

Abstract
Background
High-density oligonucleotide arrays have become a valuable tool for high-throughput gene expression profiling. Increasing the array information density and improving the analysis algorithms are two important computational research topics.
Results
A new algorithm, Match-Only Integral Distribution (MOID), was developed to analyze high-density oligonucleotide arrays. Using known data from both spiking experiments and no-change experiments performed with Affymetrix GeneChip® arrays, MOID and the Affymetrix algorithm implemented in Microarray Suite 4.0 (MAS4) were compared. While MOID gave similar performance to MAS4 in the spiking experiments, better performance was observed in the no-change experiments.
MOID also provides a set of alternative statistical analysis tools to MAS4. There are two main features that distinguish MOID from MAS4. First, MOID uses continuous P values for the likelihood of gene presence, while MAS4 resorts to discrete absolute calls. Secondly, MOID uses heuristic confidence intervals for both gene expression levels and fold change values, while MAS4 categorizes the significance of gene expression level changes into discrete fold change calls.
Conclusions
The results show that by using MOID, Affymetrix GeneChip® arrays may need as little as ten probes per gene without compromising analysis accuracy.
Background
Genomics sequencing projects have rapidly generated tremendous amount of information. At the time of writing, the NCBI UniGene database [1]http://www.ncbi.nlm.nih.gov/UniGene contained 96,109 Homo sapiens clusters and 85,047 Mus musculus clusters. Predictions from the Human Genome Project [2] and Celera Genomics [3] suggest there are about 26,000–40,000 human genes. Other recent studies suggest that these numbers may be an underestimation and that the human genome appears more complicated [4]. Understanding the functions of such a large number of genes has been an unprecedented challenge for functional genomics research. As the array of hope in recent years, gene expression array technology has quickly grown into a powerful tool to chart a gene atlas in various biological sources and under various conditions in a massively parallel manner [5-7]. Facing the challenge of annotating such a huge amount of genomic data, increasing array information density and improving analysis algorithms have become two critical research areas to ensure that gene expression profiling proceeds in an efficient and cost effective manner.
Take an Affymetrix high-density oligonucleotide GeneChip http://www.affymetrix.com for example. Firstly, its human U95 series chip consists of 5 chip types with 12,000 coding clusters each, which makes it expensive to profile all the human genes in samples of interest. Can a gene chip take more genes? Comparing its U95 chip and Human 6800 chip, Affymetrix has already increased chip information density by 20% by reducing the number of probe pairs per gene from 20 to 16. Since demand for higher information density has still not been met, it is of interest to study the probe number effect in detail. Secondly, most optional research efforts focus on the downstream statistical and clustering analysis. However, on the upstream side, Affymetrix chip users are still dependent on the Microarray Suite® software that comes with the measurement system to interpret raw data. The Affymetrix algorithm implemented in its Microarray Suite 4.0 package (referred hereafter as MAS4) uses empirical rules derived from its internal research data to assign absolute calls for the significance of gene presence and assign fold change calls for the significance of expression variations. Such discrete categorizations are not the most appropriate language to describe quantities of continuous nature. Although it is well known that fold change numbers have defined behaviors of uncertainty, there are very few studies in this area. How does one assign statistical significance to expression analysis results? This work presents our preliminary research results for the two questions raised above.
The Affymetrix gene chip layout used in this study contains the same number of perfect match (PM) probes and mismatch (MM) probes. MAS4 uses differences between these two types of probes for gene expression signals. The primary goal of Match-Only Integral Distribution (MOID) algorithm is to discard mismatch information, which allows immediate doubling of the chip information density. In this study, the performance of both algorithms were benchmarked using 366 known fold change values derived from 34 spiking experiments. Their false positive tendencies were assessed by no-change expression experiments. Computer simulations were used to study their noise tolerances, and to determine the minimum number of probes required for chip analysis.
The idea of using PM-only information is based on the following observations: MAS4 essentially discards the one-one correspondence between a PM and its MM partner (for details, see materials and methods on MAS4 algorithm for absolute analysis) and still gives satisfactory interpretation, which suggests the contribution of MM probes might be approximated in a nonspecific manner overall. After we designed the first mismatch-free gene chip (GNF-HS1) in July 1999, the match-only expression analysis idea was proposed in other independent studies as well. Li (submitted, 2001) adjusted their previous model-based analysis of oligonucleotide arrays [8] to PM-only calculations and found their results correlate well with that of using PM and MM information. In addition, the idea is endorsed by recent studies of Naef et al[9] and Irizarry et al (2002, in prepare) http://biosun01.biostat.jhsph.edu/~ririzarr/papers/.
