosoma cruzi. Front Microbiol. 2018;9:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Maeda FY, Cortez C, Yoshida N. Cell signaling during Trypanosoma cruzi invasion. Front Immunol. 2012;3:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yoshida N. Molecular basis of mammalian cell invasion by Trypanosoma cruzi. An Acad Bras Cienc. 2006;78:87–111. [DOI] [PubMed] [Google Scholar]
- [8].Magdesian MH, Giordano R, Ulrich H, et al. Infection by Trypanosoma cruzi. Identification of a parasite ligand and its host cell receptor. J Biol Chem. 2001;276:19382–19389. [DOI] [PubMed] [Google Scholar]
- [9].Martins NO, de Souza RT, Cordero EM, et al. Molecular characterization of a novel family of Trypanosoma cruzi Surface Membrane Proteins (TcSMP) involved in mammalian host cell invasion. PLoS Negl Trop Dis. 2015;9:e0004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].de Souza W, de Carvalho TMU, Barrias ES. Review on Trypanosoma cruzi: host cell interaction. Int J Cell Biol. 2010;2010:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].de Araujo-Jorge TC, Barbosa HS, Meirelles MNL, et al. Trypanosoma cruzi recognition by macrophages and muscle cells: perspectives after a 15-year study. Mem Inst Oswaldo Cruz. 1992;87:43–56. [DOI] [PubMed] [Google Scholar]
- [12].Tardieux I, Nathanson MH, Andrews NW. Role in host cell invasion of Trypanosoma cruzi-induced cytosolic-free Ca2+ transients. J Exp Med. 1994;179:1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tardieux I, Webster P, Ravesloot J, et al. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71:1117–1130. [DOI] [PubMed] [Google Scholar]
- [14].Rodríguez A, Samoff E, Rioult MG, et al. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J Cell Biol. 1996;134:349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hissa B, de Oliveira Andrade L. Trypasonoma cruzi uses a specific subset of host cell lysosomes for cell invasion. Parasitol Int. 2015;64:135–138. [DOI] [PubMed] [Google Scholar]
- [16].Romano PS, Arboit MA, Vázquez CL, et al. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy. 2009;5:6–18. [DOI] [PubMed] [Google Scholar]
- [17].Woolsey AM, Sunwoo L, Petersen CA, et al. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J Cell Sci. 2003;116:3611–3622. [DOI] [PubMed] [Google Scholar]
- [18].Wilkowsky SE, Barbieri MA, Stahl PD, et al. Regulation of Trypanosoma cruzi invasion of nonphagocytic cells by the endocytically active GTPases dynamin, Rab5, and Rab7. Biochem Biophys Res Commun. 2002;291:516–521. [DOI] [PubMed] [Google Scholar]
- [19].Barrias ES, Reignault LC, De Souza W, et al. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS One. 2010;5:e7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tam C, Idone V, Devlin C, et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. 2010;189:1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fernandes MC, Cortez M, Flannery AR, et al. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J Exp Med. 2011;208:909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cortez C, Real F, Yoshida N. Lysosome biogenesis/scattering increases host cell susceptibility to invasion by Trypanosoma cruzi metacyclic forms and resistance to tissue culture trypomastigotes. Cell Microbiol. 2016;18:748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Barrias ES, de Carvalho TMU, De Souza W. Trypanosoma cruzi: entry into mammalian host cells and parasitophorous vacuole formation. Front Immunol. 2013;4:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Andrade LO, Andrews NW. Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J Exp Med. 2004;200:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Andrade LO, Andrews NW. The Trypanosoma cruzi–host-cell interplay: location, invasion, retention. Nat Rev Microbiol. 2005;3:819–823. [DOI] [PubMed] [Google Scholar]