One difficulty in comparing algorithms for gene expression analysis is the lack of "known" results. Here we overcome the problem by resorting to a spiking set and set of no-change experiments, where results are unambiguous. Model-based methods [8] generally require a reasonable numbers of training experiments of the same chip type, among which probes under study give significantly large signals in at least some experiments. Considering the fact that the 34 spiking experiments used were obtained by three different chip types, and in addition some experiments are replicates under different concentrations and hybridization conditions, it is impractical to sufficiently train model-based algorithms in this study. We limit our study to MOID and MAS4, where training is not required.
In the materials and methods section, we summarize Affymetrix chip technologies and describe the MOID algorithms in detail. MAS4 algorithms for both absolute expression analysis and comparison analysis are included afterwards. The benchmarks used for algorithm comparison were explained and comparison results were shown in the results session. Issues such as noise-tolerance of both algorithms and further reduction of the probe set size are also included. We discuss generalization of the normalization algorithm, which may be of interest to other researchers. Finally, the main differences between MAS4 and MOID are summarized in a tabular form as conclusions.
Results and Discussion
Spiking experiments
Spiking experiments were done by adding to tissue samples a certain number of control genes with known concentrations. Since signal intensity is not exactly proportional to gene concentration across different probe sets, only the fold change values of each gene between comparisons are considered reliable and used to benchmark both algorithms. We are fortunate to have access to 34 spiking experiments done previously in GNF for a hybridization protocol study, where 366 independent known fold change numbers were derived. These experiments covered three Affymetrix chip types: Hu35kSubA, Hu6800, and Mu11kSubA. True experimental fold change values, fexp, range from 1.0 to 10.0. The raw data (34 CEL files) and experimental fold change values are available from http://carrier.gnf.org/publications/MOID.
We applied both algorithms to these array data and the fold change numbers calculated were compared to known experimental values. Because of the way we defined the base and experiment, fexp are always no less than 1.0. For MAS4, we used all the default parameters used by the Microarray Suite 4.0 program in the calculation. For MOID, we found using 70 percentile for both pct and pctf (defined in materials and methods) gave optimal performance. Figure 1 shows the comparison results (data available from http://carrier.gnf.org/publications/MOID). Two algorithms give virtually similar performance.
Figure 1.

Calculated fold change values for 366 known spiking data. MAS4 and MOID calculations are in blue and red open circles, respectively. The brightness of the circles corresponds to LogP, assigned by MOID. The darker the symbol, the better the data quality. For plotting purpose, values for MAS4 and MOID are slightly jittered horizontally to the left and the right, respectively, to avoid overlapping. The green line shows where calculation meets the experiment. Notice the scale for both coordinates are Log2 based. The relative errors of fold change numbers, 30% for MAS4 and 28% for MOID, show similar performance for both algorithms.
In the comparison, we defined relative error, Rerr, as the following to benchmark each algorithm,
Rerr1 = <|(fcalc - fexp>)/fexp|>all pairs,
Rerr2 = <|(1/fcalc - 1)/fexp) · fexp|>all pairs,
and
Rerr = <|(fexp/fcalc - fcalc/fexp)|>all pairs.
Where fexp and fcalc are fold change values from experiment and calculation, respectively. Rerr1 and Rerr2 are defined symmetrically for relative errors of fexp and 1/fexp, reflecting the fact that base and experiment can be defined arbitrarily, and the final Rerr is defined as the average of the two. Each relative error number is an average over all the 366 data points.
It seems the two algorithms have very close performance Table 1. They all have greater error margins for calculating down-regulated fold changes (captured by Rerr2) than up-regulated ones (captured by Rerr1). MAS4 is more asymmetric than MOID in this aspect.
Table 1.
Relative errors of MAS4 and MOID measured by 366 spiking fold change data points.
| Algorithm | Rerr1 | Rerr2 | Rerr |
|---|---|---|---|
| MAS4 | 0.23 | 0.37 | 0.30 |
| MOID | 0.25 | 0.32 | 0.28 |
No-Change experiments
Another set of experiments used for benchmarking algorithms is the comparison between two experiments where mRNA prepared from the same tissue sample was hybridized twice under slightly different conditions. Those fold change numbers that deviate largely from 1.0 are considered to be false positives. When we applied the algorithms to ten experiments done with either human brain or human lung replicate samples (same mRNA, slightly different hybridization conditions), the results were all similar. Figure 2 shows one of the typical comparison results. When only "Present" genes (defined by MAS4) were taken into account, 80% of fold change numbers were spread across 0.54 for MAS4 and only 0.30 for MOID. Clearly MOID assigns much less false positive fold changes than MAS4 does in this comparison. If all the genes were counted, the spreads of MAS4 and MOID results would be 0.89 and 0.29 for the same data sets. This test suggests MOID is more robust than MAS4.
Figure 2.

Histograms of fold change calculations on two human lung sample replicates. The two hybridizations obtained from the same mRNA source are supposed to have identical gene expression levels. The MAS4 and MOID calculations are in blue and red bars, respectively. The fold change values for MAS4 and MOID are shifted slightly to the left and right, respectively, to avoid overlapping. 80 percent of data are distributed in the range [0.80, 1.34] for MAS4, and [0.92, 1.22] for MOID. Only genes marked as "Present" by MAS4 were counted in the histogram. If all genes are taken into account, 80 percent of data are distributed in the range [0.59, 1.48] for MAS4 and [0.96, 1.25] for MOID.
Reduced Probe Set Simulation
In the studies above, we demonstrated the feasibility of a match-only gene chip design based on the MOID algorithm. In order to further increase chip information density, it is of common interest to understand how many probes are sufficient for expression analysis and push for the lower limit of the size of a probe set. This is done via computer simulations.
In the simulation, for each probe set, a subset of nr probes were randomly chosen to be used in the calculation. Both the spiking and no-change calculations presented above were repeated with the selected subset, as we gradually reduced nr. Figure 3 and figure 4 show the results for the spiking calculation and no-change calculation, respectively. As the graph suggested, accuracy of MOID is essentially unaffected while reducing the number of probes down to ten. This result enables us to almost triple the amount of clusters one can put on a gene chip using MOID design. Combined with other new design ideas, MOID lays the foundation for the first universal human chip that contains 75,000 UniGene clusters (release 116). The results have recently been validated and led to many interesting discoveries, which provides indirect support for MOID (to be submitted).
Figure 3.

Reduced probe set test for 366 known spiking data. The method is the same as described in Figure 1. The red and blue filled circles are the results for MAS4 and MOID, respectively. The error bars were derived from three independent simulations. As indicated, there is no significant deterioration in the performance of both methods down to 10 probe cells.
Figure 4.

Reduced probe set test for two human lung samples replicates previously described. During a test, a certain number of probe pairs were randomly selected for calculation. The computations were otherwise done is the same way as Figure 2. The error bars indicate the widths of fold change ranges where 80 percent of the fold change numbers fall. Only "Present" genes are considered here. There is no significant deterioration in the performance for both methods down to ten probes; MOID is in favor for all tests.
Noise Tolerance of both Algorithms
Computer simulations also enable us to study the behaviors of both algorithms under noise disturbance. In simulations, a subset of nn probes were randomly chosen, their PM values were multiplied by ten to mimic the effect of serious noise effect. We repeated the calculations for both spiking experiments and no-change experiments, while gradually increasing nn. Figure 5 and figure 6 show the results for spiking calculation and no-change calculation, respectively. As the graph suggested, both MAS4 and MOID can resist noise perturbation up to 2 cells out of 16, the test favors MOID slightly.
Figure 5.

Noise test for 366 known spiking data. During a test, a certain number of perfect matches were randomly selected and their intensities were multiplied by ten. The computations were otherwise done in the same way as in Figure 1. The MAS4 and MOID calculations are in blue and red filled circles, respectively. Error bars were derived from three independent simulations. Both methods are robust enough to stand for two noisy probes, MOID is slightly in favor for larger noise.
Figure 6.

Noise test for two human lung samples replicated previously described. During a test, a certain number of perfect matches were randomly selected, and their intensities were multiplied by ten. The computations were otherwise done in the same way as in Figure 2. The MAS4 and MOID calculations are in blue and red filled circles, respectively. The error bars represent the widths of intensity ranges where 80 percent of the fold change data fall. Both methods are robust enough to stand for two noisy probes; MOID is slightly in favor for larger noise.
Probe-Specific Effect
MOID is based on the assumption that the background for probes is mainly non-probe-specific. The results indicate that this assumption is sound overall. However, it is clear that by better understanding probe-specific behaviors, the analysis accuracy can be further improved. For instance, probe response factors might be derived by accumulating and studying many experiments. If there are a sufficient number of experiments, where the target gene is significantly present, probe response factors may be retrieved by some statistical modeling. A recent study [8] provides an important example in this direction. Researchers at Corimbia http://www.corimbia.com also developed some proprietary methods to identify "good" probes and assign different weights to them to improve data analysis. Efforts can be made along similar lines with the MOID algorithm in the future. If probe response factors can be calculated, the "bad" but "stable" probes can be scaled, and the distribution may be expected to be closer to normal, therefore expression levels and their uncertainties can be calculated in a more statistically sound manner.