- [26].Fernandes MC, Andrews NW. Host cell invasion by Trypanosoma cruzi : a unique strategy that promotes persistence. FEMS Microbiol Rev. 2012;36:734–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cueto JA, Vanrell MC, Salassa BN, et al. Soluble N-ethylmaleimide-sensitive factor attachment protein receptors required during Trypanosoma cruzi parasitophorous vacuole development. Cell Microbiol. 2017;19:e12713. [DOI] [PubMed] [Google Scholar]
- [28].Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. [DOI] [PubMed] [Google Scholar]
- [29].Yang Z, Huang J, Geng J, et al. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17:5094–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hayashi-Nishino M, Fujita N, Noda T, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. [DOI] [PubMed] [Google Scholar]
- [34].Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Baba M, Takeshige K, Baba N, et al. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J Cell Biol. 1994;124:903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang Z, Klionsky DJ. Permeases recycle amino acids resulting from autophagy. Autophagy. 2007;3:149–150. [DOI] [PubMed] [Google Scholar]
- [37].Itakura E, Kishi C, Inoue K, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mizushima N, Kuma A, Kobayashi Y, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–1688. [DOI] [PubMed] [Google Scholar]
- [39].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. [DOI] [PubMed] [Google Scholar]
- [42].Cheong H, Yorimitsu T, Reggiori F, et al. Atg17 regulates the magnitude of the autophagic response. Mol Biol Cell. 2005;16:3438–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. [DOI] [PubMed] [Google Scholar]
- [44].Blommaart EF, Krause U, Schellens JP, et al. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. [DOI] [PubMed] [Google Scholar]
- [45].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bauckman KA, Owusu-Boaitey N, Mysorekar IU. Selective autophagy: xenophagy. Methods. 2015;75:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pareja MEM, Colombo MI. Autophagic clearance of bacterial pathogens: molecular recognition of intracellular microorganisms. Front Cell Infect Microbiol. 2013;3:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gutierrez MG, Munafó DB, Berón W, et al. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. [DOI] [PubMed] [Google Scholar]
- [49].Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. [DOI] [PubMed] [Google Scholar]
- [50].Heckmann BL, Boada-Romero E, Cunha LD, et al. LC3-associated phagocytosis and inflammation. J Mol Biol. 2017;429:3561–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lai S, Devenish RJ. LC3-associated phagocytosis (LAP): connections with host autophagy. Cells. 2012;1:396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Veiga-Santos P, Desoti VC, Miranda N, et al. The natural compounds piperovatine and piperlonguminine induce autophagic cell death on Trypanosoma cruzi. Acta Trop. 2013;125:349–356. [DOI] [PubMed] [Google Scholar]
- [53].Fernandes MC, Da Silva EN, Pinto AV, et al. A novel triazolic naphthofuranquinone induces autophagy in reservosomes and impairment of mitosis in Trypanosoma cruzi. Parasitology. 2012;139:26–36. [DOI] [PubMed] [Google Scholar]
- [54].Dos Anjos DO, Sobral Alves ES, Gonçalves VT, et al. Effects of a novel β-lapachone derivative on Trypanosoma cruzi: parasite death involving apoptosis, autophagy and necrosis. Int J Parasitol Drugs Drug Resist. 2016;6:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lazarin-Bidóia D, Desoti VC, Martins SC, et al. Dibenzylideneacetones are potent trypanocidal compounds that affect the Trypanosoma cruzi redox system. Antimicrob Agents Chemother. 2016;60:890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Braga MV, Magaraci F, Lorente SO, et al. Effects of inhibitors of Delta24(25)-sterol methyl transferase on the ultrastructure of epimastigotes of Trypanosoma cruzi. Microsc Microanal. 2005;11:506–515. [DOI] [PubMed] [Google Scholar]
- [57].Santa-Rita RM, Lira R, Barbosa HS, et al. Anti-proliferative synergy of lysophospholipid analogues and ketoconazole against Trypanosoma cruzi (Kinetoplastida: trypanosomatidae): cellular and ultrastructural analysis. J Antimicrob Chemother. 2005;55:780–784. [DOI] [PubMed] [Google Scholar]