Future studies for more accurate background subtraction models may also improve the fold change distribution; therefore yield better statistics for fold change evaluation as well. The relative error in spiking experiments (28%) suggests that there is room for future improvement.
LogP and Absolute Calls
In the derivation of LogP (see materials and methods, Figure 7), it is assumed that the statistics of any absent probe can be described by the general background curve B. The P value should not be literally interpreted as accurate probabilities, since it is only as good as the underlining assumption. Users should also bear in mind that a P value is for the null hypothesis that the discrepancy between observables and generic mismatches is generated by noise, which is different from the likelihood of gene presence conditioned on the observed discrepancy. However, LogP values more or less serve as a statistical indicator for the sorting of genes by their significance. The discrete absolute calls in MAS4 are determined in a completely different empirically manner. Although higher LogP values have a higher tendency of being assigned "Absent" in MAS4, the exact correspondence between the two varies from experiment to experiment. Roughly, LogP values above -3.0 have a greater chance to be called "Absent" than "Present" by MAS4. Users should be aware not to over interpret this correspondence. One interesting observation we had in this study was that discrete calls by MAS4 are not as stable as LogP values. Sometimes a gene can be reassigned to "Present" from "Absent" by MAS4 due to a small perturbation in the underlying quantities without going through a "Marginal" transitional stage.
Figure 7.

Schematic diagram of the MOID algorithm. The blue curve is the integral intensity distribution of the non-probe-specific background data B(I) for the experiment. The red curve is the integral intensity distribution of a particular probe set, where each step represents the addition of one additional probe. The maximum vertical distance, dk max, is used in the P value calculation. The horizontal intensity difference, Ek, measured at pct captures the true signal for the set optimally.
Curve Normalization
It is clear by observing various experiments that the response of intensity to signal varies in different intensity regions. Occasionally, the normalization constant nf does not cover well for all intensity values. A more generalized normalization procedure should use a normalization curve instead of a constant factor. The MOID normalization algorithm can be easily modified to the following (see [10] for a similar approach):
Step 1: include all genes in the normalization gene list.
Step 2: using the Ek values for all the genes in the list, generate integral intensity distributions for both experiments. A normalization curve NF(I) is constructed in such a way that the two integral distributions are identical after normalization.
Step 3: normalize the data sets using NF obtained in the previous step; refine the gene list by further excluding those genes whose intensity values changed by more than a certain fold, >fmax or < 1/fmax, between the two data sets.
Step 4: repeat Step 2 and update NF to NF', until one of the following conditions is met:
1) the maximum number of iterations, Itrmax, is reached;
2) max(|NF(I)/NF'(I) - 1|) ≤  max, where max is a small predefined threshold;
max, where max is a small predefined threshold;
3) size of the normalization gene list drops below a predefined threshold, Szmin.
This algorithm offers a chance to correct non-linearity in the chip system to a certain extent. To demonstrate the algorithm, we applied the procedure to two measurements, where genomics DNA sample of yeast s288c strain were hybridized onto Affymetrix YG_S98 arrays. The second experiment was a repeat of the first one after about 50 days, where some of hybridization and scanning parameters had been changed over the course. It is shown in Figure 8 that the curve normalization procedure out-performed constant normalization as expected.
Figure 8.

Comparison of results between constant normalization and curve normalization. The two data sets were both genomics DNA samples of the same yeast strain (s288c) collected using the YG_S98 Affymetrix array in about 50 days period. The intensities of the 70 percentile of each probe set were used in the Log-scale plot. Results for constant normalization and curve normalization are shown in green and red pixels, respectively. Yellow dots are shown where the pixel is colored by both red and green. As it shows, the two methods give essentially the same results for lower intensity regions, while curve normalization seems to be able to correct non-linearity better in the high intensity end.
Conclusions
MOID algorithm allows at least double or even triple the information density of current (U95 human chip) Affymetrix high-density oligo nucleotide arrays without compromising analysis accuracy. Table 2 summarizes feature comparisons between MOID and MAS4.
Table 2.
Comparison between MAS4 and MOID.
| MAS4 | MOID | |
|---|---|---|
| Mismatch used | Yes | No |
| Sensitivity to probe set size | Super-Olympic filtering is turned off when remaining probe number is less than 8. | Confidence intervals gradually increase as probe set size drops. |
| Relative error (Spiking tests) | 30% | 28% |
| Minimum of probes required (Spiking tests) | 10 | 10 |
| No-change test results (True fold change = 1.0) | 80% of fold change values in [0.80, 1.34] | 80% of fold change values in [0.92,1.22] |
| Normalization | Constant based on average intensity. Normalize once. | Allow both constant and curve normalization. Iterative scheme. |
| Problematic genes (Bright mismatches) | May underestimate expression level | May overestimate expression level |
| Expression Level | Average difference (may be negative). No confidence interval evaluations | Expression value (always positive) Lower bound and upper bound (with probe set size taken into account. |
| Present Call | A, P, M (discrete, unstable). Rules were derived from 50 μm and 24 μm feature arrays, may need update for 20 μm) | Continuous LogP |
It should be noted that at the time of this study, Affymetrix U95 chip uses 16 probe pairs per set. As Affymetirx is planning on further reducing probe set size in their next design, the density improvement by using MOID may be less than the estimation given above.
Materials and Methods
The user should refer to the Affymetrix web site http://www.affymetrix.com and the documentations that come with the MAS4 software for technical details regarding Affymetrix array and MAS4 algorithm.
Affymetrix Oligonucleotide Chip
At the time of this study, Affymetrix synthesizes 25-mer oligonucleotides on a 640 × 640 array with 20 μm feature size using photolithographic fabrication techniques (some data used in this study were collected from some previous generation arrays of 540 × 540 cells and 24 μm feature size). Each cell (also called a probe) in the array contains the same oligonucleotide sequences. As shown in Figure 9, typically a set of 16–20 probes are designed for each targeting cluster (called a probe set, or sometimes a "gene".). An expression value will be derived per probe set as the result of analysis. Probes taken as fragments from a target sequence are called perfect matches (PM). Multiple matches per set serve as independent signal detectors and provide a possibility to capture statistical uncertainties. For each match, there is also a corresponding mismatch (MM) probe, whose sequence differs from its match by a single base in the middle. Mismatches are meant to detect cross hybridization components of their corresponding matches, and are used by MAS4 for probe-specific background subtraction. There are also several Affymetrix control sets on each chip used for quality references, which were used in the spiking experiments to validate and refine hybridization protocols.
Figure 9.

Schematic diagram of current Affymetrix GeneChip design.
Intensity Distribution
In an ideal case, all the probes in a set should give similar signals and serve as replicate measurements. One common rule in probe design is to select probes with similar melting temperatures to minimize the variances among probes. The chemistry involved in a hybridization experiment is often too complicated to be predicted computationally at the current stage, therefore in reality probe intensity distribution for a set is usually fairly wide and has a long tail, which causes all kinds of difficulty for data analysis.
One usual attraction in expression analysis is to assume a normal distribution for probe intensities. This hypothesis can be examined by the following calculation. For each probe set, we applied a linear transformation to probe intensities, so that the mean and the standard deviation of the intensities in the set were normalized to zero and one, respectively. Then all the resulting match intensities were used to generate an overall distribution. Our calculation shows the normal distribution assumption is not an appropriate one. When similar analysis was applied to mismatch intensities and mismatch-subtracted match intensities, the conclusion stays more or less the same.
Despite the fact that the intensity distribution of a set is non-Gaussian, it is still crucial to find out the distribution function for match or mismatch intensities to generate meaningful statistics tests, if such distributions actually exist. The authors tried several analytical functions on match and mismatch probe intensities. Unfortunately, the distributions seemed to be quite "bad", the tail portions even fade out slower than the extreme value distribution. It was found that distributions of mismatch signals also significantly depended on the sample and experimental conditions. One may reasonably suspect that cross hybridization, intensity saturation, overall sample concentration, chip production irregularities, and miscellaneous noise may all cause a change in the signal distribution. The study seems to suggest the distribution of cross hybridization should be taken from each experiment as a result of measurement instead of using an a priori determined analytical expression. This guideline is used in MOID algorithm.
MOID algorithm: hypothesis and principles
As mentioned in the intensity distribution analysis, MOID assumes cross hybridization behavior of probes is mainly non-probe-specific, and therefore all the match cells can share the same cross hybridization background. MOID primarily aims at saving 50% of the chip space currently dedicated to mismatch cells in the Affymetrix design under this study. It was also discussed before that cross hybridization distributions should be taken from experimental data instead of from some analytical function. Based upon the general belief that there is always a significant amount of genes unexpressed in any mRNA experiment, we will use the match cell signals from the 5% darkest probe sets in the current public Affymetrix arrays to prove the point that it is possible for MOID not to use any mismatch signal. For the next generation gene chip designed based on MOID algorithm, some designated probes may be used as generic mismatches to collect non-probe-specific cross hybridization data. In fact, on GNF-HS1, 16,460 probes from 476 viral genes are used for such a purpose.