- [58].Michels PAM, Bringaud F, Herman M, et al. Metabolic functions of glycosomes in trypanosomatids. Biochim Biophys Acta. 2006;1763:1463–1477. [DOI] [PubMed] [Google Scholar]
- [59].Herman M, Gillies S, Michels PA, et al. Autophagy and related processes in trypanosomatids: insights from genomic and bioinformatic analyses. Autophagy. 2006;2:107–118. [DOI] [PubMed] [Google Scholar]
- [60].Alvarez VE, Kosec G, Sant’Anna C, et al. Autophagy is involved in nutritional stress response and differentiation in Trypanosoma cruzi. J Biol Chem. 2008;283:3454–3464. [DOI] [PubMed] [Google Scholar]
- [61].Vanrell MC, Losinno AD, Cueto JA, et al. The regulation of autophagy differentially affects Trypanosoma cruzi metacyclogenesis. PLoS Negl Trop Dis. 2017;11:e0006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Eisenberg T, Knauer H, Schauer A, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. [DOI] [PubMed] [Google Scholar]
- [63].Schoijet AC, Sternlieb T, Alonso GD. The phosphatidylinositol 3-kinase class III complex containing TcVps15 and TcVps34 participates in autophagy in Trypanosoma cruzi. J Eukaryot Microbiol. 2017;64:308–321. [DOI] [PubMed] [Google Scholar]
- [64].Li F-J, He CY. Acidocalcisome is required for autophagy in Trypanosoma brucei. Autophagy. 2014;10:1978–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dorn BR, Dunn WA, Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. 2002;4:1–10. [DOI] [PubMed] [Google Scholar]
- [66].Berón W, Gutierrez MG, Rabinovitch M, et al. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70:5816–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005;7:765–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gutierrez MG, Vázquez CL, Munafó DB, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. 2005;7:981–993. [DOI] [PubMed] [Google Scholar]
- [69].Romano PS, Gutierrez MG, Berón W, et al. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol. 2007;9:891–909. [DOI] [PubMed] [Google Scholar]
- [70].Vanrell MC, Cueto JA, Barclay JJ, et al. Polyamine depletion inhibits the autophagic response modulating Trypanosoma cruzi infectivity. Autophagy. 2013;9:1080–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Romano PS, Cueto JA, Casassa AF, et al. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life. 2012;64:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Martins RM, Alves RM, Macedo S, et al. Starvation and rapamycin differentially regulate host cell lysosome exocytosis and invasion by Trypanosoma cruzi metacyclic forms. Cell Microbiol. 2011;13:943–954. [DOI] [PubMed] [Google Scholar]
- [73].Owen KA, Casanova JE. Salmonella manipulates autophagy to “serve and protect.” Cell Host Microbe. 2015;18:517–519. [DOI] [PubMed] [Google Scholar]
- [74].Onizuka Y, Takahashi C, Uematsu A, et al. Inhibition of autolysosome formation in host autophagy by Trypanosoma cruzi infection. Acta Trop. 2017;170:57–62. [DOI] [PubMed] [Google Scholar]
- [75].Krieger V, Liebl D, Zhang Y, et al. Reorganization of the endosomal system in Salmonella-infected cells: the ultrastructure of Salmonella-induced tubular compartments. PLoS Pathog. 2014;10:e1004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].López de Armentia MM, Gauron MC, Colombo MI. Staphylococcus aureus alpha-toxin induces the formation of dynamic tubules labeled with LC3 within host cells in a Rab7 and Rab1b-dependent manner. Front Cell Infect Microbiol. 2017;7:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci. 2012;109:E3168–E3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Cooper AM, Dalton DK, Stewart TA, et al. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Knop J. Immunologic effects of interferon. J Invest Dermatol. 1990;95:72S–74S. [DOI] [PubMed] [Google Scholar]
- [80].Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. [DOI] [PubMed] [Google Scholar]
- [81].Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. [DOI] [PMC free article] [PubMed] [Google Scholar]