As discussed, the distributions of quantities in expression analysis are usually asymmetric and contain a long tail, regardless whether it is match intensity, mismatch intensity, or average intensity difference. For such abnormal distributions, concepts like mathematical average and standard deviation are not the most appropriate statistical terms to use. Pre-filtering the data, like what MAS4 does, certainly helps reshape the distribution, but by no means this can bring the distribution back to normal without throwing out a significant portion of measurements. The MOID algorithm is specifically designed to avoid these problems by using percentile instead of mean and using confidence intervals instead of standard deviation in all possible cases.
MOID algorithm for significance of gene presence
A non-probe-specific integral background distribution (representing noise, cross hybridization, and possible other factors), B(I), is derived from the intensities of the 5% darkest probe sets (or from generic mismatches in the future). I stands for intensity; B satisfies boundary conditions: B(0) = 0 and B(∞) = 1. For each probe set k, the match signals are sorted and the integral distribution, Sk(I), is generated as well. Figure 7 is a schematic diagram.
If the gene represented by a probe set is absent, the intensity data from matches are due to pure background contribution, therefore Sk is likely to be close to B. We choose the Kolmogorov-Smirnov test to determine likelihood that the observed signal distribution, Sk, could be explained by B. If we define dk max as the maximum vertical distance between B and Sk, according to K-S statistics, the probability of observing discrepancies greater than dk max is determined by a P value [11],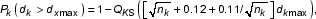
where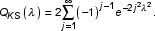
nk is the effective number of data points in the K-S statistics, which is the number of PM for gene k in our case. P carries the meaning of probability, therefore is a number between 0 and 1. Practically, Log10P, a negative value, is used as our final representation. In this way, MOID uses a continuous LogP criterion to replace MAS4 absolute calls. Those signal sets that can be easily explained by noise are assigned a LogP value closer to zero.
MOID algorithm for expression level calculation
MOID uses the horizontal distance between Sk and B to represent gene expression level (Figure 7). Since the darkest probes may be more likely caused by their poor binding properties to the target gene, and the brightest probes may be more likely caused by serious cross hybridization issues, different parts of the integral distribution tend to have different qualities. Therefore, the MOID algorithm uses the horizontal distance measured at a certain percentile, pct, instead of the whole curve. Based on the analysis of the spiking experiments discussed later, it is empirically determined that 70% is the optimal percentile for signal retrieval. We also tried to use the average of horizontal distances from several pct, but without significant improvement. Therefore the expression level Ek, related to gene concentration for gene k, is defined as
Ek = max(I|Sk = pct - I|B = pct, 0).
Instead of using the standard deviation, we take confidence intervals directly from the distribution curve Sk, with the background subtracted. E.g., 80% confidence intervals can be represented by a lower bound (Ekl) at 10 percentile and an upper bound (Eku) at 90 percentile of the distribution, i.e.,
Ekl0 (0.1) = max (I|Sk = 0.1 - I|B = pct, 0)
and
Eku0 (0.9) = max (I|Sk = 0.9 - I|B = pct, 0).
As one might expect, increment of the probe set size helps narrowing down the confidence intervals in a manner determined by the statistics of probe distribution. However, this piece of information is unknown and we took an ad hoc approach by modifying the boundaries formulae as the following: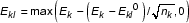
and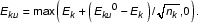
MOID algorithm for normalization
The most common way of understanding gene functions by expression profiling is to study the change of their expression levels in various disease states and cellular environments. The observed expression level is a product of complicated protocols, and is subjected to various factors from sample preparation to final image scanning. Therefore, it is a common practice to normalize expression data drawn from multiple profiles before making any reliable interpretation.
The normalization procedure in MAS4 aims at normalizing the average intensities of probe sets (excluding the top and bottom 2%). To avoid possible signal contamination, MOID takes similar approach by normalizing the probe sets between 10 and 90 percentiles. However, it is noticed that an ideal normalization factor should be derived from only those genes that do not change their expression levels between the two experiments. MAS software provides the possibility to use a list of housekeeping genes for such purposes; however, it certainly requires careful downstream research to validate any such a list. Some common normalization criteria were summarized in a recent study by Zien et al. [12]. MOID uses a heuristic bootstrap method to identify an approximate list of unchanged genes between two data sets:
Step 1: include all genes as the normalization gene list.
Step 2: sort the Ek values (already background subtracted) for all the genes in the list; the interesting portion of expression values (between 10% and 90% in our case) is retrieved and average intensity is calculated for each experiment, respectively. The initial normalization factor nf is calculated by the ratio of the two average intensities.
Step 3: normalize the data sets using nf obtained in the previous step; refine the gene list by further excluding those genes, whose intensity values changed by more than a certain fold, >fmax or < 1/fmax, between the two data sets.
Step 4: repeat Step 2 and update nf to nf', until one of the following conditions is met:
1) the maximum number of iterations, Itrmax, is reached;
2) |nf/nf' - 1| ≤ max, where max is a small predefined threshold;
3) size of the normalization gene list drops below a predefined threshold, Szmin.
In practice, a single iteration is usually sufficient for most comparisons. User can adjust threshold parameters towards their preferred stringent levels. In this study we use fmax = 2.0, max = 0.05, Itrmax = 5, Szmin = 1000.
MOID algorithm for comparison analysis
Different probes within the same probe set generally respond very differently to mRNA sample fragments. We examined various probe properties such as melting temperature, nucleotide base composition, possible sequence motifs, and potential secondary structures in order to understand the cause of such diverse response properties. That study did not find any conclusive factor that explains observed probe signal distribution satisfactory. Although it seems rather difficult to pre-compute and predict probe responses, it is possible to derive some features afterwards for a probe based on a significant amount of expression data where the target gene of interest is present [8]. Instead of using a modeling approach to explain various signal contributions, MOID uses a new approach to avoid the affect of diversities of probe response factors in calculating expression fold changes.
Let us assume any two probes in a probe set k of size nk always respond to their target gene concentration with factor r1 and r2, respectively. That is, if the gene is present at concentration c1 and c2 in two hybridizations, the two probes in average give signal intensities r1c1 and r2c1 in the first experiment, r1c2 and r2c2 in the second experiment. As in MAS4, fold change is calculated by the ratio between r1c2 + r2c2 and r1c1 + r2c1, where the result is essentially dominated by the probes with the largest response factors, and the statistics of the ratio becomes difficult to estimate. However, we observe that by taking the ratio of the two signals for each probe individually, one essentially is looking at nk independent measurements of c2/c1 for that probe set. This opens a possibility for obtaining a distribution of fold change values.
If one assumes most probes have a rather stable intrinsic response factor, r, a fold change number can be calculated from each probe i in the set independently, which hopefully removes the affect of the unknown response factors ri. However, it seems the response factor of each probe has a non-negligible intrinsic spread as well; this may be further complicated by alternative splicing and tissue-specific cross hybridization issues. In addition, the nk ratio numbers calculated for a probe set are not normally distributed (it is well known that even the ratio of two normally distributed values is no longer normally distributed). The current background subtraction algorithm may not take fully into account response factor-unrelated signals, therefore could further increase the non-normal component. To overcome this difficulty, MOID uses a percentile, pctf, of the integral distribution formed by the nk fold change numbers, Fk(f), which satisfied the boundary conditions: F(0) = 0 and F(∞) = 1. Empirically, 70% is used for pctf as determined by spiking experiments discussed later. The confidence intervals are also directly taken from relevant percentiles in the distribution corresponding to the lower bound and upper bound, respectively. The effect of the number nk is taken into account in the same heuristic manner as we did previously for absolute analysis. The final formulae are: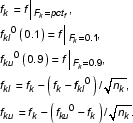
MAS4 Algorithm for Absolute Analysis
After a sample is hybridized to probes on a chip, the chip is scanned and fluorescent signals are collected and stored in an Affymetrix DAT file. For cell i, after filtering out boundary pixels, Ni numbers of pixels are used to calculate an average intensity value Ii and standard deviation σi. Hereafter we index a probe set by k, which contains nk probe pairs. The calculated intensity values for thejth pair in the kth probe set are denoted as PMkj for PM and MMkj for MM. Background intensity, bg, is derived from the average intensity of the 2% darkest cells. Noise level, Q, is determined as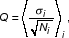
where average is done over all background cells.
MAS4 assumes that the cross hybridization component of a match is captured by its mismatch companion, therefore the difference, PMk - MMk, is used to derive expression levels. MAS4 also uses a "super-Olympic scoring" procedure to iteratively filter out outliers, defined as those probe pairs with difference values more than three times the standard deviation away from the mean. After filtering, the average difference value for a probe set, Dk, is used to represent the expression level of that particular gene.
The significance of gene presence is determined by an empirical absolute call decision matrix coupled with proprietary MAS4 algorithms, parts of which are described in the Microarray Suite documentation. As the result, a probe set is marked as either "Present", "Absent", or "Marginal".
MAS4 calculates average difference by
Dk = <PMkj - MMkj >j,
which is essentially
Dk = <PMkj >j - <MMkj >j,
where the average is taken over all the probe pairs surviving the super-Olympic filtering process mentioned above. Based on this observation, the one-one correspondences between PMkj and MMkj, typically with an average correlation coefficient around 0.8, are essentially lost during such averaging. Since MAS4 is successful in various applications despite the fact it averages out probe-specific mismatch signals, we hypothesize that contributions of the mismatch probes are mainly probe-nonspecific, if the pairwise correspondence between PMkj and MMkj are not enforced. This observation led to the idea of only using match probes in the MOID algorithm discussed above.
MAS4 Algorithm for Normalization
MAS4 introduces a normalization factor (a scaling factor in MAS4 serves the same purpose). The goal of normalization is to reduce false positives in the expression fold change calculation and ensure housekeeping genes are marked as unchanged. Since it is usually unknown a priori what genes sustain their expression level during the two experiments, different heuristic normalization schemes may be adopted and it is unclear at this point which particular approach is optimal. MAS4 normally derives the normalization factor by scaling average Dk for all genes with expression levels within the central 96% to a certain target intensity constant. This algorithm is based on the assumption that the total copy of mRNA per cell is a conserved number among experiments.
MAS4 Algorithm for Comparison Analysis
In comparison of two chips, we follow Affymetrix convention of calling the reference chip measurement as the base (b), the other one as the experiment (e). After the above normalization procedure, the fold change value for gene k, fk, is essentially calculated by dividing Dk (e) with Dk (b). MAS4 uses a filtering process to ensure that a common subset of "good" probe pairs between b and e are used to recalculate Dk. Very weak expression signals (sometimes negative) are rounded to the noise level mentioned before. The final formula for f is:
fk = [Dk(e) + max (Q - Dk(b), 0)] / max (Dk(b), Q), if Dk(e) ≥ Dk(b),
or
fk = max (Dk(e), Q) / [Dk(b) + max (Q - Dk(e), 0)], otherwise.
An empirical difference call decision matrix and a proprietary MAS4 algorithm, partly described in the Microarray Suite 4.0 documentation, determine the significance of the fold change. As a result, the probe set is marked as "Increase", "No Change", "Decrease", "Marginally Increase", or "Marginally Decrease". It is obvious that the fold change number is only meaningful if the probe set is clearly present at a level above the noise in both data sets.
List of Abbreviations
MOID - match-only integral distribution
MAS4 - Affymetrix algorithms implemented in its Microarray Suit 4.0 software
PM - perfect match
DMM - mismatch
Contributor Information
Yingyao Zhou, Email: zhou@gnf.org.
Ruben Abagyan, Email: abagyan@scripps.edu.
Acknowledgements
We thank David Lockhart for introducing us to the field of gene expression and many helpful discussions about the MAS4 analysis algorithms; Lisa Wodicka and Helin Dong for providing spiking data; David Balaban, and Paul Kaplan for stimulating discussions about the MOID algorithm; Elizabeth Winzeler for providing yeast genomics experiments for curve normalization study. Chris Benner for careful reading of the manuscript.
References
- Schuler GD. Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J. 1997;75(10):694–698. doi: 10.1007/s001090050155. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1086/172716. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA. The Sequence of the Human Genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Hogenesch JB, Ching KA, Batalov S, Su AI, Walker JR, Zhou Y, Kay SA, Schultz PG, Cooke MP. A comparison of the Celera and Ensembl predictied gene sets reveals little overlap in novel genes. Cell. 2001;106:413–415. doi: 10.1016/s0092-8674(01)00467-6. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Che MS, Mittmann M, Wang C, Kobayashi M, Horton H. Expression monitoring by hybridization to high-density oligonucleoiide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Lander ES. Array of Hope. Nature Genet. 1999;21:3–4. doi: 10.1038/4427. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. PNAS. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naef F, Lim DA, Patil N, Magnasco M, From features to expression. High-density oligonucleotide array analysis revisited (to appear), the Proceedings of the DIMACS Workshop on Analysis of Gene Expression Data. 2001. http://xxx.lanl.gov/abs/physics/0102010
- Schadt E, Li C, Su C, Wong WH. Analyzing high-density oligonucleotide gene expression array data. J. 2000;80:192–202. doi: 10.1002/1097-4644(20010201)80:2<192::AID-JCB50>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes in C. second. Cambridge University Press; 1996. pp. 623–626. [Google Scholar]
- Zien A, Aigner T, Zimmer R, Lengauer T. Centralization: a new method for the normalization of gene expression data. Bioinformatics. 2001;17 Suppl.1:S323–331. doi: 10.1093/bioinformatics/17.suppl_1.s323. [DOI] [PubMed] [Google Scholar]